

Abstract
The hepatitis C virus (HCV) is one of the most common causes of chronic liver disease and the leading indication for liver transplantation worldwide. Every aspect of the HCV life cycle is closely tied to human lipid metabolism. The virus circulates as a lipid-rich particle, utilizing lipoprotein cell receptors to gain entry into the hepatocyte. It has also been shown to upregulate lipid biosynthesis and impair lipid degradation, resulting in significant intracellular lipid accumulation and circulating hypocholesterolemia. Patients with chronic hepatitis C (CHC) are at increased risk of hepatic steatosis, fibrosis, and cardiovascular disease including accelerated atherosclerosis. HMG CoA Reductase inhibitors, or statins, have been shown to play an important role in the modulation of hepatic steatosis and fibrosis, and recent attention has focused upon their potential therapeutic role in CHC. This article reviews the hepatitis C viral life cycle as it impacts host lipoproteins and lipid metabolism. It then describes the pathogenesis of HCV-related hepatic steatosis, hypocholesterolemia and atherosclerosis, and finally describes the promising anti-viral and anti-fibrotic effects of statins, for the treatment of CHC.
Keywords: Hepatitis C virus, Lipid profiles, Cholesterol, Statin, Fibrosis, Cirrhosis
Core tip: This article reviews the complex relationship between hepatitis C virus (HCV) infection and human lipid metabolism. It discusses the aspects of the hepatitis C viral life cycle that are entwined with cholesterol homeostasis, as well as the clinical implications of HCV-mediated changes in human lipid profiles. Finally, it describes the current state of knowledge regarding the impact of statin medications on histological, virological and clinical outcomes, among patients with chronic hepatitis C.
INTRODUCTION
Hepatitis C virus (HCV) is a single-stranded RNA virus, of the genus Hepacivirus and the family Flaviviridae. Affecting 2% to 3% of the global population, HCV is one of the most common causes of chronic liver disease and the leading indication for liver transplantation worldwide[1,2]. Estimates suggest that over a period of twenty to thirty years, cirrhosis will develop in 10% to 25% of patients with untreated or relapsed chronic hepatitis C (CHC), and hepatocellular carcinoma (HCC) in 1% to 5%[2].
Hepatic steatosis is a common histopathological finding in patients with CHC. Various factors have been independently associated with steatosis, including obesity, diabetes, hyperlipidemia and alcohol consumption[3]. It has also been demonstrated that the hepatitis C virus possesses a unique relationship with host lipids and lipoproteins[4,5], and relies heavily on host lipoproteins, lipid droplets, and host co-factors for each step of the viral life cycle including the facilitation of viral replication[6-8]. At the same time, HCV causes profound lipid perturbations within the infected host, resulting in hepatic steatosis, circulating hypocholesterolemia and increased atherogenesis[6,8-12].
Statins, which inhibit the rate-limiting enzyme of the mevalonate pathway, HMG CoA Reductase, have been shown to play an important role in the modulation of hepatic steatosis and cholesterol metabolism, and recent attention has focused upon their potential therapeutic role for patients with CHC. This article reviews the molecular pathways of lipid homeostasis and the pathogenesis of hepatic steatosis as they relate to chronic hepatitis C infection, and then describes the potential impact of statin medications upon clinical, viral and histological outcomes.
HCV viral life cycle and host lipoproteins
HCV infection begins with attachment of the viral particle to the hepatocyte cell surface, in a process that requires many host proteins that are closely entwined with lipid metabolism[13-16]. To enter the cell, HCV must then associate with multiple cell surface receptors, three of which are closely linked to lipoprotein metabolism: the scavenger receptor class B member 1 (SRB1) protein, the Neimann-Pick C1 Like 1 (NPC1L1) receptor, and the low-density lipoprotein receptor (LDLR). The HCV viral life cycle is shown in Figures 1 and 2.
Figure 1.
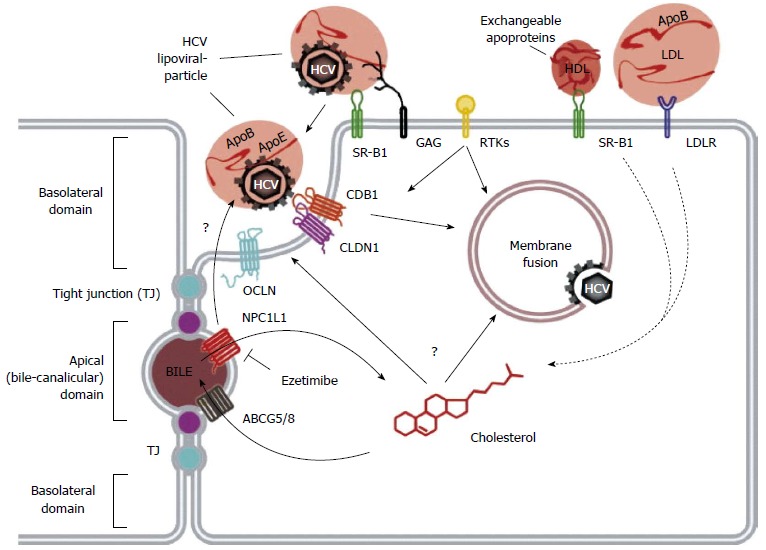
Hepatitis C virus viral life cycle. Hepatitis C virus (HCV) entry into human hepatocytes is a complex, multi-step process that takes place at the basolateral region of polarized hepatocytes. It begins when the viral particle binds surface glycosaminoglycans (GAGs) and the low-density lipoprotein receptor (LDLR) via apolipoprotein E. This is followed by a complex series of interactions mediated by cellular factors including scavenger receptor class B type I (SR-BI), the tetraspanin CD81, claudin-1 (CLDN1), occludin (OCLN), the Niemann-Pick C1-like 1 (NPC1L1) receptor, as well as receptor tyrosine kinases (RTKs) that promote CD81-CLDN1 association and membrane fusion. The HCV particle is then internalized into the hepatocyte by clathrin-mediated endocytosis. This figure is reproduced with permission from the original article, published in Journal of Hepatology, Vol 57, Issue 1, by Lupberger J, Felmlee J and Baumert TF. Cholesterol Uptake and Hepatitis C virus entry, page 215-217, Copyright Elsevier, 2012.
Figure 2.
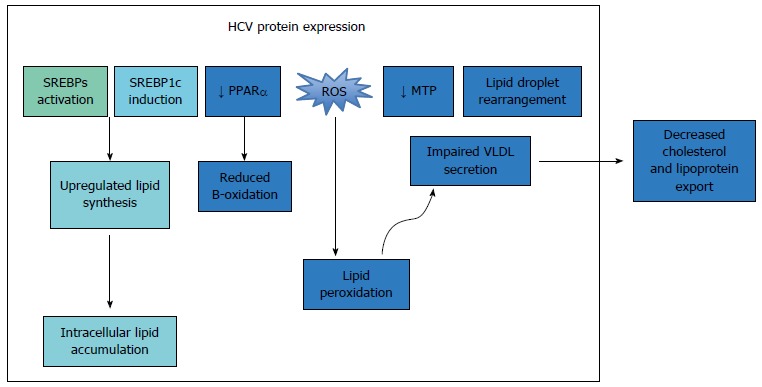
Hepatitis C virus-mediated perturbations in cholesterol metabolism. HCV: Hepatitis C virus; ROS: Reactive oxygen species; VLDL: Very low-density lipoprotein.
SRB1 is a cell surface transmembrane protein, primarily expressed in the liver and steroidogenic tissues. Although its essential function is cholesteryl ester uptake from HDL, it also serves as a multi-ligand receptor for various lipoproteins, including VLDL, LDL and HDL[17]. Oxidized LDL and VLDL have been shown to inhibit HCV cell entry[18], while HDL enhances HCV entry in an SRB1-dependent process[19-21]. Changes in circulating lipid levels have indeed been shown to impact both viremia and treatment response: increased triglyceride levels have been linked to improved viral clearance[22], while elevated LDL and total cholesterol is associated with improved treatment response to interferon-based therapy[23].
The NPC1L1 receptor is a cholesterol receptor in the intestines and the liver, essential for dietary cholesterol absorption and biliary cholesterol reabsorption. It is thought to promote HCV cell entry via interaction with cholesterol of lipoviral particles and by modulation of cholesterol homeostasis, which in turn alters membrane composition and affects HCV cell entry[24]. In vitro, inhibition of NPC1L1 blocks initiation of HCV infection, via a cholesterol-dependent mechanism occurring before virion-cell membrane fusion[25]. A recent in vivo mouse model also showed that blockade of NPC1L1 with ezetimibe blocks viral cell entry[24].
LDLR is a transmembrane glycoprotein responsible for the uptake of serum lipoproteins[26]. Transcription of LDLR is upregulated by the sterol-regulatory element binding proteins (SREBPs)[26,27], and the signaling molecules PCSK9[28,29], and inhibited by the inducible degrader of LDLR (IDOL)[30,31]. It has been shown that accumulation of HCV RNA within hepatocytes correlates with the expression of LDLR, and that antibodies directed against LDLR inhibit the cellular absorption of HCV[25,32]. HCV has also been shown to activate SREBP-mediated PI3-K/AKT and LXR pathways[10], resulting in further activation of LDLR, and thus enhancing viral infectivity.
Once inside the cytoplasm, the uncoated viral genome is translated, and the polypeptide is cleaved into 10 viral proteins. The HCV structural proteins (E1, E2 and core) play important roles in viral replication and assembly, while the non-structural proteins (p7, NS2, NS3, NS4A, NS4B, NS5A, and NS5B) are essential for the intracellular aspects of the viral life cycle[33]. Following translation, the viral genome is transcribed by the proteins NS3 and NS5B[34]. HCV core protein accumulates around lipid droplets (LD), which are stores of triacylglycerols and cholesterol esters[35], and has been shown to inhibit the activity of MTP (microsomal triacylglycerol transfer protein) and the subsequent secretion of very low-density lipoprotein (VLDL)[36]. The functions of each viral protein and their interactions with host lipid metabolism are outlined in Table 1.
Table 1.
Functions of hepatitis C virus structural and non-structural proteins
| Protein | Function | Association with Lipid Metabolism |
| Structural proteins | ||
| E1 | Surface envelope protein of LVP | Enriched around lipid droplet |
| E2 | Surface envelope protein of LVP, binds CD81 for viral fusion and cell entry | Interacts with ApoE and SRB1 facilitating lipid transfers around LD |
| Core | Important for cell surface binding, replication and assembly | Increases expression of SREBP, FASN. Colocalizes with apoB |
| Non-structural proteins | ||
| P7 | Creates transmembrane ion channel in membranous web, (viroporin) | Assists in recruitment of core protein to LD by DGAT1 |
| NS2 | Cysteine protease | Increases expression of FASN, SREBP |
| NS3 | Serine protease, RNA helicase, promotes viral protein processing with NS4A | Unknown |
| NS4A | Protease; cofactor for NS3 | Unknown |
| NS4B | Directs membrane rearrangements for formation of membranous web | Activates fatty acid synthase, associates with DGAT1 and P14KA, facilitating creation of membranous web |
| NS5A | Phosphoprotein, required for replication, forms bridge to assembly | Activates FASN; associates with P14KA, DGAT1 |
| NS5B | RNA-dependent RNA polymerase, for replication of viral genome | Direct interaction with fatty acid synthase gene |
HCV RNA replication and assembly of the LVP occur at the membranous web (MW), a specialized structure composed of clusters of viral vesicles and lipid droplets (LD)[37,38], on which the LVP and viral proteins converge[39,40]. The enzyme diacylglycerol transferase-1 (DGAT) facilitates the trafficking of core, NS5A and NS4B proteins to the LD[41,42], and results in inhibition of triglyceride lipolysis and lipid droplet turnover, thus increasing the concentration of available intracellular lipids, for the facilitation of further HCV replication[43]. To export new HCV virions, the virus co-opts the host VLDL synthesis and secretion pathways. Synthesis of VLDL involves the generation of a VLDL precursor, using the lipid transfer function of the microsomal triglyceride transfer protein (MTP), which lipidates nascent apolipoprotein B100 (apoB100)[44]. The VLDL precursor is then targeted to the Golgi apparatus for export. The viral LVP is enriched in ApoE and ApoB, and thus possesses characteristics of a VLDL particle. This enables it to utilize the lipid transfer function of the MTP, and thus co-opt the VLDL secretion pathway and facilitate virion export[6,45-48].
Lipid changes in chronic hepatitis C infection
Circulating hypocholesterolemia: Circulating lipid levels are altered in patients with HCV, regardless of the duration of infection. In a cohort of patients with acute HCV, early infection was associated with reduction in LDL and total cholesterol levels; following viral eradication through spontaneous clearance or successful anti-HCV treatment, the lipids of those patients returned to pre-infection levels[49]. Patients with CHC also have demonstrate reduced levels of circulating LDL, apolipoprotein B100 (apoB) and total cholesterol, compared to healthy controls[50]. An inverse relationship has also been described between reduction in apoB levels and HCV viral load, among those with non-genotype 1 infection[50]. These perturbations also seem to resolve after successful clearance of CHC[49], supporting the hypothesis that HCV has a direct cytopathic effect upon host lipid metabolism.
The presence and degree of hypocholesterolemia carries important prognostic implications for patients with CHC. Elevated LDL and high-density lipoprotein (HDL) levels have been associated with improved rates of sustained virologic response (SVR)[51,52]. This may be related to the dependence of HCV upon LDL cholesterol concentrations and the LDLR for both cellular entry and viral replication.
Hepatic steatosis: Hepatic steatosis is frequently observed in the setting of CHC[53], and is thought to result from a combination of viral-mediated activation of lipid biosynthesis pathways and reduced lipid export[54,55]. The presence of hepatic steatosis among patients with CHC has been associated with poor treatment response [lower sustained viral response (SVR) rates] and accelerated disease progression to advanced fibrosis and cirrhosis[56-58]. Both patient-related factors and viral factors play important roles in the modulation of HCV-related steatosis.
One important viral factor that impacts host lipid metabolism is viral genotype. Genotype 3 CHC is associated with the greatest degree of hepatic steatosis, and the most significant reductions in serum cholesterol levels[50,59]. In the fasting state, patients with genotype 3 demonstrate profoundly elevated cholesterol metabolites[60], as well as increased intracellular lipid accumulation[47,61,62], compared to all other HCV genotypes. In contrast, among patients infected with all other genotypes of CHC, metabolic risk factors, including insulin levels, diabetes and obesity, appear to play a more important role in progressive steatosis[63,64]. This was demonstrated in analyses linking hepatic steatosis to higher levels of circulating viremia, among genotype 3 patients[65]; in those cohorts, the eradication of infection resulted in improvement or resolution of steatosis, a finding not seen in other genotypes[66].
Patient factors that modulate hepatic steatosis include genetic variations and metabolic dysregulation. Patients with a single nucleotide polymorphism (SNP) in the interleukin 28B (IL28B) gene (genotype CC) possess lower serum levels of triglycerides, higher LDL-C levels[67], and an overall reduced prevalence of hepatic steatosis[68]. The IL-28B CC genotype has also been associated with increased rates of SVR[69,70]. In addition, an independent genome-wide association study also determined that a single genetic variant (I148M) in the human patatin-like phospholipase domain containing 3 (PNPLA3) rs738409 C>G SNP was the strongest genetic determinant of hepatic steatosis[71].
In multiple subsequent candidate gene studies, PNPLA3 I148M has been shown to specifically influence hepatitis C-related liver fat accumulation[72-74], as well as NASH[75,76], fibrosis progression[77] and hepatocellular carcinoma[78]. However, unlike IL28B, PNPLA3 has consistently not been shown to influence SVR[73,79]. These findings were recently confirmed in a post-hoc analysis of a large randomized trial of patients with genotype 1 CHC, where PNPLA3 again was associated with progressive steatosis and development of fibrosis, but not with SVR[73]. Interestingly, the authors observed that this was modulated by the IL28B polymorphism, suggesting new complexity to the relationship between genotype variations and disease progression[73]. Further research will be needed to fully characterize the mechanisms that underpin the associations between these risk alleles and hepatic steatosis.
Additional patient-related determinants of hepatic steatosis in CHC include alcohol use[74] and metabolic derangements, particularly insulin resistance, diabetes and the metabolic syndrome[64]. The link between hepatitis C and insulin resistance has also been supported in multiple population-based cohort studies, where adults with CHC were three to eleven times more likely to develop type 2 diabetes[80], compared to uninfected controls.
Accelerated atherogenesis: Hypocholesterolemia and lower rates of systemic hypertension do not seem to protect patients with HCV infection from atherosclerosis. Ishizaka and colleagues first demonstrated a link between HCV and carotid artery plaque formation[81], and it has since been demonstrated that HCV seropositivity is an independent risk factor for coronary artery disease (CAD), over and above traditional risk factors including age, smoking status, hypertension, diabetes and hyperlipidemia[82,83]. Although some studies have yielded conflicting results, with some confirming[84] and others refuting[85] this link, convincing recent data has nevertheless shown excess cardiovascular mortality during the course of chronic HCV infection[86,87]. Indeed, a recent review concluded that HCV infection should be considered a risk factor for the development of atherosclerosis, and argued for more vigilant preventive cardiac screening in this population[88].
Potential therapeutic role of statin medications
3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitors (statins), are among the most commonly prescribed medications worldwide, and have been shown to be safe in chronic liver disease[89,90]. There is mounting evidence that statins exert powerful pleiotropic effects via both HMG-CoA dependent and independent pathways, modulating inflammation, angiogensis, apoptosis and cell growth[89,91-98]. Several studies have also shown that statins may inhibit HCV replication, and thus may exert powerful anti-HCV effects as well.
Effect of statins on viral replication: Statins appear to block HCV replication by inhibiting de novo cholesterol and geranylgeranylated protein synthesis, thus reducing expression of key HCV viral proteins and inhibiting pro-inflammatory signaling pathways[99,100]. In early in vitro studies, cells cultured with lovastatin successfully inhibited HCV RNA replication[101,102]. This was confirmed in a high-throughput screen of small molecule modulators of HCV replication, with the strongest antiviral activity observed in atorvastatin, fluvastatin and simvastatin[103]. It is thought that the geranylgeranyl lipid product of the mevalonate pathway is necessary for HCV replication[7,104,105]. In experiments with lovastatin, the statin-mediated inhibition of HCV replication was overcome by the addition of geranylgeraniol, but not by farnesol or cholesterol[7,104], results which underscored the importance of the mevalonate pathway in HCV replication.
Despite negative results from in vivo analyses of statin monotherapy[106,107], a great deal of evidence now suggests a beneficial role of statins upon virologic outcomes in patients treated with pegylated interferon (IFN) and ribavirin[52,108,109]. In a large retrospective cohort of 8293 veterans undergoing anti-HCV therapy, statin use was an independent predictor of SVR[108]. In a subsequent uncontrolled, prospective Japanese pilot study of patients infected with genotype 1b, fluvastatin also was associated with improved SVR[110]. Since that time several randomized controlled trials have demonstrated that statins increase SVR rates when combined with peginterferon and ribavirin in genotype 1 infection[111]. Despite this compelling evidence, the future importance of statins for the enhancement of SVR is uncertain as we enter the era of second-generation and novel direct acting antiviral (DAA) therapy for CHC, which yields SVR rates of over 90%.
Anti-fibrotic effects of statins: It has also been postulated that statins may exert antifibrotic effects, although the data are more limited. Animal models show that statin use blocks the activation of hepatic myofibroblasts, inducing apoptosis and preventing both proliferation of hepatic stellate cells (HSCs) and their production of collagens[95,97,98,112-114]. Until recently, reports in humans consisted primarily of retrospective studies of laboratory markers of hepatotoxicity, and were limited by small sample sizes, lack of appropriate controls or histological data from liver biopsy, which remains the gold standard for the assessment of fibrosis[90,115,116]. However, in a recent post-hoc analysis of a large, prospective human trial of patients with advanced CHC followed with serial liver biopsies, it was demonstrated that statin use was associated with significantly reduced fibrosis scores[117,118].
HCC: Mounting evidence also suggests that statins offer chemoprevention against many malignancies, including HCC[91,119-123]. They inhibit cell growth, tumor spread, and appear to exert powerful antiproliferative, antiangiogenic and immunomodulatory effects[96-98]. One mechanism is via direct interference with lipid rafts, thus inhibiting cell signaling, tumor invasion and angiogenesis[124,125]. Via competitive inhibition of HMG-CoA reductase, statins prevent post-translational prenylation of the Ras/Rho superfamily, which are otherwise upregulated in approximately 30% of neoplasms[96,120]. By decreasing expression of MMP-14 and TIMP-2, statins also inhibit the PI3K/PTEN/AKT/mTOR pathway, blocking tumor cell spread[96-98,112].
In a recent population-based cohort of patients infected with HCV, statin users were shown to have a significant reduction in the incidence of HCC[126]. The results were statin-specific, and both dose and duration responses were seen, with a hazard ratio of 0.33 for HCC among those taking higher cumulative daily doses. These results are consistent with several other large observational studies[127,128]. Randomized data, however, does not appear to support the relationship between statins and reduced risk of HCC. In a pooled meta-analysis pooling of 7 observational cohorts and 26 randomized controlled trials, the authors found a 37% reduced risk of HCC among statin users in the observational studies (adjusted odds ratio 0.52, 95%CI: 0.42-0.64), but no benefit attributable to statins in the randomized groups[123]. Such differences between observational studies and randomized trials may reflect length of follow-up, patient selection, lack of sufficient power to detect a difference, within a selected cohort.
Statins in the era of DAA therapy: The role of statins as adjunctive therapy in HCV treatment has so far been limited to the previous standard of care, pegylated interferon and ribavirin. It is unknown what benefit, if any, statins may confer to those patients treated with the new DAA medications. Although statins have been shown to be independent predictors of SVR in both Boceprevir and Telepravir-based trials, with SVR rates > 90% in the majority of treated patients, the additive benefit of statin therapy is less substantial, than that seen with IFN and Ribavirin. Moreover, concerns have been raised about the potential for significant drug interactions between statins and DAAs[129]; at this time, simvastatin, lovastatin and atorvastatin are contraindicated for use with telaprevir[130,131], and both simvastatin and lovastatin are contraindicated with boceprevir[132]. As noted above, there is likely little additive benefit with statins for SVR, as new DAAs demonstrate SVR rates above 95%, however future studies are needed to fully characterize the role of statins for delaying or preventing fibrosis, cirrhosis or the development of HCC.
CONCLUSION
Every aspect of the HCV life cycle is closely linked to human lipid metabolism. Not only does the virus itself circulate as a lipid-rich particle that mimics VLDL, it also utilizes cell surface receptors essential for lipid metabolism to gain entry into the hepatocyte. Once inside the cell, the virus upregulates intracellular lipid synthesis, impairs lipid degradation, and decreases catabolism and export of lipoproteins. As a result, it causes significant intracellular lipid accumulation as well as a relative circulating hypocholesterolemia. Patients with chronic HCV infection are at increased risk of developing hepatic steatosis, fibrosis, and cardiovascular disease including accelerated atherosclerosis. Statins, which inhibit the rate-limiting enzyme of the mevalonate pathway, HMG CoA Reductase, have been shown to play an important role in the modulation of hepatic steatosis and fibrosis, and it is postulated that they may also possess important anti-proliferative, anti-angiogenic and antioxidant effects, with a potential protective role against the development of HCC. It remains to be seen to what degree statin medications will play a role in adjunctive management of patients with CHC in the era of new DAAs.
Footnotes
Conflict-of-interest statement: The authors declare no conflict of interests.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: February 13, 2015
First decision: March 26, 2015
Article in press: June 10, 2015
P- Reviewer: Gangl A, Said ZNA S- Editor: Ma YJ L- Editor: A E- Editor: Liu XM
References
- 1.Armstrong GL, Wasley A, Simard EP, McQuillan GM, Kuhnert WL, Alter MJ. The prevalence of hepatitis C virus infection in the United States, 1999 through 2002. Ann Intern Med. 2006;144:705–714. doi: 10.7326/0003-4819-144-10-200605160-00004. [DOI] [PubMed] [Google Scholar]
- 2.Thomas DL, Seeff LB. Natural history of hepatitis C. Clin Liver Dis. 2005;9:383–398, vi. doi: 10.1016/j.cld.2005.05.003. [DOI] [PubMed] [Google Scholar]
- 3.Chalasani N, Younossi Z, Lavine JE, Diehl AM, Brunt EM, Cusi K, Charlton M, Sanyal AJ; American Gastroenterological Association; American Association for the Study of Liver Diseases; American College of Gastroenterologyh. The diagnosis and management of non-alcoholic fatty liver disease: practice guideline by the American Gastroenterological Association, American Association for the Study of Liver Diseases, and American College of Gastroenterology. Gastroenterology. 2012;142:1592–1609. doi: 10.1053/j.gastro.2012.04.001. [DOI] [PubMed] [Google Scholar]
- 4.Wakita T, Pietschmann T, Kato T, Date T, Miyamoto M, Zhao Z, Murthy K, Habermann A, Kräusslich HG, Mizokami M, et al. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat Med. 2005;11:791–796. doi: 10.1038/nm1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lindenbach BD. Measuring HCV infectivity produced in cell culture and in vivo. Methods Mol Biol. 2009;510:329–336. doi: 10.1007/978-1-59745-394-3_24. [DOI] [PubMed] [Google Scholar]
- 6.Huang H, Sun F, Owen DM, Li W, Chen Y, Gale M, Ye J. Hepatitis C virus production by human hepatocytes dependent on assembly and secretion of very low-density lipoproteins. Proc Natl Acad Sci USA. 2007;104:5848–5853. doi: 10.1073/pnas.0700760104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ye J, Wang C, Sumpter R, Brown MS, Goldstein JL, Gale M. Disruption of hepatitis C virus RNA replication through inhibition of host protein geranylgeranylation. Proc Natl Acad Sci USA. 2003;100:15865–15870. doi: 10.1073/pnas.2237238100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ye J. Reliance of host cholesterol metabolic pathways for the life cycle of hepatitis C virus. PLoS Pathog. 2007;3:e108. doi: 10.1371/journal.ppat.0030108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Syed GH, Amako Y, Siddiqui A. Hepatitis C virus hijacks host lipid metabolism. Trends Endocrinol Metab. 2010;21:33–40. doi: 10.1016/j.tem.2009.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Syed GH, Tang H, Khan M, Hassanein T, Liu J, Siddiqui A. Hepatitis C virus stimulates low-density lipoprotein receptor expression to facilitate viral propagation. J Virol. 2014;88:2519–2529. doi: 10.1128/JVI.02727-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gastaminza P, Cheng G, Wieland S, Zhong J, Liao W, Chisari FV. Cellular determinants of hepatitis C virus assembly, maturation, degradation, and secretion. J Virol. 2008;82:2120–2129. doi: 10.1128/JVI.02053-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chang KS, Jiang J, Cai Z, Luo G. Human apolipoprotein e is required for infectivity and production of hepatitis C virus in cell culture. J Virol. 2007;81:13783–13793. doi: 10.1128/JVI.01091-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ploss A, Dubuisson J. New advances in the molecular biology of hepatitis C virus infection: towards the identification of new treatment targets. Gut. 2012;61 Suppl 1:i25–i35. doi: 10.1136/gutjnl-2012-302048. [DOI] [PubMed] [Google Scholar]
- 14.Jiang J, Cun W, Wu X, Shi Q, Tang H, Luo G. Hepatitis C virus attachment mediated by apolipoprotein E binding to cell surface heparan sulfate. J Virol. 2012;86:7256–7267. doi: 10.1128/JVI.07222-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dao DY, Seremba E, Ajmera V, Sanders C, Hynan LS, Lee WM; Acute Liver Failure Study Group. Use of nucleoside (tide) analogues in patients with hepatitis B-related acute liver failure. Dig Dis Sci. 2012;57:1349–1357. doi: 10.1007/s10620-011-2013-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ploss A, Evans MJ. Hepatitis C virus host cell entry. Curr Opin Virol. 2012;2:14–19. doi: 10.1016/j.coviro.2011.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rhainds D, Brissette L. The role of scavenger receptor class B type I (SR-BI) in lipid trafficking. defining the rules for lipid traders. Int J Biochem Cell Biol. 2004;36:39–77. doi: 10.1016/s1357-2725(03)00173-0. [DOI] [PubMed] [Google Scholar]
- 18.von Hahn T, Rice CM. Hepatitis C virus entry. J Biol Chem. 2008;283:3689–3693. doi: 10.1074/jbc.R700024200. [DOI] [PubMed] [Google Scholar]
- 19.Zeisel MB, Koutsoudakis G, Schnober EK, Haberstroh A, Blum HE, Cosset FL, Wakita T, Jaeck D, Doffoel M, Royer C, et al. Scavenger receptor class B type I is a key host factor for hepatitis C virus infection required for an entry step closely linked to CD81. Hepatology. 2007;46:1722–1731. doi: 10.1002/hep.21994. [DOI] [PubMed] [Google Scholar]
- 20.Voisset C, Callens N, Blanchard E, Op De Beeck A, Dubuisson J, Vu-Dac N. High density lipoproteins facilitate hepatitis C virus entry through the scavenger receptor class B type I. J Biol Chem. 2005;280:7793–7799. doi: 10.1074/jbc.M411600200. [DOI] [PubMed] [Google Scholar]
- 21.Dreux M, Pietschmann T, Granier C, Voisset C, Ricard-Blum S, Mangeot PE, Keck Z, Foung S, Vu-Dac N, Dubuisson J, et al. High density lipoprotein inhibits hepatitis C virus-neutralizing antibodies by stimulating cell entry via activation of the scavenger receptor BI. J Biol Chem. 2006;281:18285–18295. doi: 10.1074/jbc.M602706200. [DOI] [PubMed] [Google Scholar]
- 22.Marzouk D, Sass J, Bakr I, El Hosseiny M, Abdel-Hamid M, Rekacewicz C, Chaturvedi N, Mohamed MK, Fontanet A. Metabolic and cardiovascular risk profiles and hepatitis C virus infection in rural Egypt. Gut. 2007;56:1105–1110. doi: 10.1136/gut.2006.091983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gopal K, Johnson TC, Gopal S, Walfish A, Bang CT, Suwandhi P, Pena-Sahdala HN, Clain DJ, Bodenheimer HC, Min AD. Correlation between beta-lipoprotein levels and outcome of hepatitis C treatment. Hepatology. 2006;44:335–340. doi: 10.1002/hep.21261. [DOI] [PubMed] [Google Scholar]
- 24.Sainz B, Barretto N, Martin DN, Hiraga N, Imamura M, Hussain S, Marsh KA, Yu X, Chayama K, Alrefai WA, et al. Identification of the Niemann-Pick C1-like 1 cholesterol absorption receptor as a new hepatitis C virus entry factor. Nat Med. 2012;18:281–285. doi: 10.1038/nm.2581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Albecka A, Belouzard S, Op de Beeck A, Descamps V, Goueslain L, Bertrand-Michel J, Tercé F, Duverlie G, Rouillé Y, Dubuisson J. Role of low-density lipoprotein receptor in the hepatitis C virus life cycle. Hepatology. 2012;55:998–1007. doi: 10.1002/hep.25501. [DOI] [PubMed] [Google Scholar]
- 26.Goldstein JL, Brown MS. The LDL receptor. Arterioscler Thromb Vasc Biol. 2009;29:431–438. doi: 10.1161/ATVBAHA.108.179564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chappell DA, Medh JD. Receptor-mediated mechanisms of lipoprotein remnant catabolism. Prog Lipid Res. 1998;37:393–422. doi: 10.1016/s0163-7827(98)00017-4. [DOI] [PubMed] [Google Scholar]
- 28.Molina S, Castet V, Fournier-Wirth C, Pichard-Garcia L, Avner R, Harats D, Roitelman J, Barbaras R, Graber P, Ghersa P, et al. The low-density lipoprotein receptor plays a role in the infection of primary human hepatocytes by hepatitis C virus. J Hepatol. 2007;46:411–419. doi: 10.1016/j.jhep.2006.09.024. [DOI] [PubMed] [Google Scholar]
- 29.Carrière M, Rosenberg AR, Conti F, Chouzenoux S, Terris B, Sogni P, Soubrane O, Calmus Y, Podevin P. Low density lipoprotein receptor transcripts correlates with liver hepatitis C virus RNA in patients with alcohol consumption. J Viral Hepat. 2006;13:633–642. doi: 10.1111/j.1365-2893.2006.00737.x. [DOI] [PubMed] [Google Scholar]
- 30.Maxwell KN, Fisher EA, Breslow JL. Overexpression of PCSK9 accelerates the degradation of the LDLR in a post-endoplasmic reticulum compartment. Proc Natl Acad Sci USA. 2005;102:2069–2074. doi: 10.1073/pnas.0409736102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang DW, Lagace TA, Garuti R, Zhao Z, McDonald M, Horton JD, Cohen JC, Hobbs HH. Binding of proprotein convertase subtilisin/kexin type 9 to epidermal growth factor-like repeat A of low density lipoprotein receptor decreases receptor recycling and increases degradation. J Biol Chem. 2007;282:18602–18612. doi: 10.1074/jbc.M702027200. [DOI] [PubMed] [Google Scholar]
- 32.Agnello V, Abel G, Elfahal M, Knight GB, Zhang QX. Hepatitis C virus and other flaviviridae viruses enter cells via low density lipoprotein receptor. Proc Natl Acad Sci USA. 1999;96:12766–12771. doi: 10.1073/pnas.96.22.12766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Moradpour D, Penin F, Rice CM. Replication of hepatitis C virus. Nat Rev Microbiol. 2007;5:453–463. doi: 10.1038/nrmicro1645. [DOI] [PubMed] [Google Scholar]
- 34.Lohmann V, Körner F, Koch J, Herian U, Theilmann L, Bartenschlager R. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science. 1999;285:110–113. doi: 10.1126/science.285.5424.110. [DOI] [PubMed] [Google Scholar]
- 35.Moradpour D, Englert C, Wakita T, Wands JR. Characterization of cell lines allowing tightly regulated expression of hepatitis C virus core protein. Virology. 1996;222:51–63. doi: 10.1006/viro.1996.0397. [DOI] [PubMed] [Google Scholar]
- 36.Perlemuter G, Sabile A, Letteron P, Vona G, Topilco A, Chrétien Y, Koike K, Pessayre D, Chapman J, Barba G, et al. Hepatitis C virus core protein inhibits microsomal triglyceride transfer protein activity and very low density lipoprotein secretion: a model of viral-related steatosis. FASEB J. 2002;16:185–194. doi: 10.1096/fj.01-0396com. [DOI] [PubMed] [Google Scholar]
- 37.Miyanari Y, Atsuzawa K, Usuda N, Watashi K, Hishiki T, Zayas M, Bartenschlager R, Wakita T, Hijikata M, Shimotohno K. The lipid droplet is an important organelle for hepatitis C virus production. Nat Cell Biol. 2007;9:1089–1097. doi: 10.1038/ncb1631. [DOI] [PubMed] [Google Scholar]
- 38.Ohsaki Y, Cheng J, Suzuki M, Shinohara Y, Fujita A, Fujimoto T. Biogenesis of cytoplasmic lipid droplets: from the lipid ester globule in the membrane to the visible structure. Biochim Biophys Acta. 2009;1791:399–407. doi: 10.1016/j.bbalip.2008.10.002. [DOI] [PubMed] [Google Scholar]
- 39.Ogawa K, Hishiki T, Shimizu Y, Funami K, Sugiyama K, Miyanari Y, Shimotohno K. Hepatitis C virus utilizes lipid droplet for production of infectious virus. Proc Jpn Acad Ser B Phys Biol Sci. 2009;85:217–228. doi: 10.2183/pjab.85.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Depla M, Uzbekov R, Hourioux C, Blanchard E, Le Gouge A, Gillet L, Roingeard P. Ultrastructural and quantitative analysis of the lipid droplet clustering induced by hepatitis C virus core protein. Cell Mol Life Sci. 2010;67:3151–3161. doi: 10.1007/s00018-010-0373-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tanaka T, Kuroda K, Ikeda M, Wakita T, Kato N, Makishima M. Hepatitis C virus NS4B targets lipid droplets through hydrophobic residues in the amphipathic helices. J Lipid Res. 2013;54:881–892. doi: 10.1194/jlr.M026443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Camus G, Herker E, Modi AA, Haas JT, Ramage HR, Farese RV, Ott M. Diacylglycerol acyltransferase-1 localizes hepatitis C virus NS5A protein to lipid droplets and enhances NS5A interaction with the viral capsid core. J Biol Chem. 2013;288:9915–9923. doi: 10.1074/jbc.M112.434910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Harris C, Herker E, Farese RV, Ott M. Hepatitis C virus core protein decreases lipid droplet turnover: a mechanism for core-induced steatosis. J Biol Chem. 2011;286:42615–42625. doi: 10.1074/jbc.M111.285148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hussain MM, Rava P, Walsh M, Rana M, Iqbal J. Multiple functions of microsomal triglyceride transfer protein. Nutr Metab (Lond) 2012;9:14. doi: 10.1186/1743-7075-9-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nahmias Y, Goldwasser J, Casali M, van Poll D, Wakita T, Chung RT, Yarmush ML. Apolipoprotein B-dependent hepatitis C virus secretion is inhibited by the grapefruit flavonoid naringenin. Hepatology. 2008;47:1437–1445. doi: 10.1002/hep.22197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Goldwasser J, Cohen PY, Lin W, Kitsberg D, Balaguer P, Polyak SJ, Chung RT, Yarmush ML, Nahmias Y. Naringenin inhibits the assembly and long-term production of infectious hepatitis C virus particles through a PPAR-mediated mechanism. J Hepatol. 2011;55:963–971. doi: 10.1016/j.jhep.2011.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Counihan NA, Rawlinson SM, Lindenbach BD. Trafficking of hepatitis C virus core protein during virus particle assembly. PLoS Pathog. 2011;7:e1002302. doi: 10.1371/journal.ppat.1002302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jiang J, Luo G. Apolipoprotein E but not B is required for the formation of infectious hepatitis C virus particles. J Virol. 2009;83:12680–12691. doi: 10.1128/JVI.01476-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Corey KE, Mendez-Navarro J, Barlow LL, Patwardhan V, Zheng H, Kim AY, Lauer GM, Chung RT. Acute hepatitis C infection lowers serum lipid levels. J Viral Hepat. 2011;18:e366–e371. doi: 10.1111/j.1365-2893.2011.01434.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lambert JE, Bain VG, Ryan EA, Thomson AB, Clandinin MT. Elevated lipogenesis and diminished cholesterol synthesis in patients with hepatitis C viral infection compared to healthy humans. Hepatology. 2013;57:1697–1704. doi: 10.1002/hep.25990. [DOI] [PubMed] [Google Scholar]
- 51.Sheridan DA, Price DA, Schmid ML, Toms GL, Donaldson P, Neely D, Bassendine MF. Apolipoprotein B-associated cholesterol is a determinant of treatment outcome in patients with chronic hepatitis C virus infection receiving anti-viral agents interferon-alpha and ribavirin. Aliment Pharmacol Ther. 2009;29:1282–1290. doi: 10.1111/j.1365-2036.2009.04012.x. [DOI] [PubMed] [Google Scholar]
- 52.Harrison SA, Rossaro L, Hu KQ, Patel K, Tillmann H, Dhaliwal S, Torres DM, Koury K, Goteti VS, Noviello S, et al. Serum cholesterol and statin use predict virological response to peginterferon and ribavirin therapy. Hepatology. 2010;52:864–874. doi: 10.1002/hep.23787. [DOI] [PubMed] [Google Scholar]
- 53.Goodman ZD, Ishak KG. Histopathology of hepatitis C virus infection. Semin Liver Dis. 1995;15:70–81. doi: 10.1055/s-2007-1007264. [DOI] [PubMed] [Google Scholar]
- 54.Fujino T, Nakamuta M, Yada R, Aoyagi Y, Yasutake K, Kohjima M, Fukuizumi K, Yoshimoto T, Harada N, Yada M, et al. Expression profile of lipid metabolism-associated genes in hepatitis C virus-infected human liver. Hepatol Res. 2010;40:923–929. doi: 10.1111/j.1872-034X.2010.00700.x. [DOI] [PubMed] [Google Scholar]
- 55.Abid K, Pazienza V, de Gottardi A, Rubbia-Brandt L, Conne B, Pugnale P, Rossi C, Mangia A, Negro F. An in vitro model of hepatitis C virus genotype 3a-associated triglycerides accumulation. J Hepatol. 2005;42:744–751. doi: 10.1016/j.jhep.2004.12.034. [DOI] [PubMed] [Google Scholar]
- 56.Castéra L, Hézode C, Roudot-Thoraval F, Bastie A, Zafrani ES, Pawlotsky JM, Dhumeaux D. Worsening of steatosis is an independent factor of fibrosis progression in untreated patients with chronic hepatitis C and paired liver biopsies. Gut. 2003;52:288–292. doi: 10.1136/gut.52.2.288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cross TJ, Quaglia A, Hughes S, Joshi D, Harrison PM. The impact of hepatic steatosis on the natural history of chronic hepatitis C infection. J Viral Hepat. 2009;16:492–499. doi: 10.1111/j.1365-2893.2009.01098.x. [DOI] [PubMed] [Google Scholar]
- 58.Lok AS, Everhart JE, Chung RT, Kim HY, Everson GT, Hoefs JC, Greenson JK, Sterling RK, Lindsay KL, Lee WM, et al. Evolution of hepatic steatosis in patients with advanced hepatitis C: results from the hepatitis C antiviral long-term treatment against cirrhosis (HALT-C) trial. Hepatology. 2009;49:1828–1837. doi: 10.1002/hep.22865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rubbia-Brandt L, Quadri R, Abid K, Giostra E, Malé PJ, Mentha G, Spahr L, Zarski JP, Borisch B, Hadengue A, et al. Hepatocyte steatosis is a cytopathic effect of hepatitis C virus genotype 3. J Hepatol. 2000;33:106–115. doi: 10.1016/s0168-8278(00)80166-x. [DOI] [PubMed] [Google Scholar]
- 60.Clark PJ, Thompson AJ, Vock DM, Kratz LE, Tolun AA, Muir AJ, McHutchison JG, Subramanian M, Millington DM, Kelley RI, et al. Hepatitis C virus selectively perturbs the distal cholesterol synthesis pathway in a genotype-specific manner. Hepatology. 2012;56:49–56. doi: 10.1002/hep.25631. [DOI] [PubMed] [Google Scholar]
- 61.Roingeard P, Hourioux C. Hepatitis C virus core protein, lipid droplets and steatosis. J Viral Hepat. 2008;15:157–164. doi: 10.1111/j.1365-2893.2007.00953.x. [DOI] [PubMed] [Google Scholar]
- 62.Piodi A, Chouteau P, Lerat H, Hézode C, Pawlotsky JM. Morphological changes in intracellular lipid droplets induced by different hepatitis C virus genotype core sequences and relationship with steatosis. Hepatology. 2008;48:16–27. doi: 10.1002/hep.22288. [DOI] [PubMed] [Google Scholar]
- 63.Hui JM, Kench J, Farrell GC, Lin R, Samarasinghe D, Liddle C, Byth K, George J. Genotype-specific mechanisms for hepatic steatosis in chronic hepatitis C infection. J Gastroenterol Hepatol. 2002;17:873–881. doi: 10.1046/j.1440-1746.2002.02813.x. [DOI] [PubMed] [Google Scholar]
- 64.Fartoux L, Chazouillères O, Wendum D, Poupon R, Serfaty L. Impact of steatosis on progression of fibrosis in patients with mild hepatitis C. Hepatology. 2005;41:82–87. doi: 10.1002/hep.20519. [DOI] [PubMed] [Google Scholar]
- 65.Lonardo A, Lombardini S, Scaglioni F, Carulli L, Ricchi M, Ganazzi D, Adinolfi LE, Ruggiero G, Carulli N, Loria P. Hepatic steatosis and insulin resistance: does etiology make a difference? J Hepatol. 2006;44:190–196. doi: 10.1016/j.jhep.2005.06.018. [DOI] [PubMed] [Google Scholar]
- 66.Kumar D, Farrell GC, Fung C, George J. Hepatitis C virus genotype 3 is cytopathic to hepatocytes: Reversal of hepatic steatosis after sustained therapeutic response. Hepatology. 2002;36:1266–1272. doi: 10.1053/jhep.2002.36370. [DOI] [PubMed] [Google Scholar]
- 67.Li JH, Lao XQ, Tillmann HL, Rowell J, Patel K, Thompson A, Suchindran S, Muir AJ, Guyton JR, Gardner SD, et al. Interferon-lambda genotype and low serum low-density lipoprotein cholesterol levels in patients with chronic hepatitis C infection. Hepatology. 2010;51:1904–1911. doi: 10.1002/hep.23592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tillmann HL, Patel K, Muir AJ, Guy CD, Li JH, Lao XQ, Thompson A, Clark PJ, Gardner SD, McHutchison JG, et al. Beneficial IL28B genotype associated with lower frequency of hepatic steatosis in patients with chronic hepatitis C. J Hepatol. 2011;55:1195–1200. doi: 10.1016/j.jhep.2011.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Thompson AJ, Muir AJ, Sulkowski MS, Ge D, Fellay J, Shianna KV, Urban T, Afdhal NH, Jacobson IM, Esteban R, et al. Interleukin-28B polymorphism improves viral kinetics and is the strongest pretreatment predictor of sustained virologic response in genotype 1 hepatitis C virus. Gastroenterology. 2010;139:120–129.e18. doi: 10.1053/j.gastro.2010.04.013. [DOI] [PubMed] [Google Scholar]
- 70.Asselah T, De Muynck S, Broët P, Masliah-Planchon J, Blanluet M, Bièche I, Lapalus M, Martinot-Peignoux M, Lada O, Estrabaud E, et al. IL28B polymorphism is associated with treatment response in patients with genotype 4 chronic hepatitis C. J Hepatol. 2012;56:527–532. doi: 10.1016/j.jhep.2011.09.008. [DOI] [PubMed] [Google Scholar]
- 71.Romeo S, Kozlitina J, Xing C, Pertsemlidis A, Cox D, Pennacchio LA, Boerwinkle E, Cohen JC, Hobbs HH. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat Genet. 2008;40:1461–1465. doi: 10.1038/ng.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cai T, Dufour JF, Muellhaupt B, Gerlach T, Heim M, Moradpour D, Cerny A, Malinverni R, Kaddai V, Bochud M, Negro F, Bochud PY; Swiss Hepatitis C Cohort Study Group. Viral genotype-specific role of PNPLA3, PPARG, MTTP, and IL28B in hepatitis C virus-associated steatosis. J Hepatol. 2011;55:529–535. doi: 10.1016/j.jhep.2010.12.020. [DOI] [PubMed] [Google Scholar]
- 73.Clark PJ, Thompson AJ, Zhu Q, Vock DM, Zhu M, Patel K, Harrison SA, Naggie S, Ge D, Tillmann HL, et al. The association of genetic variants with hepatic steatosis in patients with genotype 1 chronic hepatitis C infection. Dig Dis Sci. 2012;57:2213–2221. doi: 10.1007/s10620-012-2171-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Valenti L, Colombo M, Fargion S. Modulation of the effect of PNPLA3 I148M mutation on steatosis and liver damage by alcohol intake in patients with chronic hepatitis C. J Hepatol. 2011;55:1470–1471; author reply 1471-1472. doi: 10.1016/j.jhep.2011.04.032. [DOI] [PubMed] [Google Scholar]
- 75.Sookoian S, Pirola CJ. Meta-analysis of the influence of I148M variant of patatin-like phospholipase domain containing 3 gene (PNPLA3) on the susceptibility and histological severity of nonalcoholic fatty liver disease. Hepatology. 2011;53:1883–1894. doi: 10.1002/hep.24283. [DOI] [PubMed] [Google Scholar]
- 76.Ampuero J, Del Campo JA, Rojas L, García-Lozano JR, Solá R, Andrade R, Pons JA, Navarro JM, Calleja JL, Buti M, et al. PNPLA3 rs738409 causes steatosis according to viral & amp; IL28B genotypes in hepatitis C. Ann Hepatol. 2014;13:356–363. [PubMed] [Google Scholar]
- 77.Petit JM, Guiu B, Masson D, Duvillard L, Jooste V, Buffier P, Bouillet B, Brindisi MC, Robin I, Gambert P, et al. PNPLA3 polymorphism influences liver fibrosis in unselected patients with type 2 diabetes. Liver Int. 2011;31:1332–1336. doi: 10.1111/j.1478-3231.2011.02566.x. [DOI] [PubMed] [Google Scholar]
- 78.Guyot E, Sutton A, Rufat P, Laguillier C, Mansouri A, Moreau R, Ganne-Carrié N, Beaugrand M, Charnaux N, Trinchet JC, et al. PNPLA3 rs738409, hepatocellular carcinoma occurrence and risk model prediction in patients with cirrhosis. J Hepatol. 2013;58:312–318. doi: 10.1016/j.jhep.2012.09.036. [DOI] [PubMed] [Google Scholar]
- 79.Valenti L, Aghemo A, Stättermayer AF, Maggioni P, De Nicola S, Motta BM, Rumi MG, Dongiovanni P, Ferenci P, Colombo M, et al. Implications of PNPLA3 polymorphism in chronic hepatitis C patients receiving peginterferon plus ribavirin. Aliment Pharmacol Ther. 2012;35:1434–1442. doi: 10.1111/j.1365-2036.2012.05109.x. [DOI] [PubMed] [Google Scholar]
- 80.Mehta SH, Brancati FL, Strathdee SA, Pankow JS, Netski D, Coresh J, Szklo M, Thomas DL. Hepatitis C virus infection and incident type 2 diabetes. Hepatology. 2003;38:50–56. doi: 10.1053/jhep.2003.50291. [DOI] [PubMed] [Google Scholar]
- 81.Ishizaka N, Ishizaka Y, Takahashi E, Tooda Ei, Hashimoto H, Nagai R, Yamakado M. Association between hepatitis C virus seropositivity, carotid-artery plaque, and intima-media thickening. Lancet. 2002;359:133–135. doi: 10.1016/s0140-6736(02)07339-7. [DOI] [PubMed] [Google Scholar]
- 82.Vassalle C, Masini S, Bianchi F, Zucchelli GC. Evidence for association between hepatitis C virus seropositivity and coronary artery disease. Heart. 2004;90:565–566. doi: 10.1136/hrt.2003.018937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Butt AA, Xiaoqiang W, Budoff M, Leaf D, Kuller LH, Justice AC. Hepatitis C virus infection and the risk of coronary disease. Clin Infect Dis. 2009;49:225–232. doi: 10.1086/599371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Targher G, Bertolini L, Padovani R, Rodella S, Arcaro G, Day C. Differences and similarities in early atherosclerosis between patients with non-alcoholic steatohepatitis and chronic hepatitis B and C. J Hepatol. 2007;46:1126–1132. doi: 10.1016/j.jhep.2007.01.021. [DOI] [PubMed] [Google Scholar]
- 85.Arcari CM, Nelson KE, Netski DM, Nieto FJ, Gaydos CA. No association between hepatitis C virus seropositivity and acute myocardial infarction. Clin Infect Dis. 2006;43:e53–e56. doi: 10.1086/507031. [DOI] [PubMed] [Google Scholar]
- 86.Guiltinan AM, Kaidarova Z, Custer B, Orland J, Strollo A, Cyrus S, Busch MP, Murphy EL. Increased all-cause, liver, and cardiac mortality among hepatitis C virus-seropositive blood donors. Am J Epidemiol. 2008;167:743–750. doi: 10.1093/aje/kwm370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.El-Kamary SS, Jhaveri R, Shardell MD. All-cause, liver-related, and non-liver-related mortality among HCV-infected individuals in the general US population. Clin Infect Dis. 2011;53:150–157. doi: 10.1093/cid/cir306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Adinolfi LE, Zampino R, Restivo L, Lonardo A, Guerrera B, Marrone A, Nascimbeni F, Florio A, Loria P. Chronic hepatitis C virus infection and atherosclerosis: clinical impact and mechanisms. World J Gastroenterol. 2014;20:3410–3417. doi: 10.3748/wjg.v20.i13.3410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Schuppan D, Afdhal NH. Liver cirrhosis. Lancet. 2008;371:838–851. doi: 10.1016/S0140-6736(08)60383-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lewis JH, Mortensen ME, Zweig S, Fusco MJ, Medoff JR, Belder R; Pravastatin in Chronic Liver Disease Study Investigators. Efficacy and safety of high-dose pravastatin in hypercholesterolemic patients with well-compensated chronic liver disease: Results of a prospective, randomized, double-blind, placebo-controlled, multicenter trial. Hepatology. 2007;46:1453–1463. doi: 10.1002/hep.21848. [DOI] [PubMed] [Google Scholar]
- 91.Kuoppala J, Lamminpää A, Pukkala E. Statins and cancer: A systematic review and meta-analysis. Eur J Cancer. 2008;44:2122–2132. doi: 10.1016/j.ejca.2008.06.025. [DOI] [PubMed] [Google Scholar]
- 92.Kalluri R, Neilson EG. Epithelial-mesenchymal transition and its implications for fibrosis. J Clin Invest. 2003;112:1776–1784. doi: 10.1172/JCI20530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kisseleva T, Brenner DA. Mechanisms of fibrogenesis. Exp Biol Med (Maywood) 2008;233:109–122. doi: 10.3181/0707-MR-190. [DOI] [PubMed] [Google Scholar]
- 94.Iwaisako K, Brenner DA, Kisseleva T. What’s new in liver fibrosis? The origin of myofibroblasts in liver fibrosis. J Gastroenterol Hepatol. 2012;27 Suppl 2:65–68. doi: 10.1111/j.1440-1746.2011.07002.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Shirin H, Sharvit E, Aeed H, Gavish D, Bruck R. Atorvastatin and rosuvastatin do not prevent thioacetamide induced liver cirrhosis in rats. World J Gastroenterol. 2013;19:241–248. doi: 10.3748/wjg.v19.i2.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Sun HY, Singh N. Antimicrobial and immunomodulatory attributes of statins: relevance in solid-organ transplant recipients. Clin Infect Dis. 2009;48:745–755. doi: 10.1086/597039. [DOI] [PubMed] [Google Scholar]
- 97.Wu J, Wong WW, Khosravi F, Minden MD, Penn LZ. Blocking the Raf/MEK/ERK pathway sensitizes acute myelogenous leukemia cells to lovastatin-induced apoptosis. Cancer Res. 2004;64:6461–6468. doi: 10.1158/0008-5472.CAN-04-0866. [DOI] [PubMed] [Google Scholar]
- 98.Rao S, Porter DC, Chen X, Herliczek T, Lowe M, Keyomarsi K. Lovastatin-mediated G1 arrest is through inhibition of the proteasome, independent of hydroxymethyl glutaryl-CoA reductase. Proc Natl Acad Sci USA. 1999;96:7797–7802. doi: 10.1073/pnas.96.14.7797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Dimitroulakos J, Lorimer IA, Goss G. Strategies to enhance epidermal growth factor inhibition: targeting the mevalonate pathway. Clin Cancer Res. 2006;12:4426s–4431s. doi: 10.1158/1078-0432.CCR-06-0089. [DOI] [PubMed] [Google Scholar]
- 100.Zhao TT, Le Francois BG, Goss G, Ding K, Bradbury PA, Dimitroulakos J. Lovastatin inhibits EGFR dimerization and AKT activation in squamous cell carcinoma cells: potential regulation by targeting rho proteins. Oncogene. 2010;29:4682–4692. doi: 10.1038/onc.2010.219. [DOI] [PubMed] [Google Scholar]
- 101.Goldstein JL, Brown MS. Regulation of the mevalonate pathway. Nature. 1990;343:425–430. doi: 10.1038/343425a0. [DOI] [PubMed] [Google Scholar]
- 102.Peng LF, Schaefer EA, Maloof N, Skaff A, Berical A, Belon CA, Heck JA, Lin W, Frick DN, Allen TM, et al. Ceestatin, a novel small molecule inhibitor of hepatitis C virus replication, inhibits 3-hydroxy-3-methylglutaryl-coenzyme A synthase. J Infect Dis. 2011;204:609–616. doi: 10.1093/infdis/jir303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kim SS, Peng LF, Lin W, Choe WH, Sakamoto N, Kato N, Ikeda M, Schreiber SL, Chung RT. A cell-based, high-throughput screen for small molecule regulators of hepatitis C virus replication. Gastroenterology. 2007;132:311–320. doi: 10.1053/j.gastro.2006.10.032. [DOI] [PubMed] [Google Scholar]
- 104.Zhang FL, Casey PJ. Protein prenylation: molecular mechanisms and functional consequences. Annu Rev Biochem. 1996;65:241–269. doi: 10.1146/annurev.bi.65.070196.001325. [DOI] [PubMed] [Google Scholar]
- 105.Wang C, Gale M, Keller BC, Huang H, Brown MS, Goldstein JL, Ye J. Identification of FBL2 as a geranylgeranylated cellular protein required for hepatitis C virus RNA replication. Mol Cell. 2005;18:425–434. doi: 10.1016/j.molcel.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 106.Patel K, Jhaveri R, George J, Qiang G, Kenedi C, Brown K, Cates C, Zekry A, Tillmann HL, Elliott L, et al. Open-label, ascending dose, prospective cohort study evaluating the antiviral efficacy of Rosuvastatin therapy in serum and lipid fractions in patients with chronic hepatitis C. J Viral Hepat. 2011;18:331–337. doi: 10.1111/j.1365-2893.2010.01310.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.O’Leary JG, Chan JL, McMahon CM, Chung RT. Atorvastatin does not exhibit antiviral activity against HCV at conventional doses: a pilot clinical trial. Hepatology. 2007;45:895–898. doi: 10.1002/hep.21554. [DOI] [PubMed] [Google Scholar]
- 108.Rao GA, Pandya PK. Statin therapy improves sustained virologic response among diabetic patients with chronic hepatitis C. Gastroenterology. 2011;140:144–152. doi: 10.1053/j.gastro.2010.08.055. [DOI] [PubMed] [Google Scholar]
- 109.Kondo C, Atsukawa M, Tsubota A, Itokawa N, Fukuda T, Matsushita Y, Kidokoro H, Kobayashi T, Narahara Y, Nakatsuka K, et al. An open-label randomized controlled study of pegylated interferon/ribavirin combination therapy for chronic hepatitis C with versus without fluvastatin. J Viral Hepat. 2012;19:615–622. doi: 10.1111/j.1365-2893.2011.01584.x. [DOI] [PubMed] [Google Scholar]
- 110.Sezaki H, Suzuki F, Akuta N, Yatsuji H, Hosaka T, Kobayashi M, Suzuki Y, Arase Y, Ikeda K, Miyakawa Y, et al. An open pilot study exploring the efficacy of fluvastatin, pegylated interferon and ribavirin in patients with hepatitis C virus genotype 1b in high viral loads. Intervirology. 2009;52:43–48. doi: 10.1159/000213504. [DOI] [PubMed] [Google Scholar]
- 111.Zhu Q, Li N, Han Q, Zhang P, Yang C, Zeng X, Chen Y, Lv Y, Liu X, Liu Z. Statin therapy improves response to interferon alfa and ribavirin in chronic hepatitis C: a systematic review and meta-analysis. Antiviral Res. 2013;98:373–379. doi: 10.1016/j.antiviral.2013.04.009. [DOI] [PubMed] [Google Scholar]
- 112.Trebicka J, Hennenberg M, Odenthal M, Shir K, Klein S, Granzow M, Vogt A, Dienes HP, Lammert F, Reichen J, et al. Atorvastatin attenuates hepatic fibrosis in rats after bile duct ligation via decreased turnover of hepatic stellate cells. J Hepatol. 2010;53:702–712. doi: 10.1016/j.jhep.2010.04.025. [DOI] [PubMed] [Google Scholar]
- 113.Miyaki T, Nojiri S, Shinkai N, Kusakabe A, Matsuura K, Iio E, Takahashi S, Yan G, Ikeda K, Joh T. Pitavastatin inhibits hepatic steatosis and fibrosis in non-alcoholic steatohepatitis model rats. Hepatol Res. 2011;41:375–385. doi: 10.1111/j.1872-034X.2010.00769.x. [DOI] [PubMed] [Google Scholar]
- 114.Marcelli M, Cunningham GR, Haidacher SJ, Padayatty SJ, Sturgis L, Kagan C, Denner L. Caspase-7 is activated during lovastatin-induced apoptosis of the prostate cancer cell line LNCaP. Cancer Res. 1998;58:76–83. [PubMed] [Google Scholar]
- 115.Avins AL, Manos MM, Ackerson L, Zhao W, Murphy R, Levin TR, Watson DJ, Hwang PM, Replogle A, Levine JG. Hepatic effects of lovastatin exposure in patients with liver disease: a retrospective cohort study. Drug Saf. 2008;31:325–334. doi: 10.2165/00002018-200831040-00006. [DOI] [PubMed] [Google Scholar]
- 116.Henderson LM, Patel S, Giordano TP, Green L, El-Serag HB. Statin therapy and serum transaminases among a cohort of HCV-infected veterans. Dig Dis Sci. 2010;55:190–195. doi: 10.1007/s10620-009-0959-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Simon TG, King LY, Zheng H, Chung RT. Statin use is associated with a reduced risk of fibrosis progression in chronic hepatitis C. J Hepatol. 2015;62:18–23. doi: 10.1016/j.jhep.2014.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Butt AA, Shaikh O, Rogal S. Statins improve SVR, reduce fibrosis progression and HCC among HCV persons. 22nd Conference on Retroviruses and Opportunistic Infections. Seattle: WA; 2015. [Google Scholar]
- 119.Kisseleva T, Brenner DA. Anti-fibrogenic strategies and the regression of fibrosis. Best Pract Res Clin Gastroenterol. 2011;25:305–317. doi: 10.1016/j.bpg.2011.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Demierre MF, Higgins PD, Gruber SB, Hawk E, Lippman SM. Statins and cancer prevention. Nat Rev Cancer. 2005;5:930–942. doi: 10.1038/nrc1751. [DOI] [PubMed] [Google Scholar]
- 121.Welzel TM, Graubard BI, Zeuzem S, El-Serag HB, Davila JA, McGlynn KA. Metabolic syndrome increases the risk of primary liver cancer in the United States: a study in the SEER-Medicare database. Hepatology. 2011;54:463–471. doi: 10.1002/hep.24397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Bonovas S, Filioussi K, Tsavaris N, Sitaras NM. Statins and cancer risk: a literature-based meta-analysis and meta-regression analysis of 35 randomized controlled trials. J Clin Oncol. 2006;24:4808–4817. doi: 10.1200/JCO.2006.06.3560. [DOI] [PubMed] [Google Scholar]
- 123.Singh S, Singh PP, Singh AG, Murad MH, Sanchez W. Statins are associated with a reduced risk of hepatocellular cancer: a systematic review and meta-analysis. Gastroenterology. 2013;144:323–332. doi: 10.1053/j.gastro.2012.10.005. [DOI] [PubMed] [Google Scholar]
- 124.Ishihara Y, Ohmori K, Mizukawa M, Hasan AU, Noma T, Kohno M. Beneficial direct adipotropic actions of pitavastatin in vitro and their manifestations in obese mice. Atherosclerosis. 2010;212:131–138. doi: 10.1016/j.atherosclerosis.2010.04.019. [DOI] [PubMed] [Google Scholar]
- 125.Clendening JW, Penn LZ. Targeting tumor cell metabolism with statins. Oncogene. 2012;31:4967–4978. doi: 10.1038/onc.2012.6. [DOI] [PubMed] [Google Scholar]
- 126.Tsan YT, Lee CH, Ho WC, Lin MH, Wang JD, Chen PC. Statins and the risk of hepatocellular carcinoma in patients with hepatitis C virus infection. J Clin Oncol. 2013;31:1514–1521. doi: 10.1200/JCO.2012.44.6831. [DOI] [PubMed] [Google Scholar]
- 127.El-Serag HB, Johnson ML, Hachem C, Morgana RO. Statins are associated with a reduced risk of hepatocellular carcinoma in a large cohort of patients with diabetes. Gastroenterology. 2009;136:1601–1608. doi: 10.1053/j.gastro.2009.01.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Nielsen SF, Nordestgaard BG, Bojesen SE. Statin use and reduced cancer-related mortality. N Engl J Med. 2012;367:1792–1802. doi: 10.1056/NEJMoa1201735. [DOI] [PubMed] [Google Scholar]
- 129.Poordad F, McCone J, Bacon BR, Bruno S, Manns MP, Sulkowski MS, Jacobson IM, Reddy KR, Goodman ZD, Boparai N, et al. Boceprevir for untreated chronic HCV genotype 1 infection. N Engl J Med. 2011;364:1195–1206. doi: 10.1056/NEJMoa1010494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Kiser JJ, Burton JR, Anderson PL, Everson GT. Review and management of drug interactions with boceprevir and telaprevir. Hepatology. 2012;55:1620–1628. doi: 10.1002/hep.25653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Lee JE, van Heeswijk R, Alves K, Smith F, Garg V. Effect of the hepatitis C virus protease inhibitor telaprevir on the pharmacokinetics of amlodipine and atorvastatin. Antimicrob Agents Chemother. 2011;55:4569–4574. doi: 10.1128/AAC.00653-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Seden K, Back D. Directly acting antivirals for hepatitis C and antiretrovirals: potential for drug-drug interactions. Curr Opin HIV AIDS. 2011;6:514–526. doi: 10.1097/COH.0b013e32834b54dc. [DOI] [PubMed] [Google Scholar]