

Abstract
This paper reviews the distinctive roles played by the transcriptional coactivators CREB-binding protein (CBP) and p300 in Wnt/β-catenin signaling and cell physiology in colorectal cancer (CRC). Specifically, we focus on the effects of CBP- and p300-mediated Wnt activity on (1) neoplastic progression; (2) the activities of butyrate, a breakdown product of dietary fiber, on cell signaling and colonic cell physiology; (3) the development of resistance to histone deacetylase inhibitors (HDACis), including butyrate and synthetic HDACis, in colonic cells; and (4) the physiology and number of cancer stem cells. Mutations of the Wnt/β-catenin signaling pathway initiate the majority of CRC cases, and we have shown that hyperactivation of this pathway by butyrate and other HDACis promotes CRC cell apoptosis. This activity by butyrate may in part explain the preventive action of fiber against CRC. However, individuals with a high-fiber diet may still develop neoplasia; therefore, resistance to the chemopreventive action of butyrate likely contributes to CRC. CBP or p300 may modify the ability of butyrate to influence colonic cell physiology since the two transcriptional coactivators affect Wnt signaling, and likely, its hyperactivation by butyrate. Also, CBP and p300 likely affect colonic tumorigenesis, as well as stem cell pluripotency. Improvement of CRC prevention and therapy requires a better understanding of the alterations in Wnt signaling and gene expression that underlie neoplastic progression, stem cell fate, and the development of resistance to butyrate and clinically relevant HDACis. Detailed knowledge of how CBP- and p300 modulate colonic cell physiology may lead to new approaches for anti-CRC prevention and therapeutics, particularly with respect to combinatorial therapy of CBP/p300 inhibitors with HDACis.
Keywords: CREB-binding protein, p300, Wnt, Colorectal cancer, Butyrate, Stem cells
Core tip: Deregulated Wnt/β-catenin signaling is responsible for initiating most human colorectal cancer (CRC), and hyperactivation of this pathway by histone deacetylase inhibitors such as butyrate, derived from dietary fiber, promotes CRC cell death. The transcriptional cofactors CREB-binding protein (CBP) or p300 affect Wnt signaling and the hyperactivation of this pathway by butyrate. CBP and p300 likely affect colonic tumorigenesis, stem cell fate, and butyrate resistance in CRC. Therefore, pharmacological or genetic methodologies that target CBP- and p300-mediated Wnt activity are possible preventive or therapeutic approaches against CRC.
WNT SIGNALING, BUTYRATE, AND CREB-BINDING PROTEIN /P300
Dietary fiber, colorectal cancer, and Wnt signaling
The anti-colorectal cancer (CRC) protective effect[1-5] of dietary fiber has been attributed to its fermentation product butyrate[6-10], a histone deacetylase inhibitor (HDACi)[11]. HDACis, which are current or proposed chemotherapeutic agents, induce apoptosis[1,2,12-14], CRC cell cycle arrest, and/or differentiation in vitro[1,2,15-17]. Deregulated Wnt/β-catenin signaling[18-23], involving β-catenin and Tcf/Lef factors[23,24], is responsible for most CRCs[25-27], and in these cells HDACis hyperactivate the Wnt/β-catenin pathway[1,2,17]. Exciting new findings[18] suggest one mechanism whereby this hyperactivation takes place at Wnt target genes. Thus, the repressor p15RS inhibits Wnt/β-catenin activity by recruiting HDAC2 to Wnt target gene promoters, where it associates with Tcf4 and maintains histone H3 in a deacetylated state. In this scenario, β-catenin-Tcf4 association is disrupted and transcription from Wnt target gene promoters is repressed; HDACis interfere with the activity of p15RS and HDAC2 at these promoters, upregulating canonical Wnt transcriptional activity[18]. Wnt hyperactivation has important consequences for CRC cell physiology. Butyrate and other HDACis induce CRC cell apoptosis, and we have shown that Wnt signaling hyperactivation promotes high levels of apoptosis[1,2,17]. Thus, there is a causative linear relationship between fold-induction of Wnt activity and the degree of apoptosis in CRC cells exposed to butyrate. In agreement with these findings, abrogation of Wnt/β-catenin activity in CRC cells markedly reduces the levels of butyrate-induced apoptosis[1]. To summarize, Wnt/β-catenin signaling, which is deregulated by mutations, leads to moderate Wnt activity levels that promote colonic cell proliferation and tumorigenesis; however, relatively high and relatively low levels of the same signaling activity lead to increased CRC cell apoptosis and repressed cell proliferation[1,2,17,28,29].
CREB-binding protein, p300, butyrate, and Wnt signaling
The physical association between β-catenin and the histone acetylases CREB-binding protein (CBP) and p300[30-34], which are cotranscriptional factors[35-39], influences Wnt/β-catenin signaling[30-33]. CBP-mediated Wnt signaling has been associated with colonic cell proliferation, and p300-mediated Wnt activity promotes differentiation[30-39]. The interaction between CBP and Wnt signaling can be evaluated utilizing the small molecule inhibitor ICG-001 that binds to CBP but not to p300[30]. ICG-001 downregulates Wnt transcriptional activity in CRC cells by suppressing the association between CBP and β-catenin without blocking p300/β-catenin association[30]. Treatment of cells with ICG-001 enhances p300/β-catenin association at the expense of CBP/β-catenin association, promoting cellular pathways that favor differentiation and/or apoptosis and repress cell proliferation[30-33]. Thus, ICG-001 inhibits CRC cell proliferation, increases apoptosis as measured by caspase activity, and inhibits expression of the anti-apoptotic factor survivin[30,33]. A water soluble version of ICG-001 reduced the formation of intestinal neoplasms in the APCMin mouse model of CRC[30], and the second generation CBP-Wnt inhibitor PRI-724 is in clinical trials[31,40]. Thus, although CBP and p300 have pleiotropic effects on gene expression and cell physiology, these findings clearly indicate differential roles for CBP- and p300-mediated Wnt signaling in colonic cell physiology. In summary, these studies suggest that Wnt signaling can be divided into CBP-mediated and p300-mediated components, with CBP-Wnt activity promoting cell proliferation, while p300-Wnt activity is associated with cell differentiation and, possibly, apoptosis.
HDACis are promising agents against malignancies[15,16], including CRC. In addition to the already recognized therapeutic uses of synthetic HDACis, the production of the HDACi butyrate from dietary fiber in the human colon and the effects of butyrate on CRC cells suggest that dietary HDACis play a role in CRC prevention[1,2,17,28,29]. HDACis may influence the ability of CBP and p300 to modulate Wnt/β-catenin activity and affect colonic cell physiology. Therefore, studies have been conducted to evaluate how CBP and p300 activity influence butyrate-mediated effects on Wnt activity and cell physiology in colonic adenoma and butyrate-sensitive and butyrate-resistant colonic carcinoma cells[41-43].
While all studied human CRC cells expressed CBP[41,42], differences in p300 expression have been detected. Thus, butyrate-sensitive HCT-116 and SW620 CRC cells express p300; butyrate-resistant HCT-R cells, which were derived from HCT-116 cells, do not[41,42]. Treatment with ICG-001, which specifically targets the association of CBP, but not of p300, with β-catenin, inhibited basal and butyrate-induced Wnt signaling in all CRC cell lines studied[41-43]. Both ICG-001 and butyrate were shown to reduce HCT-116 and SW620 cell proliferation; however, ICG-001 interfered with butyrate-induced apoptosis in SW620, but not HCT-116, CRC cells[41]. Butyrate-mediated apoptosis in SW620 cells was reduced by ICG-001, but it was not completely abrogated, since (1) p300-mediated Wnt activity remains intact after treatment with ICG-001[30]; and (2) butyrate exerts both Wnt signaling-dependent and Wnt signaling-independent effects on CRC cell apoptosis[1,2]. Most likely, the differential modulation of butyrate-mediated apoptosis by ICG-001 in the CRC cell lines is at least partially due to the relative utilization of CBP-mediated vs p300-mediated Wnt activity in each CRC cell line.
Modulation of cell cycle progression and proliferation can also affect apoptosis. Therefore, the differential effects of ICG-001 on butyrate-induced apoptosis in CRC cells could be due to the variable effects of ICG-001/butyrate treatment on cell proliferation, through modulated expression of the cell cycle inhibitor p21 and the anti-apoptotic factor survivin (a gene targeted by CBP-mediated Wnt signaling)[33,41]. Significantly, ICG-001 has more marked effects on proliferation and cell cycle arrest in SW620 cells than in HCT-116 cells, and the effects of butyrate and ICG-001 on p21 and survivin expression differ between these two cell lines[41].
ICG-001 was shown to repress butyrate-induced Wnt activity, and knockdown of p300 with siRNA also repressed butyrate-mediated Wnt signaling[42]; therefore, CBP and p300 activities are both required for the hyperactivation of Wnt signaling by butyrate. It is likely that butyrate upregulates separate CBP- and p300-mediated components of Wnt activity, which both contribute to the overall hyperactivation of Wnt signaling observed in butyrate-treated CRC cells. Targeting CBP vs p300 activity can have different effects on butyrate-mediated changes in CRC cell physiology. For example, unlike ICG-001 treatment, partial p300 knockdown did not affect HCT-116 or SW620 CRC cell proliferation in the presence or absence of butyrate[42]; this lack of effect may be due to differences in the function of CBP and p300 in these cell lines. However, the role of p300 in butyrate-mediated CRC cell proliferation is likely cell type-specific; thus, findings from CRC cells that naturally lack p300 expression suggest that p300-Wnt activity is required for optimal butyrate-mediated repression of cell proliferation (see below)[42]. Thus, upregulation of the specific p300-mediated component of Wnt signaling by butyrate may be responsible for that agent’s activity in promoting CRC cell apoptosis, consistent with previous reports that p300-Wnt activity is associated with differentiation (and possibly apoptosis), while CBP-Wnt activity is more pro-proliferative[30-33]. The relative effects of butyrate upregulation of CBP-Wnt vs p300-Wnt activities likely determine the final cell fate, and would differ on a cell-type dependent basis. Thus, in summary, modulating distinct CBP-mediated and p300-mediated components of Wnt/β-catenin activity (e.g., by ICG-001, siRNA, HDACis) may differentially affect CRC cell physiology (e.g., proliferation vs differentiation/apoptosis cell fate) in a cell type-specific manner[41-43].
Combinatorial therapy and effects on apoptosis
Both relatively high and low levels of Wnt/β-catenin activity can promote apoptosis of colonic cells with activating mutations in the pathway[1,2,17,28,29]. Interference between butyrate and ICG-001 on apoptosis in certain CRC cells can be explained by the fact that butyrate upregulates Wnt activity; whereas, ICG-001 suppresses that activity. The hyperactivation of Wnt signaling by butyrate is responsible for part, albeit not all, of that agent’s ability to promote CRC cell apoptosis[1]; on the other hand, ICG-001 can activate apoptosis by repressing Wnt activity[30-33]. However, in certain CRC cell lines butyrate and ICG-001 cooperate to enhance Wnt activity-dependent effects on apoptosis and proliferation, and this requires explanation.
One possibility is that ICG-001 has both Wnt activity-independent as well as Wnt activity-dependent effects on colonic cell physiology. Thus, one study suggested that ICG-001 may enhance apoptosis in multiple myeloma cells in a Wnt activity-independent manner[44], although that study did not distinguish between the effects of CBP-Wnt and p300-Wnt activity. However, it has been shown that in colonic cells ICG-001 has significant Wnt-dependent effects[30-43]. Thus, a Wnt activity-specific mechanism to explain our findings is that ICG-001, a specific inhibitor of CBP-mediated Wnt signaling, represses the CBP-mediated Wnt activity required for cell growth; whereas, leaving unaffected p300-mediated Wnt signaling that promotes colonic cell differentiation and apoptosis[41-43]. Further, it is the fold-change in Wnt activity, and not its absolute levels, which correlates to butyrate’s effects on cell proliferation and apoptosis[1]. Thus, whereas ICG-001 suppresses the absolute level of Wnt/β-catenin signaling, butyrate retains the ability to induce a fold-increase in Wnt activity even in the presence of ICG-001[41-43].
Thus, combinatorial treatment of colonic cells with ICG-001 and butyrate inhibits CBP-mediated Wnt signaling, a pathway responsible for cell proliferation, whereas, it maintains fold-induction of p300-mediated Wnt signaling, a pathway that enhances differentiation[30-33] and apoptosis in certain cell types[41-43]. On the other hand, in butyrate-resistant CRC cells in which butyrate does not increase Wnt signaling levels and does not induce apoptosis, ICG-001-like agents enhance apoptosis[42] by repressing CBP-mediated Wnt activity below the threshold required for CRC cell viability. Finally, both butyrate and ICG-001 can have Wnt-independent, as well as Wnt-dependent, effects on cell apoptosis and proliferation. Therefore, in some cell lines the Wnt-independent effects of butyrate/HDACis and ICG-001 on apoptosis may be additive while, at the same time, the Wnt-dependent effects of these agents may counteract each other.
ICG-001 and high/low-Wnt activity fractions in CRC
Modulation of the relative levels of CBP vs p300 activity can affect tumor behavior by influencing the cell populations within a neoplasm. CRC cell populations in culture can be divided into fractions with high and low Wnt activity, and the number of cells with high Wnt activity is increased by butyrate treatment[1,2]. CRC cells with hyperactivated Wnt signaling commit to apoptosis[1,2]; whereas, cells with moderate but constitutive levels of Wnt signaling have been associated with more aggressive CRC phenotypes[45]. An EGFP reporter for Wnt activity combined with flow cytometry analysis of the transfected cells was utilized to evaluate the effects of ICG-001 treatment on Wnt activity fractions in CRC cell populations. This experimental approach revealed that CBP-mediated Wnt signaling is absolutely required for the maintenance of high Wnt activity and low Wnt activity fractions in CRC cell lines, and that ICG-001 abrogates the ability of butyrate to increase the number of CRC cells with high Wnt signaling levels[41]. These findings suggest a possible CRC therapy approach of modulating CBP-mediated Wnt activity in order to transition CRC cells to less tumorigenic types.
NEOPLASTIC PROGRESSION
Repression of CBP-mediated Wnt/β-catenin activity by ICG-001 has greater effects on butyrate action on apoptosis and proliferation in SW620 cells than in HCT-116 cells[41]. SW620 cells are derived from a CRC metastasis and HCT-116 cells are derived from a primary CRC. One possibility is that CBP-mediated Wnt activity, which is associated with cell proliferation and pluripotency[30-33,40], has a more central role (i.e., it is utilized to a greater extent) in later, advanced stages of neoplastic development. Although studies evaluating different forms of cancer come to variable conclusions about the prognostic value of p300 expression, the most recent study involving colon cancer showed that overexpression of nuclear p300 was associated with a favorable prognosis (disease-free survival rate for patients with colon cancer)[46]. This finding is generally supportive of the general hypothesis that p300-mediated Wnt activity is associated with a lesser degree of neoplastic progression, while CBP-mediated Wnt activity is associated with more aggressive and advanced cancer. The differential effects of CBP and p300 on cell physiology during neoplastic progression may derive from altered expression of genes targeted by CBP-mediated and p300-mediated Wnt activity. For example, survivin, c-myc, and cyclin D1 are all genes targeted by Wnt/β-catenin signaling, and their expression is controlled by the relative levels of CBP vs p300 activity; furthermore, the products of these genes directly influence decisions of proliferation, differentiation, and apoptosis. Thus, survivin expression is stimulated by CBP and repressed by p300, expression of cyclin D1 is stimulated by CBP and unaffected by p300, and expression of c-myc is stimulated by p300[33]. The anti-apoptotic protein survivin and the pro-proliferative cyclin D1 may mediate more tumorigenic/aggressive cell phenotypes associated with CBP-mediated Wnt activity[30,33]. The role of c-myc expression in the downstream consequences of p300 activity is uncertain, as increased expression of c-myc is usually associated with tumorigenesis. However, c-myc can, in certain contexts, promote differentiation and apoptosis, consistent with the pro-differentiation role suggested for p300-mediated Wnt activity[30,33].
In vitro studies evaluating the effects of butyrate have typically utilized CRC cells; however, diet-derived butyrate is likely most effective against early stage colonic neoplasia[1,2]. In this context, it is important to note that microadenoma LT97 cells, isolated from the earliest stage of colonic neoplasia[3,4], have been found to be extremely sensitive to the growth-suppressing[4] and apoptosis-inducing[43] effects of butyrate. These cells were used to determine the role of CBP- and p300-mediated Wnt signaling in modulating the effects of butyrate. Similar to what was observed in colon carcinoma cells, ICG-001 repressed the butyrate-induced hyperactivation of Wnt signaling in LT97 adenoma cells. Unlike late-stage metastatic SW620 CRC cells, cotreatment of LT97 cells with ICG-001 does not repress butyrate-mediated apoptosis. Proliferation of LT97 cells was more affected by butyrate than by ICG-001, and expression of the anti-apoptotic product of the survivin gene was unaffected by ICG-001 treatment.
Differences in apoptotic levels between ICG-001/butyrate-treated LT97 adenoma cells and metastatic SW620 CRC cells may be due to: (1) the relative role of CBP-mediated and p300-mediated Wnt signaling; and/or (2) the levels of factors such as survivin (a target gene of CBP-mediated Wnt signaling), p21, and other Wnt signaling-targeted genes. Thus, the repression of butyrate-induced apoptosis in ICG-001 – treated SW620 cells, compared to LT97 cells, is consistent with a more prominent role of CBP-mediated Wnt activity in SW620 CRC cells, and a more prominent role of p300-mediated Wnt activity (not affected by ICG-001) in LT97 cells. ICG-001 represses butyrate-induced Wnt transcriptional activity in both SW620 and LT97 cells, and this finding could be reconciled by the possibility that p300-mediated Wnt activity (that is not affected by ICG-001) is utilized to a greater extent in LT97 than in SW620 cells. In addition, the lack of effect of ICG-001 on the expression of survivin, a gene targeted by CBP-mediated Wnt activity[33], in LT97 cells[43] also suggests that CBP-mediated Wnt signaling has a lesser role in these cells compared to later-stage, metastatic SW620 cells. This explanation is supported by the higher RNA expression levels of survivin in SW620 cells compared to LT97 cells, as established by microarray analysis[43]. In addition, exogenous overexpression of CBP stimulates the butyrate-induced hyperactivation of Wnt signaling in LT97 cells[43], but not in SW620 cells[42], a finding that suggests saturation of CBP-mediated Wnt activity in SW620 but not in LT97 cells.
In summary, findings reported in the literature are consistent with neoplastic progression being affected by the relative role/activity of CBP-mediated and p300-mediated Wnt signaling. This possibility is supported by the following findings: (1) most of the analyzed CRC cell lines express CBP but some do not express p300[41-43]; (2) p300-mediated Wnt signaling is associated with cell differentiation; whereas, CBP-mediated Wnt signaling is associated with cell proliferation[30,33]; (3) CBP is the preferred binding partner for β-catenin in CRC cell lines[30,41]; (4) expression of survivin, a gene targeted by CBP-mediated Wnt activity, is inhibited by ICG-001 in CRC cells but not in LT97 cells, suggesting a greater utilization of CBP-mediated Wnt activity in later stages of colonic neoplasia; and (5) overexpression of CBP in LT97 cells enhances butyrate-induced hyperactivation of Wnt signaling[43]; whereas, no similar effect is observed in CRC cells[42], suggesting that endogenous CBP-mediated Wnt signaling is saturated in CRC cells, but not in LT97 adenoma cells. In addition, a recent report[47] suggests that CBP can interact with thymine DNA glycosylase to enhance Wnt signaling and proliferation of colonic cells. This finding suggests that it is likely that a number of signaling pathways and enzymatic factors cross-talk with CBP and p300 to influence decisions of proliferation vs differentiation/apoptosis, and underscores the importance of CBP activity in promoting Wnt activity-driven proliferation of human colon cancer cells. An interpretation of these findings is that a more advanced neoplastic phenotype is characterized by enhanced expression of genes targeted by CBP-mediated Wnt activity (e.g., survivin, cyclin D1). Transcription reporter assays and microarray findings[1,2,45,48,49] clearly have established that HDACis hyperactivate Wnt signaling; however, certain Wnt activity-targeted genes exhibit downregulated expression due to both Wnt signaling-dependent and Wnt signaling-independent mechanisms[45,48,49]. For example, expression of survivin, c-myc, and cyclin D1 is typically repressed in HDACi-treated CRC cells[48]. Therefore, one possibility is that both up- and downregulated expression of Wnt activity-targeted genes in HDACi-treated colonic neoplastic cells is influenced by the relative levels of CBP-mediated vs p300-mediated Wnt activity, and the relative levels of these activities in different colonic cells determine the direction and magnitude of the effects of HDACis on gene expression. These conjectures need to be carefully evaluated in future studies.
Such additional studies will require use of other colonic cell lines, particularly those intermediate on the neoplastic spectrum between the LT97 and SW620 lines. Another cell line useful for evaluating neoplastic progression is SW480. The SW480 cell line was derived from a primary tumor from the same patient from whom the SW620 metastatic line was isolated, and we have shown that these two cell lines differ in the degree of response to butyrate[1]. Thus, future studies should utilize cell lines such as SW480, in addition to LT97 and SW620, to further dissect the role of CBP and p300 in colonic tumorigenesis. In general, studies that compare cells from matched primary vs metastatic tumors from the same patient would assist in identifying the roles played by CBP and p300 in neoplastic progression. We would expect that cell lines derived from metastases would exhibit a greater degree of CBP-Wnt activity and less p300-Wnt activity compared to matched primary tumor samples from the same patient.
Butyrate/HDACis resistance
Preliminary analyses of the mechanisms underlying butyrate resistance in CRC cells[49] utilized HCT-R cells that were derived from HCT-116 cells exposed to increasing concentrations of butyrate (up to 5 mmol/L). Microarray and western blot analyses demonstrated altered gene expression in HCT-R cells, compared to HCT-116 cells[47]. Importantly, HCT-R cells exhibit a markedly decreased expression of p300 compared to parental HCT-116 CRC cells. ICG-001, an inhibitor of CBP-mediated Wnt activity, was shown to repress Wnt activity in HCT-R cells, and these cells are highly sensitive to the apoptosis-inducing effects of ICG-001, suggesting that survival of butyrate resistant cells is associated with CBP-mediated Wnt activity.
Butyrate does not affect HCT-R cell proliferation, unlike the sharp repression of cell growth caused by the agent in butyrate-sensitive CRC cells expressing p300[41]. HCT-15 CRC cells that, similar to HCT-R cells, lack expression of p300 also exhibit repressed hyperactivation of Wnt signaling upon exposure to butyrate[42]. Also similar to HCT-R cells, the proliferation of HCT-15 cells is not influenced by butyrate[42]. Thus, both HCT-15 CRC cells that are deficient in p300 expression, and HCT-R cells with repressed p300 expression, exhibit relative resistance to the effects of butyrate on Wnt signaling and cell proliferation. Unlike HCT-R cells, HCT-15 cells respond to butyrate treatment with a modest upregulation of apoptosis and lack the heightened sensitivity to the apoptosis-inducing action of ICG-001 displayed by the HCT-R line[42]. These findings suggest that p300-mediated Wnt signaling is required for optimal hyperactivation of Wnt signaling and repression of cell proliferation by butyrate in at least some CRC cell lines. This possible role for p300 in mediating butyrate-modulated CRC cell apoptosis may be related to the association of p300-mediated Wnt signaling in colonic cell differentiation and apoptosis[30-33]. In butyrate-resistant HCT-R cells, CBP-mediated Wnt signaling likely promotes cell proliferation[42], and the downregulation of this signaling by ICG-001 induces apoptosis[42]. Thus, resistance to butyrate and other HDACis might be associated with a shift of the cancer cells utilizing more CBP-mediated than p300-mediated Wnt signaling.
Summary: Neoplastic progression and resistance to HDACis
Further studies should address the possibility that the simultaneous upregulation of p300-mediated Wnt signaling and downregulation of CBP-mediated Wnt signaling represent a therapeutic approach against both neoplastic progression and the development of resistance to butyrate and other HDACis (Figure 1).
Figure 1.
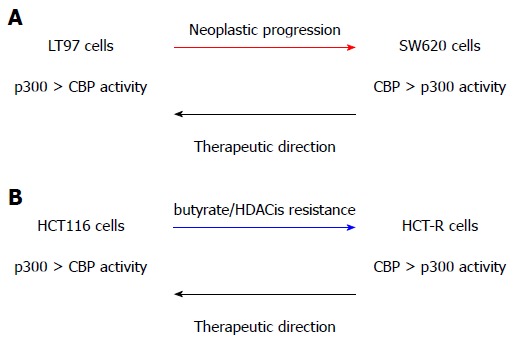
CREB-binding protein/p300 in neoplastic progression and Histone deacetylase inhibitors resistance, and a possible therapeutic approach. We posit that neoplastic progression (A) and resistance to HDACis (B) are associated with downregulated p300-mediated Wnt activity and relatively increased CBP-mediated Wnt signaling. Gene expression programs dependent upon CBP activity > p300 activity drive cell phenotype changes characteristic of greater neoplastic progression and HDACi resistance. Therefore, therapeutic approaches to prevent and/or reverse progression and resistance will depend upon enhancing p300-mediated Wnt activity at the expense of that mediated by CBP. Adapted from[43]. CBP: CREB-binding protein; HDACis: Histone deacetylase inhibitors.
CBP/p300-Wnt activity and cancer stem cell therapeutics
Differential modulation of CBP and p300 activity can target the cancer stem cell (CSC) fraction of a tumor, while sparing normal somatic stem cells (SSCs). The Wnt signaling pathway is associated with promotion of pluripotency/stemness; enhanced nuclear translocation of β-catenin (a marker for Wnt transcriptional activity) is most evident at the invasive front of metastatic solid tumors where epithelial to mesenchymal transition (EMT) takes place[31]. However, Wnt activity has also been associated with differentiation; therefore Wnt activity can trigger distinct physiological pathways in normal and neoplastic cells. These reports are consistent with the proposed specific role of CBP-mediated Wnt activity in proliferation vs the role of p300-mediated Wnt activity in differentiation. Thus, blocking the CBP-β-catenin interaction (e.g., with ICG-001) promotes p300-mediated Wnt activity and differentiation of stem and progenitor cells; conversely, inhibiting the p300-β-catenin interaction enhances CBP-mediated Wnt activity, promoting pluripotency and multipotency[31]. Therefore, repression of CBP-mediated Wnt signaling would seem to be an effective approach to eliminate CSCs through enforced differentiation.
However, this leads to the question of whether normal SSCs will be also targeted by this CBP-Wnt inhibitory approach. Lenz and Kahn[31] argue that negative effects on SSCs may be evaded due to the tendency of SSCs to multiply through asymmetric division (producing one daughter stem cell and one daughter differentiated cell); whereas, CSCs, due to various mutations, tend to divide symmetrically (producing either two daughter stem cells or two daughter differentiated cells). Agents such as ICG-001 would not change the differences between these types of stem cells regarding division; SSCs and CDCs exposed to these agents will continue to divide asymmetrically and symmetrically, respectively. However, the objective of treatment would be to force the CSC symmetrical cell division to produce two differentiated cells, abrogating the symmetrical production of daughter stem cells. The CSC symmetrical division that produces stem cells would be repressed by CBP-Wnt inhibitors, reducing the fraction of stem cells in the tumor. Thus, when exposed to a pro-differentiation CBP-Wnt inhibitor (such as ICG-001), CSCs would undergo a symmetrical division that only produces differentiated cells. In contrast, when exposed to the same agent, SSCs continue to normally divide asymmetrically, producing daughter stem cells that allow for the maintenance of the stem cell niche[31]. Effects of CBP and p300 on stem cell dynamics have a number of implications for cancer. For example, in human CML cell lines, hypoxia enhances the leukemia stem/initiating-like fraction of the cell population via increased CBP-mediated Wnt signaling[50]. ICG-001 can reverse the effects, emphasizing the role played by CBP in CSC maintenance and suggesting that repression of CBP-Wnt activity is a therapeutic option against the relatively larger pool of CSCs maintained in hypoxic conditions.
Therapeutic implications of findings involving HDACis and modulation of CBP and p300 activity
Findings[41-43] suggest that combinatorial therapy utilizing HDACis and ICG-001-like agents is likely to be effective in suppressing CRC cell growth. However, since apoptosis is a preferable outcome to repressed cell growth in therapy, cotreatment with HDACis and CBP-Wnt signaling inhibitors (such as ICG-001) should be avoided in CRCs in which these two types of agents interfere with each other’s effect on apoptosis. A challenge would be identifying which subset of CRCs fall into this category. In addition, the effect of diet on anti-CRC chemotherapeutics must be considered. Physiologically relevant concentrations of butyrate are produced in the colonic lumen from the fermentation of dietary fiber, and may also interfere with the activity of ICG-001-like agents on apoptosis. Therefore, the effects of a fiber-rich diet on the therapeutic activity of ICG-001-like agents against CRC needs to be investigated through animal studies. To evade undesirable interactions between dietary factors such as butyrate and CBP-Wnt inhibitors, CRC patients who are treated with ICG-001-like agents may need to fast or consume a low-fiber diet[41]. Modulation of p300-mediated Wnt activity represents another potential therapeutic approach, although further studies evaluating of the association of p300 with β-catenin are required in order to fully evaluate the role of p300 in colonic cell physiology.
Compared to HDACi-sensitive CRC cell lines, HDACi-resistant CRC cells exhibited greater apoptotic response to ICG-001[41,42]. Possibly, CBP-Wnt signaling inhibitors exert their greatest therapeutic effects against CRCs that are resistant to dietary and pharmacological HDACis. We theorize that right-sided colonic tumors may be particularly sensitive to CBP-Wnt signaling inhibitors since these neoplasms are more likely to be butyrate resistant since they have developed in the proximal colon where diet-derived butyrate is at its highest levels.
The anti-CRC preventive action of diet-derived butyrate is most likely exerted during early stage disease. Consistent with this, LT97 microadenoma cells are highly sensitive to the anti-proliferative and pro-apoptotic effects of butyrate[3,4,43]. Thus, growth arrest is an important mechanism whereby HDACis, combined with CBP-Wnt signaling inhibitors, affect cells in early stage colonic neoplasia[43]. With respect to apoptosis, both ICG-001 and butyrate each stimulated apoptosis of LT97 cells, and there was no interference between these agents in the induction of LT97 cell death; whereas, this type of interaction was observed in some CRC cell lines[41-43]. These findings suggest that CBP-Wnt signaling inhibitors, alone or in combination with HDACis, are effective in suppressing the earliest stages of colonic tumorigenesis. Practically speaking, application of this combinatorial treatment approach would be most useful in patients who are at particularly high risk for developing CRC (e.g., patients who have already had CRC and are at risk for recurrence).
Suppression of CBP-mediated Wnt activity may also have a therapeutic effect by stimulating differentiation of cancer stem cells[31,40]. How this activity of ICG-001 - like agents is influenced by dietary or pharmacological HDACis remains to be determined. The therapeutic choice of enhancing or suppressing Wnt signaling activity in CRC could be based upon (1) levels of the Wnt pathway, CBP and p300 expression levels in neoplastic cells ascertained through biopsy of patient samples; and (2) identification of specific gene mutation profiles associated with the HDACi-resistant and HDACi-sensitive phenotypes.
WHAT NEXT?
CBP-Wnt signaling inhibitors are in clinical trial for cancer and are specifically being investigated as differentiating agents for normal and cancer stem cells[31,40]. We believe that modulation of CBP- and p300-mediated Wnt activity has preventive and therapeutic potential for CRC, particularly with respect to controlling neoplastic progression and preventing/reversing resistance to HDACis. Combinatorial therapy with such agents and HDACis remains a possibility. However, additional in vitro and in vivo studies are required to further evaluate these possibilities. The unexplored question of how the combination of CBP-Wnt signaling inhibitors and HDACis influences cancer stem cell phenotypes remains to be determined.
The dissection of the relative roles of CBP and p300 in Wnt activity-mediated CRC would be advanced by the development of new specific inhibitors of p300-mediated Wnt activity in human cells, analogous to the CBP-Wnt inhibitor ICG-001. One promising development in this regard is the identification of windorphen as an agent which has greater activity against p300 HAT activity and the p300-β-catenin association compared to its effects on CBP; in addition, windorphen treatment also stimulates apoptosis of CRC cells[51]. The activity and specificity of this agent, alone or in combination with other agents (e.g., HDACis) requires further investigation. Future studies can utilize primary patient samples and animal models of CRC to validate in vitro findings. Comparison of adenoma vs carcinoma stage in patients and animal models may recapitulate the differences observed between LT97 vs SW620 cells with respect to CBP and p300 expression and activity, response to butyrate and/or ICG-001, as well as expression of target genes.
Further, primary neoplastic samples acquired along the length of the large bowel should be evaluated for relative butyrate resistance in ex vivo experiments. For example, CRCs from the right colon have been hypothesized to exhibit more butyrate resistance than those from the left colon[52,53], due to intra-colonic differences in luminal butyrate concentrations. We also note that CRCs with microsatellite instability (MSI+) are typically found in right-sided colon cancer; importantly, these tumors, which display differences in cell signaling and therapeutic response compared to non-MSI CRC, frequently exhibit mutations in CBP and, particularly, p300[54]. A number of cell lines, including HCT-116, that are characterized by p300 mutation, are MSI+[54], and one can hypothesize a link between MSI+ status, p300 expression, butyrate-resistance, and the development of right-sided CRC in the context of higher levels of butyrate in the proximal colon[53]. Whether agents such as ICG-001 would be especially efficacious against MSI+ colonic neoplasms (Figure 2) needs to be evaluated, including through the use of animal models of mismatch repair-deficient CRC (see below). Thus, variation in butyrate sensitivity in primary patient samples may reflect differences in CBP-mediated and p300-mediated Wnt activity, and the downstream consequences with respect to effects of butyrate.
Figure 2.
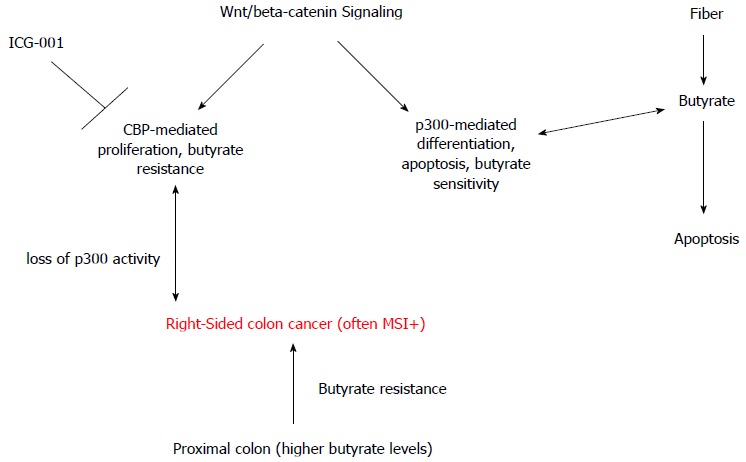
Hypothetical relationship between p300 loss, butyrate-resistance, and right-sided colon cancer. Wnt/β-catenin signaling has both CBP-mediated and p300-mediated components. CBP-Wnt activity, which is repressed by the clinically relevant agent ICG-001, likely mediates colonic cell proliferation and resistance to butyrate (and other HDACis). In contrast, p300-Wnt activity likely mediates differentiation, apoptosis, and sensitivity to butyrate. Butyrate, derived from dietary fiber, enhances apoptosis of colon cancer cells, possibly acting through the p300-mediated component of Wnt/β-catenin activity. Neoplasms that arise in the proximal colon, where butyrate concentrations are highest, may be resistant to butyrate, and this resistance may arise through loss of p300 expression and activity and a greater reliance on CBP-mediated Wnt signaling. Development of these right-sided colon cancers may be prevented/threated via agents such as ICG-001 and/or other approaches that can promote p300-Wnt > CBP-Wnt activity. CBP: CREB-binding protein; HDACis: Histone deacetylase inhibitors.
Mouse xenograft models can be utilized to determine how alteration in CBP- and p300-mediated Wnt activity correlates to in vivo tumorigenicity. Thus, adenoma cells engineered to exhibit greater neoplastic potential in vitro can be tested for their ability to form tumors in vivo; conversely, carcinoma cells with modified CBP- and p300-mediated Wnt activity and reduced neoplastic potential in vitro can be compared to parental cells with respect to tumorigenicity in vivo. The same principle can be applied in evaluating how modulation of CBP- and p300-mediated Wnt signaling in butyrate-resistant cells affects the ability of these cells to form tumors and the sensitivity of the resulting tumors to butyrate/HDACis.
The findings generated by these studies can be subsequently utilized to evaluate approaches that target earlier stages in tumor development using genetic Apc mutant mouse models. Perhaps a more optimal set of mouse models for these studies are those mismatch repair-deficient strains developed by the Edelmann laboratory[55]. Given the prevalence of MSI+ tumors in right-sided colon cancer, the increased possibility of butyrate resistance in such tumors, and the frequency of CBP and p300 mutations in MSI+ CRC, murine models of mismatch repair-deficient CRC should also be utilized to understand the role of CBP and p300 in colonic neoplasia, possible connections between MSI+ status, p300 expression, and butyrate-resistance, as well as the utility of using ICG-001-like agents for the treatment of such tumors. Finally, we note that many of the issues discussed in this review are likely not confined to CRC; for example, the CBP-Wnt inhibitor ICG-001 has shown efficacy in repressing pancreatic cell growth[56]. Further, inhibition of p300-mediated Wnt signaling in mouse models indicate a role for p300-Wnt activity in proximalizing lung epithelium[57], raising the possibility that perturbation of CBP vs p300 activity influences lung carcinogenesis. These findings, and the work on effects of CBP and p300 on normal vs cancer stem cells, suggest that modulation of CBP vs p300 activity can be a common therapeutic tool against many forms of human cancer.
CONCLUSION
A more comprehensive understanding of how CBP and p300 influence colonic cell physiology will allow for the development of anti-CRC preventive and therapeutic approaches that target CBP and p300 activity in order to repress neoplastic progression, prevent or reverse the development of resistance to butyrate and other HDACis, and target colon cancer stem cells. Further studies are required to generate data that may lead to novel chemopreventive and therapeutic approaches against CRC.
Footnotes
Supported by National Institutes of Health (Bethesda, MD) National Cancer Institute, No. 1R15CA149589-01.
Conflict-of-interest statement: The authors have no conflicts of interest.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: March 31, 2015
First decision: April 23, 2015
Article in press: May 27, 2015
P- Reviewer: Kadiyska TK, Linnebacher M, Zouiten-Mekki L S- Editor: Ma YJ L- Editor: A E- Editor: Liu XM
References
- 1.Lazarova DL, Bordonaro M, Carbone R, Sartorelli AC. Linear relationship between Wnt activity levels and apoptosis in colorectal carcinoma cells exposed to butyrate. Int J Cancer. 2004;110:523–531. doi: 10.1002/ijc.20152. [DOI] [PubMed] [Google Scholar]
- 2.Bordonaro M, Lazarova DL, Sartorelli AC. The activation of beta-catenin by Wnt signaling mediates the effects of histone deacetylase inhibitors. Exp Cell Res. 2007;313:1652–1666. doi: 10.1016/j.yexcr.2007.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Richter M, Jurek D, Wrba F, Kaserer K, Wurzer G, Karner-Hanusch J, Marian B. Cells obtained from colorectal microadenomas mirror early premalignant growth patterns in vitro. Eur J Cancer. 2002;38:1937–1945. doi: 10.1016/s0959-8049(02)00158-2. [DOI] [PubMed] [Google Scholar]
- 4.Kautenburger T, Beyer-Sehlmeyer G, Festag G, Haag N, Kühler S, Küchler A, Weise A, Marian B, Peters WH, Liehr T, et al. The gut fermentation product butyrate, a chemopreventive agent, suppresses glutathione S-transferase theta (hGSTT1) and cell growth more in human colon adenoma (LT97) than tumor (HT29) cells. J Cancer Res Clin Oncol. 2005;131:692–700. doi: 10.1007/s00432-005-0013-4. [DOI] [PubMed] [Google Scholar]
- 5.Brabletz T, Jung A, Spaderna S, Hlubek F, Kirchner T. Opinion: migrating cancer stem cells - an integrated concept of malignant tumour progression. Nat Rev Cancer. 2005;5:744–749. doi: 10.1038/nrc1694. [DOI] [PubMed] [Google Scholar]
- 6.Bingham SA, Day NE, Luben R, Ferrari P, Slimani N, Norat T, Clavel-Chapelon F, Kesse E, Nieters A, Boeing H, et al. Dietary fibre in food and protection against colorectal cancer in the European Prospective Investigation into Cancer and Nutrition (EPIC): an observational study. Lancet. 2003;361:1496–1501. doi: 10.1016/s0140-6736(03)13174-1. [DOI] [PubMed] [Google Scholar]
- 7.Peters U, Sinha R, Chatterjee N, Subar AF, Ziegler RG, Kulldorff M, Bresalier R, Weissfeld JL, Flood A, Schatzkin A, et al. Dietary fibre and colorectal adenoma in a colorectal cancer early detection programme. Lancet. 2003;361:1491–1495. doi: 10.1016/S0140-6736(03)13173-X. [DOI] [PubMed] [Google Scholar]
- 8.Bingham SA, Norat T, Moskal A, Ferrari P, Slimani N, Clavel-Chapelon F, Kesse E, Nieters A, Boeing H, Tjønneland A, et al. Is the association with fiber from foods in colorectal cancer confounded by folate intake? Cancer Epidemiol Biomarkers Prev. 2005;14:1552–1556. doi: 10.1158/1055-9965.EPI-04-0891. [DOI] [PubMed] [Google Scholar]
- 9.Bingham SA. Mechanisms and experimental and epidemiological evidence relating dietary fibre (non-starch polysaccharides) and starch to protection against large bowel cancer. Proc Nutr Soc. 1990;49:153–171. doi: 10.1079/pns19900021. [DOI] [PubMed] [Google Scholar]
- 10.Cummings JH, Pomare EW, Branch WJ, Naylor CP, Macfarlane GT. Short chain fatty acids in human large intestine, portal, hepatic and venous blood. Gut. 1987;28:1221–1227. doi: 10.1136/gut.28.10.1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Boffa LC, Lupton JR, Mariani MR, Ceppi M, Newmark HL, Scalmati A, Lipkin M. Modulation of colonic epithelial cell proliferation, histone acetylation, and luminal short chain fatty acids by variation of dietary fiber (wheat bran) in rats. Cancer Res. 1992;52:5906–5912. [PubMed] [Google Scholar]
- 12.Heerdt BG, Houston MA, Augenlicht LH. Potentiation by specific short-chain fatty acids of differentiation and apoptosis in human colonic carcinoma cell lines. Cancer Res. 1994;54:3288–3293. [PubMed] [Google Scholar]
- 13.Hague A, Elder DJ, Hicks DJ, Paraskeva C. Apoptosis in colorectal tumour cells: induction by the short chain fatty acids butyrate, propionate and acetate and by the bile salt deoxycholate. Int J Cancer. 1995;60:400–406. doi: 10.1002/ijc.2910600322. [DOI] [PubMed] [Google Scholar]
- 14.Medina V, Edmonds B, Young GP, James R, Appleton S, Zalewski PD. Induction of caspase-3 protease activity and apoptosis by butyrate and trichostatin A (inhibitors of histone deacetylase): dependence on protein synthesis and synergy with a mitochondrial/cytochrome c-dependent pathway. Cancer Res. 1997;57:3697–3707. [PubMed] [Google Scholar]
- 15.Marks PA, Dokmanovic M. Histone deacetylase inhibitors: discovery and development as anticancer agents. Expert Opin Investig Drugs. 2005;14:1497–1511. doi: 10.1517/13543784.14.12.1497. [DOI] [PubMed] [Google Scholar]
- 16.Lin HY, Chen CS, Lin SP, Weng JR, Chen CS. Targeting histone deacetylase in cancer therapy. Med Res Rev. 2006;26:397–413. doi: 10.1002/med.20056. [DOI] [PubMed] [Google Scholar]
- 17.Bordonaro M, Mariadason JM, Aslam F, Heerdt BG, Augenlicht LH. Butyrate-induced apoptotic cascade in colonic carcinoma cells: modulation of the beta-catenin-Tcf pathway and concordance with effects of sulindac and trichostatin A but not curcumin. Cell Growth Differ. 1999;10:713–720. [PubMed] [Google Scholar]
- 18.Liu C, Zhang Y, Li J, Wang Y, Ren F, Zhou Y, Wu Y, Feng Y, Zhou Y, Su F, et al. p15RS/RPRD1A (p15INK4b-related sequence/regulation of nuclear pre-mRNA domain-containing protein 1A) interacts with HDAC2 in inhibition of the Wnt/β-catenin signaling pathway. J Biol Chem. 2015;290:9701–9713. doi: 10.1074/jbc.M114.620872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cook D, Fry MJ, Hughes K, Sumathipala R, Woodgett JR, Dale TC. Wingless inactivates glycogen synthase kinase-3 via an intracellular signalling pathway which involves a protein kinase C. EMBO J. 1996;15:4526–4536. [PMC free article] [PubMed] [Google Scholar]
- 20.Rubinfeld B, Souza B, Albert I, Müller O, Chamberlain SH, Masiarz FR, Munemitsu S, Polakis P. Association of the APC gene product with beta-catenin. Science. 1993;262:1731–1734. doi: 10.1126/science.8259518. [DOI] [PubMed] [Google Scholar]
- 21.Su LK, Vogelstein B, Kinzler KW. Association of the APC tumor suppressor protein with catenins. Science. 1993;262:1734–1737. doi: 10.1126/science.8259519. [DOI] [PubMed] [Google Scholar]
- 22.Munemitsu S, Albert I, Souza B, Rubinfeld B, Polakis P. Regulation of intracellular beta-catenin levels by the adenomatous polyposis coli (APC) tumor-suppressor protein. Proc Natl Acad Sci USA. 1995;92:3046–3050. doi: 10.1073/pnas.92.7.3046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Behrens J, von Kries JP, Kühl M, Bruhn L, Wedlich D, Grosschedl R, Birchmeier W. Functional interaction of beta-catenin with the transcription factor LEF-1. Nature. 1996;382:638–642. doi: 10.1038/382638a0. [DOI] [PubMed] [Google Scholar]
- 24.Molenaar M, van de Wetering M, Oosterwegel M, Peterson-Maduro J, Godsave S, Korinek V, Roose J, Destrée O, Clevers H. XTcf-3 transcription factor mediates beta-catenin-induced axis formation in Xenopus embryos. Cell. 1996;86:391–399. doi: 10.1016/s0092-8674(00)80112-9. [DOI] [PubMed] [Google Scholar]
- 25.Korinek V, Barker N, Morin PJ, van Wichen D, de Weger R, Kinzler KW, Vogelstein B, Clevers H. Constitutive transcriptional activation by a beta-catenin-Tcf complex in APC-/- colon carcinoma. Science. 1997;275:1784–1787. doi: 10.1126/science.275.5307.1784. [DOI] [PubMed] [Google Scholar]
- 26.Morin PJ, Sparks AB, Korinek V, Barker N, Clevers H, Vogelstein B, Kinzler KW. Activation of beta-catenin-Tcf signaling in colon cancer by mutations in beta-catenin or APC. Science. 1997;275:1787–1790. doi: 10.1126/science.275.5307.1787. [DOI] [PubMed] [Google Scholar]
- 27.Bienz M, Clevers H. Linking colorectal cancer to Wnt signaling. Cell. 2000;103:311–320. doi: 10.1016/s0092-8674(00)00122-7. [DOI] [PubMed] [Google Scholar]
- 28.Albuquerque C, Breukel C, van der Luijt R, Fidalgo P, Lage P, Slors FJ, Leitão CN, Fodde R, Smits R. The ‘just-right’ signaling model: APC somatic mutations are selected based on a specific level of activation of the beta-catenin signaling cascade. Hum Mol Genet. 2002;11:1549–1560. doi: 10.1093/hmg/11.13.1549. [DOI] [PubMed] [Google Scholar]
- 29.Bordonaro M, Lazarova DL, Sartorelli AC. Hyperinduction of Wnt activity: a new paradigm for the treatment of colorectal cancer? Oncol Res. 2008;17:1–9. doi: 10.3727/096504008784046108. [DOI] [PubMed] [Google Scholar]
- 30.Emami KH, Nguyen C, Ma H, Kim DH, Jeong KW, Eguchi M, Moon RT, Teo JL, Kim HY, Moon SH, et al. A small molecule inhibitor of beta-catenin/CREB-binding protein transcription [corrected] Proc Natl Acad Sci USA. 2004;101:12682–12687. doi: 10.1073/pnas.0404875101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lenz HJ, Kahn M. Safely targeting cancer stem cells via selective catenin coactivator antagonism. Cancer Sci. 2014;105:1087–1092. doi: 10.1111/cas.12471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Teo JL, Ma H, Nguyen C, Lam C, Kahn M. Specific inhibition of CBP/beta-catenin interaction rescues defects in neuronal differentiation caused by a presenilin-1 mutation. Proc Natl Acad Sci USA. 2005;102:12171–12176. doi: 10.1073/pnas.0504600102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ma H, Nguyen C, Lee KS, Kahn M. Differential roles for the coactivators CBP and p300 on TCF/beta-catenin-mediated survivin gene expression. Oncogene. 2005;24:3619–3631. doi: 10.1038/sj.onc.1208433. [DOI] [PubMed] [Google Scholar]
- 34.Miyabayashi T, Teo JL, Yamamoto M, McMillan M, Nguyen C, Kahn M. Wnt/beta-catenin/CBP signaling maintains long-term murine embryonic stem cell pluripotency. Proc Natl Acad Sci USA. 2007;104:5668–5673. doi: 10.1073/pnas.0701331104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Teo JL, Kahn M. The Wnt signaling pathway in cellular proliferation and differentiation: A tale of two coactivators. Adv Drug Deliv Rev. 2010;62:1149–1155. doi: 10.1016/j.addr.2010.09.012. [DOI] [PubMed] [Google Scholar]
- 36.Waltzer L, Bienz M. Drosophila CBP represses the transcription factor TCF to antagonize Wingless signalling. Nature. 1998;395:521–525. doi: 10.1038/26785. [DOI] [PubMed] [Google Scholar]
- 37.Hecht A, Vleminckx K, Stemmler MP, van Roy F, Kemler R. The p300/CBP acetyltransferases function as transcriptional coactivators of beta-catenin in vertebrates. EMBO J. 2000;19:1839–1850. doi: 10.1093/emboj/19.8.1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li J, Sutter C, Parker DS, Blauwkamp T, Fang M, Cadigan KM. CBP/p300 are bimodal regulators of Wnt signaling. EMBO J. 2007;26:2284–2294. doi: 10.1038/sj.emboj.7601667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lévy L, Wei Y, Labalette C, Wu Y, Renard CA, Buendia MA, Neuveut C. Acetylation of beta-catenin by p300 regulates beta-catenin-Tcf4 interaction. Mol Cell Biol. 2004;24:3404–3414. doi: 10.1128/MCB.24.8.3404-3414.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Takahashi-Yanaga F, Kahn M. Targeting Wnt signaling: can we safely eradicate cancer stem cells? Clin Cancer Res. 2010;16:3153–3162. doi: 10.1158/1078-0432.CCR-09-2943. [DOI] [PubMed] [Google Scholar]
- 41.Lazarova DL, Chiaro C, Wong T, Drago E, Rainey A, O’Malley S, Bordonaro M. CBP Activity Mediates Effects of the Histone Deacetylase Inhibitor Butyrate on WNT Activity and Apoptosis in Colon Cancer Cells. J Cancer. 2013;4:481–490. doi: 10.7150/jca.6583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lazarova DL, Wong T, Chiaro C, Drago E, Bordonaro M. p300 Influences Butyrate-Mediated WNT Hyperactivation In Colorectal Cancer Cells. J Cancer. 2013;4:491–501. doi: 10.7150/jca.6582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lazarova D, Lee A, Wong T, Marian B, Chiaro C, Rainey C, Bordonaro M. Modulation of Wnt Activity and Cell Physiology by Butyrate in LT97 Microadenoma Cells. J Cancer. 2014;5:203–213. doi: 10.7150/jca.8569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Grigson ER, Ozerova M, Pisklakova A, Liu H, Sullivan DM, Nefedova Y. Canonical Wnt pathway inhibitor ICG-001 induces cytotoxicity of multiple myeloma cells in Wnt-independent manner. PLoS One. 2015;10:e0117693. doi: 10.1371/journal.pone.0117693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fodde R, Brabletz T. Wnt/beta-catenin signaling in cancer stemness and malignant behavior. Curr Opin Cell Biol. 2007;19:150–158. doi: 10.1016/j.ceb.2007.02.007. [DOI] [PubMed] [Google Scholar]
- 46.Huh JW, Kim HC, Kim SH, Park YA, Cho YB, Yun SH, Lee WY, Chun HK. Prognostic impact of p300 expression in patients with colorectal cancer. J Surg Oncol. 2013;108:374–377. doi: 10.1002/jso.23405. [DOI] [PubMed] [Google Scholar]
- 47.Jia Y, Nie F, Du A, Chen Z, Qin Y, Huang T, Song X, Li L. Thymine DNA glycosylase promotes transactivation of β-catenin/TCFs by cooperating with CBP. J Mol Cell Biol. 2014;6:231–239. doi: 10.1093/jmcb/mju014. [DOI] [PubMed] [Google Scholar]
- 48.Götze S, Coersmeyer M, Müller O, Sievers S. Histone deacetylase inhibitors induce attenuation of Wnt signaling and TCF7L2 depletion in colorectal carcinoma cells. Int J Oncol. 2014;45:1715–1723. doi: 10.3892/ijo.2014.2550. [DOI] [PubMed] [Google Scholar]
- 49.Chiaro C, Lazarova DL, Bordonaro M. Tcf3 and cell cycle factors contribute to butyrate resistance in colorectal cancer cells. Biochem Biophys Res Commun. 2012;428:121–126. doi: 10.1016/j.bbrc.2012.10.018. [DOI] [PubMed] [Google Scholar]
- 50.Kida A, Kahn M. Hypoxia selects for a quiescent, CML stem/leukemia initiating-like population dependent on CBP/catenin transcription. Curr Mol Pharmacol. 2013;6:204–210. doi: 10.2174/1874467207666140219121219. [DOI] [PubMed] [Google Scholar]
- 51.Hao J, Ao A, Zhou L, Murphy CK, Frist AY, Keel JJ, Thorne CA, Kim K, Lee E, Hong CC. Selective small molecule targeting β-catenin function discovered by in vivo chemical genetic screen. Cell Rep. 2013;4:898–904. doi: 10.1016/j.celrep.2013.07.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bordonaro M, Tewari S, Cicco CE, Atamna W, Lazarova DL. A switch from canonical to noncanonical Wnt signaling mediates drug resistance in colon cancer cells. PLoS One. 2011;6:e27308. doi: 10.1371/journal.pone.0027308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lazarova DL, Bordonaro M. Extreme fluctuations in Wnt/beta-catenin signaling as an approach for colon cancer prevention and therapy. Adv Stud Biol. 2012;4:351–362. [Google Scholar]
- 54.Ionov Y, Matsui S, Cowell JK. A role for p300/CREB binding protein genes in promoting cancer progression in colon cancer cell lines with microsatellite instability. Proc Natl Acad Sci USA. 2004;101:1273–1278. doi: 10.1073/pnas.0307276101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Woerner SM, Tosti E, Yuan YP, Kloor M, Bork P, Edelmann W, Gebert J. Detection of coding microsatellite frameshift mutations in DNA mismatch repair-deficient mouse intestinal tumors. Mol Carcinog. 2014:Epub ahead of print. doi: 10.1002/mc.22213. [DOI] [PubMed] [Google Scholar]
- 56.Arensman MD, Telesca D, Lay AR, Kershaw KM, Wu N, Donahue TR, Dawson DW. The CREB-binding protein inhibitor ICG-001 suppresses pancreatic cancer growth. Mol Cancer Ther. 2014;13:2303–2314. doi: 10.1158/1535-7163.MCT-13-1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sasaki T, Kahn M. Inhibition of β-catenin/p300 interaction proximalizes mouse embryonic lung epithelium. Transl Respir Med. 2014;2:8. doi: 10.1186/s40247-014-0008-1. [DOI] [PMC free article] [PubMed] [Google Scholar]