

Abstract
AIM: To explore the effect of the histone deacetylase inhibitor givinostat on proteins related to regulation of hepatic stellate cell proliferation.
METHODS: The cell counting kit-8 assay and flow cytometry were used to observe changes in proliferation, apoptosis, and cell cycle in hepatic stellate cells treated with givinostat. Western blot was used to observe expression changes in p21, p57, CDK4, CDK6, cyclinD1, caspase-3, and caspase-9 in hepatic stellate cells exposed to givinostat. The scratch assay was used to analyze the effect of givinostat on cell migration. Effects of givinostat on the reactive oxygen species profile, mitochondrial membrane potential, and mitochondrial permeability transition pore opening in JS-1 cells were observed by laser confocal microscopy.
RESULTS: Givinostat significantly inhibited JS-1 cell proliferation and promoted cell apoptosis, leading to cell cycle arrest in G0/G1 phases. Treatment with givinostat downregulated protein expression of CDK4, CDK6, and cyclin D1, whereas expression of p21 and p57 was significantly increased. The givinostat-induced apoptosis of hepatic stellate cells was mainly mediated through p38 and extracellular signal-regulated kinase 1/2. Givinostat treatment increased intracellular reactive oxygen species production, decreased mitochondrial membrane potential, and promoted mitochondrial permeability transition pore opening. Acetylation of superoxide dismutase (acetyl K68) and nuclear factor-κB p65 (acetyl K310) was upregulated, while there was no change in protein expression. Moreover, the notable beneficial effect of givinostat on liver fibrosis was also confirmed in the mouse models.
CONCLUSION: Givinostat has antifibrotic activities via regulating the acetylation of nuclear factor-κB and superoxide dismutase 2, thus inhibiting hepatic stellate cell proliferation and inducing apoptosis.
Keywords: Givinostat, Hepatic stellate cells, Histone deacetylase inhibitor, Nuclear factor-κB, Superoxide dismutase
Core tip: There is currently no effective therapeutic treatment for liver fibrosis. Inhibition of hepatic stellate cell activation and proliferation or induction of apoptosis is the mainstream strategy for the treatment of liver fibrosis. This study demonstrates that a histone deacetylase inhibitor, givinostat, has antifibrotic activities both in vivo and in vitro, which might be achieved by regulating the acetylation of nuclear factor-κB and superoxide dismutase, thus stimulating oxidative stress, activating mitochondrial pathways, inhibiting hepatic stellate cell proliferation, and inducing apoptosis. These results may provide new directions and evidence in the research and development of novel drugs for liver fibrosis.
INTRODUCTION
Liver fibrosis is a major cause of morbidity and mortality worldwide due to chronic viral hepatitis and, more recently, from fatty liver disease associated with obesity[1]. Activation of hepatic stellate cells (HSCs) is the central event in liver fibrosis and often serves as a trigger[1,2].
HSCs are activated and transdifferentiated into myofibroblasts in event of hepatic injury. The main changes include proliferation, migration, and enhanced contractility, as well as synthesis of large amounts of cytokines and extracellular matrix[3-5]. Persistent injury can cause chronic inflammation, resulting in replacement of liver parenchymal cells by scar tissue. Moreover, long-term fibrotic reaction leads to end-stage liver disease, cirrhosis, and hepatocellular carcinoma. In fact, the incidence of hepatocellular carcinoma has steadily increased worldwide. Thus, in addition to management of the causes of disease, inhibition of HSC activation and proliferation, as well as induction of apoptosis, has been the mainstream strategy for the treatment of liver fibrosis. HSC activation after stimulation is recognized as a process involving multiple cytokines and is coregulated by various cell-signaling pathways at different levels[6,7]. Although no well-established medication or regimen has been available for the treatment of liver fibrosis, an increasing number of molecularly targeted drugs have promising efficacy[8].
Recent studies[9-11] have shown that protein acetylation plays an important role during HSC activation; imbalanced histone acetylation due to histone deacetylase (HDAC) overexpression is closely associated with the occurrence and development of liver fibrosis[9]. The HDAC inhibitors trichostatin A[10] and valproic acid[9] can prevent the transdifferentiation of static HSCs into myofibroblasts and suppress α-smooth muscle actin (SMA) and collagen I gene expression in vitro.
As a novel hydroxamate-derived HDAC inhibitor[12], givinostat (also known as ITF2357) is characterized by its significant targeted anticancer activities and low toxicity profiles[13-15]. Moreover, givinostat has anti-inflammatory activity and can suppress lipopolysaccharide (LPS)-induced cytokines such as tumor necrosis factor-α, interleukin-1 and -6, and interferon[16-18]. The phase II clinical trials on the roles of givinostat in treating juvenile idiopathic arthritis, Hodgkin’s lymphoma, and polycythemia vera were finished in 2002. In the current study, the antifibrotic activities of givinostat were assessed both in vivo and in vitro to understand the mechanism of liver fibrosis and to provide new directions and evidence for novel drug development.
MATERIALS AND METHODS
Reagents
The murine HSC line JS-1 was provided courtesy of Xu Lieming from Shanghai University of Traditional Chinese Medicine. Givinostat was purchased from Selleck (Houston, TX, United States). The following were purchased from Thermo Fisher Scientific (Waltham, MA, United States): Ham’s F12 medium, Dulbecco’s Modified Eagle’s medium (DMEM), trypsin-EDTA solution, fetal bovine serum, and the Pierce BCA Protein Assay Kit. The Cell Counting Kit-8 (CCK-8) was purchased from Dojindo (Kumamoto, Japan). JC-1 staining solution, 2′,7′-dichlorofluorescein diacetate (DCFH-DA), calcein-AM, and CoCl2 were obtained from Sigma-Aldrich (St. Louis, MO, United States). The Annexin V-FITC Apoptosis Detection Kit and FACSCalibur Flow Cytometer were purchased from BD Pharmingen (San Diego, CA, United States), and Amersham ECL plus Western Blotting Detection System was purchased from GE (Little Chalfont, United Kingdom. The confocal laser-scanning microscope used was the FluoView FV1200 from Olympus (Tokyo, Japan). Other reagents were from Abcam (Cambridge, United Kingdom).
CCK-8 assay
After the JS-1 cell line was cultured in DMEM with 10% fetal bovine serum for 24 h, 30 wells of JS-1 cells were divided into two groups. In the first group, the culture medium was replaced by complete medium with final givinostat concentrations of 0 nmol/L, 125 nmol/L, 250 nmol/L, 500 nmol/L, and 1000 nmol/L. In the second group, givinostat of relevant concentrations was added concomitantly with 100 nmol/L of LPS solution. Three replicates were performed for each group. After inoculation at 37 °C and 5% CO2 for 24 h, each well (100 μL) was incubated with 10 μL of CCK-8 solution. The plates were incubated at 37 °C for 1 h and the absorbance was measured at 450 nm using a microplate reader.
Detection of apoptosis and cell cycle by flow cytometry
The JS-1 cells were inoculated in 10 mL complete medium in three 100-mm culture dishes (1 × 106 cells/well). After incubation for 24 h, the medium was changed to complete medium with final concentrations of 0 nmol/L, 125 nmol/L, and 250 nmol/L givinostat if normal cell growth was observed. Following incubation for another 48 h, the cells were harvested and treated thoroughly with the appropriate amount of tryptic digestion to afford a single-cell suspension. Then, 1 × 105 resuspended cells were collected and centrifuged at 1000 rpm for 5 min. The supernatant was discarded. The residue was resuspended with 100 μL Annexin V binding buffer, and then transferred into a 5-mL culture tube. Then, 5 μL Annexin V-FITC and propidium iodide (PI) was added, and the mixture was incubated at 20 °C-25 °C in darkness for 15 min. Next, 400 μL of Annexin V binding buffer was added immediately before flow cytometry. The Annexin V-FITC showed green fluorescence, while PI showed red fluorescence. Flow cytometry with 488-nm laser excitation was used. The FITC fluorescein was detected using a 515-nm long-pass filter, and the PI fluorescein was detected using a filter at a wavelength > 560 nm. Moreover, after treatment with 1 mL of prechilled 70% ethanol for cell immobilization, the cell pellet was washed and centrifuged twice in 0.5 mL PBS containing 50 μg/mL PI. The cells were resuspended with 100 μg/mL RNase A, and then inoculated in the dark at 37 °C for 30 min before the flow cytometer was used to determine the cell cycles.
Western blotting
The JS-1 cells were inoculated in 100-mm culture dishes containing complete medium (1 × 1010 cells/well). After 24 h, the cells in one dish were treated with tryptic digestion and harvested as the 0-h sample if normal cell growth was observed. The other four dishes were incubated with complete medium containing a final concentration of 250 nmol/L givinostat. After the culture medium was replaced by serum-free culture medium, the mixtures were inoculated for 12 h using the above described method. Two dishes were incubated with givinostat (final concentration: 50-100 nmol/L), and the resultant mixtures were incubated for 1 h before addition of LPS (final concentration: 100 ng/mL). One of the other two dishes was incubated with LPS (final concentration: 100 ng/mL), and the cells were harvested after 15 min. All preparations were subjected to Western blotting. The preparation was incubated in a 37 °C/5% CO2 incubator for 48 h, treated with tryptic digestion to harvest the cells, and then washed twice with PBS. The mixture was centrifuged to remove the supernatant, and the collected cells were placed on ice before lysis. The proteins were quantified using the BCA method. SDS-PAGE, membrane transfer, immunoreactions, development, and gel electrophoresis image analysis was performed for the target genes.
Determination of the effect of givinostat on cell migration by scratch assay
JS-1 cells were inoculated on a 12-well plate (2 × 105 cells/well) and then incubated in a 37 °C/5% CO2 incubator. After incubation for 24 h, if the cell morphology was normal, a single-cell suspension was prepared and scratches were made using the tip of a 200-μL pipette tip. Complete medium containing serum was replaced by serum-free DMEM. The cells were pretreated with 100 nmol/L givinostat for 2 h, and LPS solution (100 ng/mL) was added for the cell migration assay. The preparation was incubated for another 24 h before analysis.
Laser confocal microscopy
Detection of reactive oxygen species (ROS) using fluorescent probe DCFH-DA: JS-1 cells were inoculated in two 20-mm Falcon confocal dishes containing 2 mL of relevant complete medium (5 × 103 cells/dish). After 24 h, if normal cell growth was observed, the medium was removed and 1 mL of diluted DCFH-DA was added. The preparation was incubated in a 37 °C incubator for 15 min and washed three times with serum-free medium. Then, 2 mL of complete medium containing givinostat (0 nmol/L or 500 nmol/L final concentration) was added to these two dishes, which were then incubated in a 37 °C/5% CO2 incubator for 30 min. Under an excitation wavelength of 488 nm and an emission wavelength of 525 nm, the fluorescence intensity of the two dishes was determined at time intervals of 0 min, 10 min, 20 min, and 30 min using a laser confocal microscope.
Detection of mitochondrial membrane potential using the JC-1 method: Reference ROS were prepared before the assay. Then, 2 mL of complete medium containing givinostat (final concentrations: 0 nmol/L, 250 nmol/L, 500 nmol/L, or 1000 nmol/L) was added to each of the four dishes. The mixture was incubated for another 2 h, washed, 1 mL of JC-1 staining solution (final concentration: 2 μg/mL) was added, and the mixture was incubated at 37 °C for 20 min. The supernatant was discarded and the residue was washed twice. After 2 mL of DMEM complete medium was added, red and green fluorescence was detected through the Cy3 and FITC channels, respectively, and photographed within 20 min using a laser confocal microscope.
Detection of mitochondrial permeability transition pore (mPTP) opening by coloading with calcein-AM and CoCl2: Reference ROS were prepared before the assay. After the cells were washed with serum-free DMEM medium once, 1 mL of serum-free DMEM staining solution containing calcein-AM (final concentration: 1 μmol/L) and CoCl2 (final concentration: 5 mmol/L) was added before incubation at 37 °C for 15 min. The supernatant was discarded and the residue was washed twice. Two dishes were incubated with 2 mL of CoCl2 (final concentration: 5 mmol/L), followed by complete medium containing 0 nmol/or 500 L givinostat. The green fluorescence was examined using the FITC channel using a laser confocal microscope. The fluorescence intensities of two dishes were determined at 0 min, 10 min, 20 min, and 30 min. The fluorescence intensity of the regions of interest randomly selected from multiple intracellular mitochondria were analyzed by Image J to quantitate any mPTP opening events.
Establishment of animal models
Fifty 5-wk-old specific-pathogen-free Balb/C male mice weighing 20 ± 5 g were supplied by Shanghai Experimental Animal Center, Chinese Academy of Sciences [License No.: SCXK (Shanghai) 2012-0005]. They were randomly divided into three groups: control group (n = 10), liver fibrosis group (n = 20), and givinostat treatment group (n = 20). In the control group, mice were fed a normal diet and administered saline (i.p., amounts equivalent to other groups) every 3 d. In the liver fibrosis group, mice were fed a high-fat diet containing 0.5% cholesterol and received i.p. administration of the working solution prepared from four portions of CCl4 and six of olive oil and sacrificed to harvest the liver tissues. In the givinostat treatment group, mice were fed a high-fat diet containing 0.5% cholesterol and received ip administration of the working solution prepared from four portions of CCl4 and six of olive oil (at a dose of 3 mL/kg CCl4), and givinostat (5 mg/kg) was administered by gavage (0.1 mL) on the next day until week 5, when the mice were sacrificed to harvest liver tissues.
For liver histopathologic examination, the liver tissues were fixed in formalin, embedded in paraffin, and thin sections were stained with the Masson’s Trichrome Stain Kit. Damage to liver cells and deposition of collagen fibers were observed under a light microscope. Collagen fibers, muscle fibers, and red blood cells were stained green, red and orange, respectively. In vivo experiments were approved by the Shanghai Tongren Hospital affiliated to Shanghai Jiao Tong University School of Medicine Ethics Committee.
Statistical analysis
The data were analyzed with SPSS version 13.0 statistical software (SPSS Inc., Chicago, IL, United States), and expressed as mean ± SD. Multiple groups were analyzed with one-way analysis of variance, pairwise comparison was conducted using a least significant difference t-test, and different groups were compared using a t-test. P < 0.05 was considered statistically significant.
RESULTS
Suppressive effects of givinostat on proliferation and migration of JS-1 cells
As shown by the CCK-8 assay, givinostat inhibited JS-1 cell proliferation in a concentration-dependent manner. The cell suppression rates markedly differed after treatment with givinostat at different concentrations (ranging from 0 nmol/L to 1000 nmol/L) (Figure 1A). Treatment with givinostat ≥ 500 nmol/L was associated with significant inhibition of JS-1 cell proliferation (P < 0.01). Also, the cell inhibition rate significantly differed between the group cotreated with givinostat ≥ 250 nmol/L plus LPS and the group without LPS treatment (same givinostat concentration) (P < 0.05). As demonstrated by scratch assay (Figure 1B), the area of cell migration was significantly reduced in JS-1 cells cotreated with givinostat plus LPS in comparison to the controls (P < 0.05), suggesting givinostat treatment markedly decreased LPS-activated JS-1 cell migration. Western blotting showed that the protein expression of α-SMA, transforming growth factor β 1, and vascular endothelial growth factor in JS-1 cells significantly decreased 24 h and 48 h after givinostat treatment (all P < 0.05) (Figure 1C).
Figure 1.

Effects of givinostat in suppressing proliferation and migration of JS-1 cells. A: Givinostat inhibits JS-1 cell proliferation in a concentration-dependent manner. The cell inhibition rate significantly differed between the group cotreated with givinostat ≥ 250 nmol/L plus lipopolysaccharide (LPS) and the group without LPS treatment (same givinostat concentration) (P < 0.05); B: The cell migration area significantly decreased in the group cotreated with givinostat plus LPS (P < 0.05); C: Compared with the control group, treatment with givinostat for 24 h or 48 h significantly suppressed expression of α-smooth muscle actin (SMA), transforming growth factor (TGF)-β1 and Vascular endothelial growth factor (VEGF). aP < 0.05, bP < 0.01 vs control group.
Effects of givinostat on apoptosis and cell cycle of JS-1 cells
In the bivariate scatter plots of flow cytometry, after treatment with givinostat (concentration gradient: 0 nmol/L, 125 nmol/L, and 250 nmol/L), the numbers of apoptotic and necrotic JS-1 cells markedly increased, suggesting givinostat induced JS-1 cell apoptosis in a concentration-dependent manner (Figure 2A). According to the cell cycle plot of flow cytometry, the JS-1 cells were arrested at the G0/G1 phase after givinostat treatment (Figure 2A). Western blotting showed that prolonged givinostat treatment was significantly associated with decreased protein expression of cyclin-dependent kinase (CDK)4, CDK 6, and cyclin D1, and with increased expression of p21 and p57 (compared with the untreated group) (all P < 0.05) (Figure 2B). As shown by Western blotting, givinostat activated caspase-3 and caspase-9 in JS-1 cells in a concentration-dependent manner (Figure 2C), without causing any changes in the precursors of these two proteins.
Figure 2.
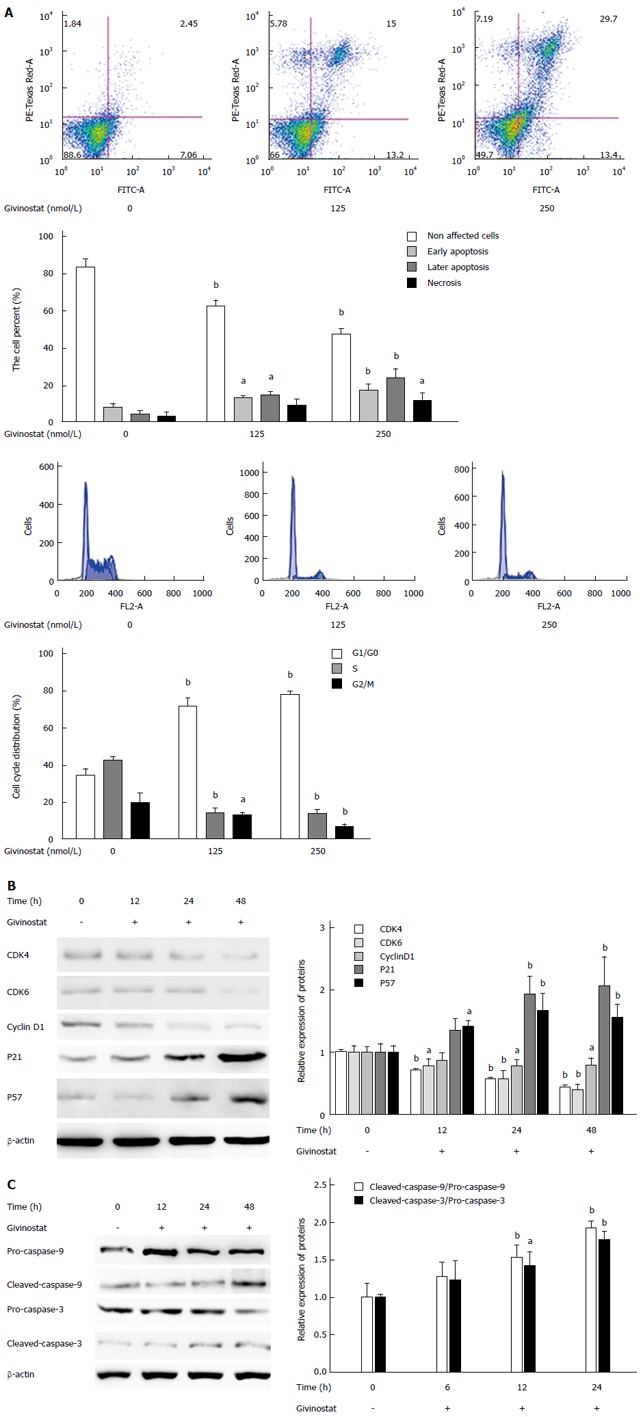
Effects of givinostat on apoptosis and cell cycle of JS-1 cells. A: Flow cytometry shows that the givinostat treatment induces JS-1 cell apoptosis in a concentration-dependent manner. The numbers of apoptotic and necrotic cells progressively increased when the concentration of givinostat increased; B: Givinostat arrested the JS-1 cells at G0/G1 phase; C: Givinostat activated caspase-3 and caspase-9 in JS-1 cells without causing changes in the precursors of these two proteins. aP < 0.05, bP < 0.01 vs the control group.
Effects of givinostat on the ROS profile, mitochondrial membrane potential, and mPTP opening in JS-1 cells
The effects of givinostat treatment on the ROS profile of JS-1 cells were examined using laser confocal microscopy (Figure 3A). As shown by the green fluorescence that represented intracellular ROS, givinostat treatment markedly increased ROS production. Changes in mitochondrial membrane potential were examined using JC-1 as the fluorescent probe and laser confocal microscopy (Figure 3B), with green and red fluorescence denoting low and high membrane potentials, respectively. It was found that givinostat treatment induced a concentration-dependent decrease in mitochondrial membrane potential. The mPTP opening was detected by coloading with calcein-AM and CoCl2 (Figure 3C). The mitochondria of normal JS-1 cells had intense green fluorescence, with a granular appearance. In the givinostat treatment group, however, there was considerable mPTP opening, along with decreased intensity of fluorescence in comparison to that of the control group.
Figure 3.
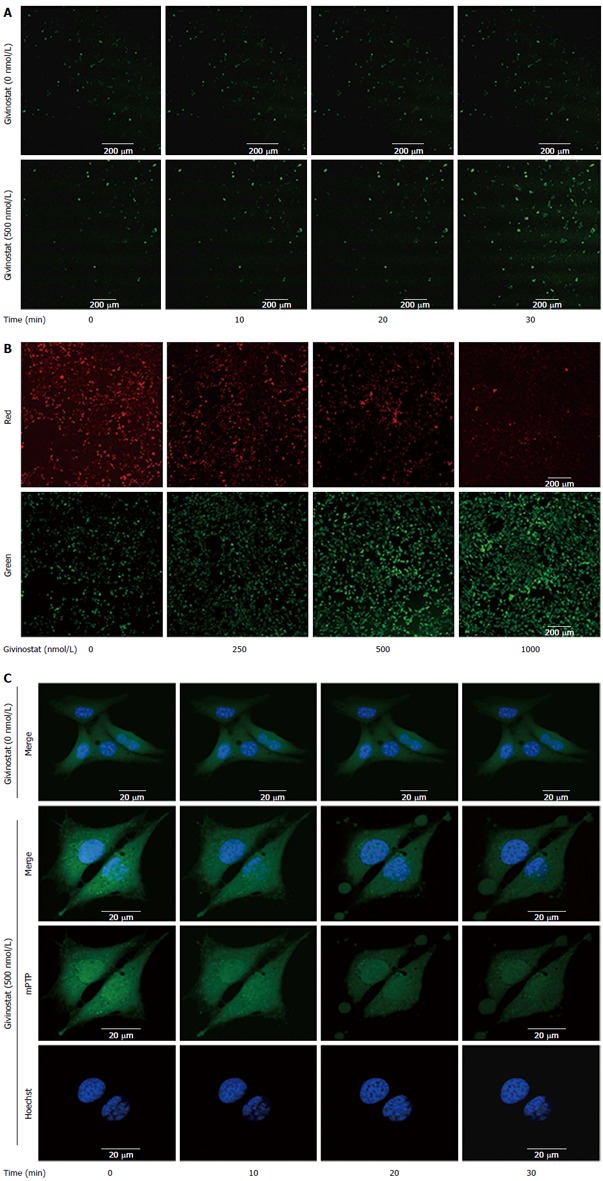
Effects of givinostat on the reactive oxygen species profile, mitochondrial membrane potential, and mitochondrial permeability transition pore opening in JS-1 cells. A: Effects of givinostat on reactive oxygen species (ROS) in JS-1 cells were determined by 2’,7’-dichlorofluorescein diacetate. Green fluorescence represents the intracellular ROS. Givinostat significantly increased ROS production, in particular at 30 min (P < 0.01); B: Mitochondrial membrane potential was detected using the fluorescent probe JC-1. The green and red fluorescence represented the low and high membrane potentials, respectively. Givinostat reduced mitochondrial membrane potential in a concentration-dependent manner, particularly at 1000 nmol/L (P < 0.01); C: The mitochondrial permeability transition pore (mPTP) opening was detected by coloading with calcein-AM and CoCl2. In the givinostat treatment group, considerable mPTP opening was observed, along with decreased intensity of fluorescence in comparison to that of the control group.
Changes in histone acetylation and regulation of LPS-activated mitogen-activated protein kinase signaling in givinostat-treated JS-1 cells
The effects of givinostat on the post-translational modifications of superoxide dismutase (SOD)2, p53, nuclear factor (NF)-κB, and p65 were analyzed by Western blotting (Figure 4A), which showed that SOD2 (acetyl K68) acetylation was upregulated, while the expression profile of SOD2 protein showed no significant change. Acetylation of NF-κB p65 (acetyl K310) was upregulated, while its protein expression showed no significant change. There were no obvious changes in the expression profiles of p53, p53 (acetyl K382), and p53 (acetyl K120).
Figure 4.
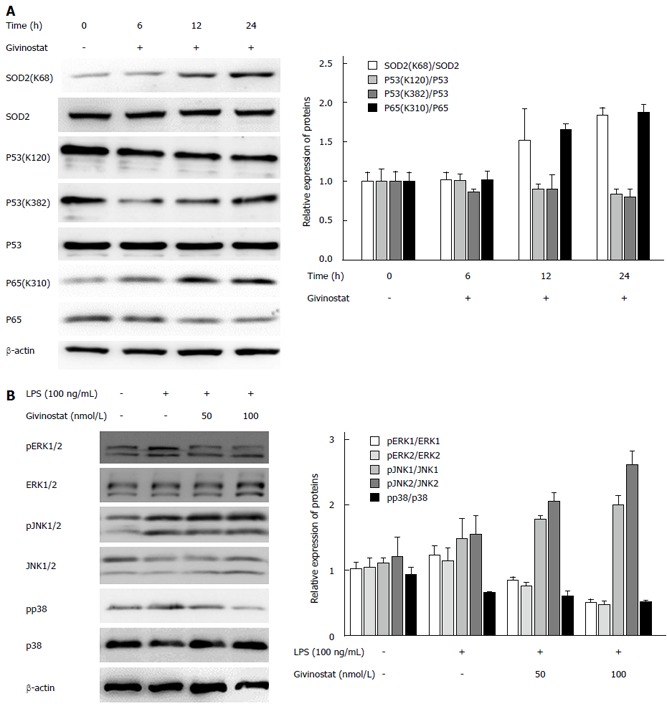
Changes in histone acetylation and regulation of lipopolysaccharide-activated mitogen-activated protein kinase signaling in givinostat-treated JS-1 cells. A: Effects of givinostat on the post-translational modifications of superoxide dismutase (SOD)2, p53, nuclear factor (NF)-κB, and p65 were analyzed by Western blotting. SOD2 (acetyl K68) acetylation was upregulated, while the expression profile of SOD2 protein showed no significant change. The ratios show a significant difference when compared with those in the control group, in particular at 24 h. Similarly, acetylation of NF-κB p65 (acetyl K310) was upregulated, while its protein expression showed no significant change. There were no obvious changes in the expression profiles of p53, p53 (acetyl K382), and p53 (acetyl K120); B: Expression of extracellular signal-regulated kinase (ERK)1/2, phosphorylated ERK1/2, p38, and phosphorylated p38 was upregulated after lipopolysaccharide (LPS) treatment; in contrast, givinostat inhibited the upregulated expression of phosphorylated ERK1/2 and phosphorylated P38 induced by LPS in a time-dependent manner, while no obvious effect on c-Jun N-terminal kinase (JNK)1/2 and phosphorylated JNK1/2 was found.
Change in the mitogen-activated protein kinase (MAPK) signaling-pathway-related protein expression was examined after givinostat treatment of LPS-activated JS-1 cells, which showed that expression of extracellular signal-regulated kinase (ERK)1/2, phosphorylated ERK1/2, p38, and phosphorylated p38 increased following LPS activation; givinostat suppressed LPS-induced upregulation of ERK1/2 and phosphorylated p38 in a time-dependent manner, but showed no effect on the expression of C-Jun N-terminal kinase (JNK)1/2 and phosphorylated JNK1/2 (Figure 4B).
Pathologic changes of liver tissue in mouse models of liver fibrosis induced by givinostat treatment
The hepatic lobules of healthy mice were clear in structure, with only a small amount of collagen deposited in the portal area. In the mouse models of liver fibrosis induced by CCl4 treatment, however, liver cell degeneration was concomitant with a large amount of collagen deposition in the portal area. Some of them exhibited fiber spacing, with visible pseudolobules. In contrast, the givinostat treatment group had markedly less collagen deposition and improved fibrosis. Collagen area percent was significantly reduced in givinostat group compared to the model group (P < 0.01) (Figure 5).
Figure 5.
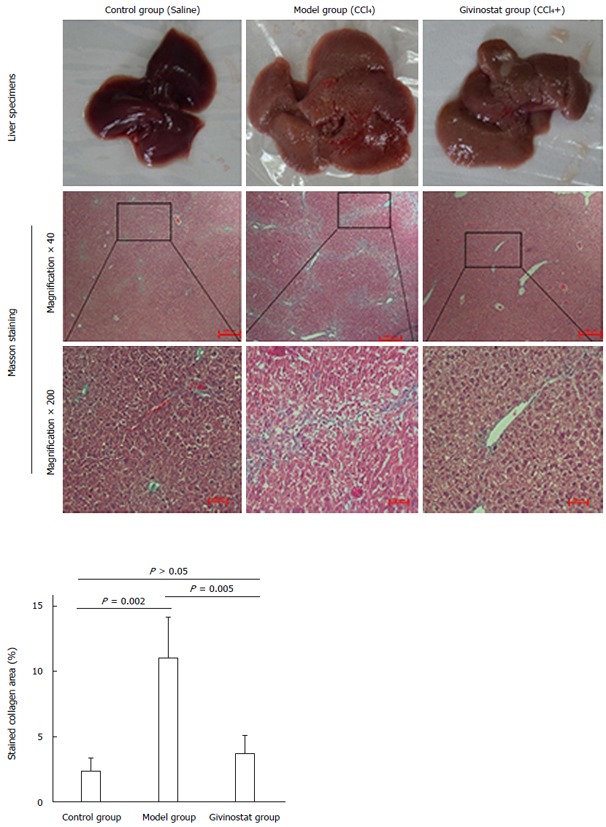
Pathologic changes of liver tissue in mouse models of liver fibrosis induced by CCl4 treatment. Control group: The hepatic lobules had integrated structures, with only a small number of stained collagen fibers in the portal area. There was no obvious collagen deposition, and no degeneration, necrosis, or inflammatory cell infiltration. Model group: The normal hepatic lobules were damaged, along with the markedly increased collagen in liver tissue. The collagen fibers extended along the portal area or outwards to the inflammatory necrosis area, forming fiber spacing with varied thickness. Pseudolobules were visible. Givinostat group: compared with the model group, the structure of liver tissue was markedly improved, along with the obviously decreased collagen deposition. Collagen area percent was significantly reduced in givinostat group compared to the model group (P = 0.005).
DISCUSSION
As demonstrated in the current study, givinostat, an HDAC inhibitor, has marked antifibrotic activity. In the in vitro experiment, givinostat markedly suppressed proliferation of HSCs (JS-1 cell line), and such an inhibitory effect was even more obvious among LPS-activated HSCs. Givinostat induced apoptosis of mouse HSCs and arrested these cells at G0/G1 phase. The protein expression profiles of α-SMA, transforming growth factor β 1, and vascular endothelial growth factor in HSCs significantly decreased after givinostat treatment. Moreover, the notable beneficial effect of givinostat on liver fibrosis was also confirmed in the mouse models.
Further study on the G0/G1 arrest suggested that such an effect was achieved by regulating the expression of some cell-cycle-related proteins. Givinostat downregulated protein expression of CDK4, CDK6, and cyclin D1, whereas expression of p21 and p57 was significantly increased. These findings are consistent with the results of previous HDAC-inhibitor-related studies[19-22]. For the induction of mouse HSC apoptosis, givinostat activates caspase-3 and caspase-9 in JS-1 cells, which is consistent with previous studies[23-25]. As suggested by laser confocal microscopy in the current study, givinostat treatment increased intracellular ROS production, decreased mitochondrial membrane potential, and promoted mPTP opening. Moreover, mPTP opening promotes the uncoupling between mitochondrial electron transport chain and oxidative phosphorylation, decreases membrane potential, reduces ATP production and glutathione, and increases intracellular ROS. As a result, the mitochondrial matrix becomes swollen, along with a decrease in its outer membrane folds and surface area; thus, the mitochondria become fragile and susceptible to rupture, causing the release of proapoptotic proteins from the intermembrane spaces. Ultimately, apoptosis occurs.
As a signaling molecule, ROS is involved in the regulation of multiple cellular functions such as cell growth, differentiation, apoptosis, and immune responses[26]. ROS has both positive and negative effects on cell survival and apoptosis. ROS can exert an antiapoptotic effect by activating NF-κB or Akt/activation of apoptosis signal-regulating kinase[27,28]. However, it is also a key link in the apoptosis process[29]. ROS can induce apoptosis through multiple mechanisms. For instance, they can cause damage to DNA, lipids and proteins, or they can regulate apoptosis via the redox-insensitive MAPK pathways including JNK, p38, and ERK1/2[30]. In the current study, the givinostat-induced HSC apoptosis was mainly mediated through p38 and ERK1/2 rather than JNK.
The mitochondrial respiratory chain is a key site responsible for the production of free radicals. As a SOD in mitochondria, SOD2 is a key enzyme responsible for eliminating mitochondrial superoxide anions in the mitochondria[31]. Approximately 90% of intracellular ROS can be explained by the electron leakage that occurs in the mitochondrial electron transport chain, while SIRT3-induced deacetylation of SOD2 can eliminate most of the intracellular ROS. Apoptosis induced by mitochondrial damage may be associated with a decrease in oxygen radical scavenging capacity[32,33]. SIRT3 deacetylates two critical lysine residues on SOD2 and promotes its antioxidative activity[34,35]. In our current study, however, upregulation of SOD2 (acetyl K68) acetylation induced by givinostat might be one of the key mechanisms responsible for the decrease in oxygen radical scavenging capacity.
Many studies have found that the NF-κB activity is correlated with liver fibrosis[36,37]. Suppression of NF-κB binding affinity or blockade of NF-κB stimulation may induce HSC apoptosis, thus alleviating the liver fibrosis caused by multiple etiologies; therefore, the NF-κB pathway may be a useful way for drugs to exert their efficacy in fighting against injuries or fibrosis. In the nucleus, the inactivated or activated status of NF-κB is determined by its acetylation. By regulating histone acetylation, the HDAC controls a series of intranuclear biologic processes induced by NF-κB. Kuo et al[38] have reported that acetylation and deacetylation of histones are closely correlated to regulation of gene transcription in eukaryotic cells. A study on synovial fibroblasts found that HDAC inhibitors block the activation of NF-κB p65 and thus induce apoptosis[39]. Similarly, in the current study, givinostat treatment upregulated NF-κB p65 (acetyl K310) acetylation but showed no obvious effect on protein expression of NF-κB p65.
In summary, as an HDAC inhibitor, givinostat has antifibrotic activities both in vivo and in vitro, which might be achieved by regulating the acetylation of NF-κB and SOD2, stimulating oxidative stress, activating mitochondrial pathways, inhibiting proliferation of mouse HSCs (JS-1 cell line), and inducing apoptosis. The current study may provide new directions and evidence in the research and development of novel drugs for liver fibrosis.
COMMENTS
Background
At present, there is no effective therapeutic means for the treatment of liver fibrosis. Activation of hepatic stellate cells (HSCs) is the central event in liver fibrosis and often serves as a trigger. Inhibition of HSC activation and proliferation or induction of apoptosis is the mainstream strategy for the treatment of liver fibrosis.
Research frontiers
Recent studies have shown that protein acetylation plays an important role during HSC activation; imbalanced histone acetylation due to histone deacetylase overexpression is closely correlated to the occurrence and development of liver fibrosis.
Innovations and breakthroughs
Givinostat has antifibrotic activities, which might be achieved by regulating the acetylation of nuclear factor-κB (NF-κB) and superoxide dismutase 2, inhibiting proliferation of HSCs, and inducing apoptosis. The study is the first to examine the differential acetylation of specific proteins before and after HSCs are exposed to givinostat and to explore the effect on regulating HSC proliferation.
Applications
The study provides an experimental basis for future studies on exploring additional histone deacetylase inhibitors and their targets for the development of new drugs for liver fibrosis.
Terminology
Acetylation is an important modification of proteins in cell biology, and proteomics studies have identified thousands of acetylated mammalian proteins.
Peer-review
These authors explored givinostat and found it can inhibit HSC proliferation. NF-κB and superoxide dismutase 2 were acetylated after givinostat treatment. Simultaneously, the authors imaged the acetylated proteins and modified sites in HSC cells. The paper is well presented and the results are interesting.
Footnotes
Supported by Shanghai Municipal Health Bureau Key Disciplines Grant, No. ZK2012A05; Bootstrap Class Project Foundation of the Science and Technology Commission of Shanghai Municipality, No. 14411973700; and Shanghai Municipal Health Bureau, No. 20134100.
Institutional review board statement: In vivo experiments were approved by the Shanghai Tongren Hospital affiliated to Shanghai Jiao Tong University School of Medicine Ethics Committee.
Institutional animal care and use committee statement: In vivo experiments were approved by the Shanghai Tongren Hospital affiliated to Shanghai Jiao Tong University School of Medicine Institutional animal care and use committee.
Conflict-of-interest statement: The authors declare no conflicts of interest.
Data sharing statement: No additional unpublished data are available.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: January 20, 2015
First decision: February 26, 2015
Article in press: May 21, 2015
P- Reviewer: Atta H, Marcos R S- Editor: Ma YJ L- Editor: AmEditor E- Editor: Ma S
References
- 1.Hernandez-Gea V, Friedman SL. Pathogenesis of liver fibrosis. Annu Rev Pathol. 2011;6:425–456. doi: 10.1146/annurev-pathol-011110-130246. [DOI] [PubMed] [Google Scholar]
- 2.Kocabayoglu P, Friedman SL. Cellular basis of hepatic fibrosis and its role in inflammation and cancer. Front Biosci (Schol Ed) 2013;5:217–230. doi: 10.2741/s368. [DOI] [PubMed] [Google Scholar]
- 3.Sato M, Suzuki S, Senoo H. Hepatic stellate cells: unique characteristics in cell biology and phenotype. Cell Struct Funct. 2003;28:105–112. doi: 10.1247/csf.28.105. [DOI] [PubMed] [Google Scholar]
- 4.Friedman SL. Hepatic stellate cells: protean, multifunctional, and enigmatic cells of the liver. Physiol Rev. 2008;88:125–172. doi: 10.1152/physrev.00013.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Atta H, El-Rehany M, Hammam O, Abdel-Ghany H, Ramzy M, Roderfeld M, Roeb E, Al-Hendy A, Raheim SA, Allam H, et al. Mutant MMP-9 and HGF gene transfer enhance resolution of CCl4-induced liver fibrosis in rats: role of ASH1 and EZH2 methyltransferases repression. PLoS One. 2014;9:e112384. doi: 10.1371/journal.pone.0112384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen SL, Zheng MH, Shi KQ, Yang T, Chen YP. A new strategy for treatment of liver fibrosis: letting MicroRNAs do the job. BioDrugs. 2013;27:25–34. doi: 10.1007/s40259-012-0005-2. [DOI] [PubMed] [Google Scholar]
- 7.Sánchez-Valle V, Chávez-Tapia NC, Uribe M, Méndez-Sánchez N. Role of oxidative stress and molecular changes in liver fibrosis: a review. Curr Med Chem. 2012;19:4850–4860. doi: 10.2174/092986712803341520. [DOI] [PubMed] [Google Scholar]
- 8.Shiha G, Sarin SK, Ibrahim AE, Omata M, Kumar A, Lesmana LA, Leung N, Tozun N, Hamid S, Jafri W, et al. Liver fibrosis: consensus recommendations of the Asian Pacific Association for the Study of the Liver (APASL) Hepatol Int. 2009;3:323–333. doi: 10.1007/s12072-008-9114-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mannaerts I, Nuytten NR, Rogiers V, Vanderkerken K, van Grunsven LA, Geerts A. Chronic administration of valproic acid inhibits activation of mouse hepatic stellate cells in vitro and in vivo. Hepatology. 2010;51:603–614. doi: 10.1002/hep.23334. [DOI] [PubMed] [Google Scholar]
- 10.Niki T, Rombouts K, De Bleser P, De Smet K, Rogiers V, Schuppan D, Yoshida M, Gabbiani G, Geerts A. A histone deacetylase inhibitor, trichostatin A, suppresses myofibroblastic differentiation of rat hepatic stellate cells in primary culture. Hepatology. 1999;29:858–867. doi: 10.1002/hep.510290328. [DOI] [PubMed] [Google Scholar]
- 11.Qin L, Han YP. Epigenetic repression of matrix metalloproteinases in myofibroblastic hepatic stellate cells through histone deacetylases 4: implication in tissue fibrosis. Am J Pathol. 2010;177:1915–1928. doi: 10.2353/ajpath.2010.100011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Leoni F, Fossati G, Lewis EC, Lee JK, Porro G, Pagani P, Modena D, Moras ML, Pozzi P, Reznikov LL, et al. The histone deacetylase inhibitor ITF2357 reduces production of pro-inflammatory cytokines in vitro and systemic inflammation in vivo. Mol Med. 2005;11:1–15. doi: 10.2119/2006-00005.Dinarello. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Armeanu S, Pathil A, Venturelli S, Mascagni P, Weiss TS, Göttlicher M, Gregor M, Lauer UM, Bitzer M. Apoptosis on hepatoma cells but not on primary hepatocytes by histone deacetylase inhibitors valproate and ITF2357. J Hepatol. 2005;42:210–217. doi: 10.1016/j.jhep.2004.10.020. [DOI] [PubMed] [Google Scholar]
- 14.Pathil A, Armeanu S, Venturelli S, Mascagni P, Weiss TS, Gregor M, Lauer UM, Bitzer M. HDAC inhibitor treatment of hepatoma cells induces both TRAIL-independent apoptosis and restoration of sensitivity to TRAIL. Hepatology. 2006;43:425–434. doi: 10.1002/hep.21054. [DOI] [PubMed] [Google Scholar]
- 15.Golay J, Cuppini L, Leoni F, Micò C, Barbui V, Domenghini M, Lombardi L, Neri A, Barbui AM, Salvi A, et al. The histone deacetylase inhibitor ITF2357 has anti-leukemic activity in vitro and in vivo and inhibits IL-6 and VEGF production by stromal cells. Leukemia. 2007;21:1892–1900. doi: 10.1038/sj.leu.2404860. [DOI] [PubMed] [Google Scholar]
- 16.Leoni F, Zaliani A, Bertolini G, Porro G, Pagani P, Pozzi P, Donà G, Fossati G, Sozzani S, Azam T, et al. The antitumor histone deacetylase inhibitor suberoylanilide hydroxamic acid exhibits antiinflammatory properties via suppression of cytokines. Proc Natl Acad Sci USA. 2002;99:2995–3000. doi: 10.1073/pnas.052702999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Carta S, Tassi S, Semino C, Fossati G, Mascagni P, Dinarello CA, Rubartelli A. Histone deacetylase inhibitors prevent exocytosis of interleukin-1beta-containing secretory lysosomes: role of microtubules. Blood. 2006;108:1618–1626. doi: 10.1182/blood-2006-03-014126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Furlan A, Monzani V, Reznikov LL, Leoni F, Fossati G, Modena D, Mascagni P, Dinarello CA. Pharmacokinetics, safety and inducible cytokine responses during a phase 1 trial of the oral histone deacetylase inhibitor ITF2357 (givinostat) Mol Med. 2011;17:353–362. doi: 10.2119/molmed.2011.00020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hirsch CL, Bonham K. Histone deacetylase inhibitors regulate p21WAF1 gene expression at the post-transcriptional level in HepG2 cells. FEBS Lett. 2004;570:37–40. doi: 10.1016/j.febslet.2004.06.018. [DOI] [PubMed] [Google Scholar]
- 20.Anh TD, Ahn MY, Kim SA, Yoon JH, Ahn SG. The histone deacetylase inhibitor, Trichostatin A, induces G2/M phase arrest and apoptosis in YD-10B oral squamous carcinoma cells. Oncol Rep. 2012;27:455–460. doi: 10.3892/or.2011.1496. [DOI] [PubMed] [Google Scholar]
- 21.Wang YG, Wang N, Li GM, Fang WL, Wei J, Ma JL, Wang T, Shi M. Mechanisms of trichostatin A inhibiting AGS proliferation and identification of lysine-acetylated proteins. World J Gastroenterol. 2013;19:3226–3240. doi: 10.3748/wjg.v19.i21.3226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.An JH, Jang SM, Kim JW, Kim CH, Song PI, Choi KH. The expression of p21 is upregulated by forkhead box A1/2 in p53-null H1299 cells. FEBS Lett. 2014;588:4065–4070. doi: 10.1016/j.febslet.2014.09.033. [DOI] [PubMed] [Google Scholar]
- 23.Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov. 2006;5:769–784. doi: 10.1038/nrd2133. [DOI] [PubMed] [Google Scholar]
- 24.Maiso P, Carvajal-Vergara X, Ocio EM, López-Pérez R, Mateo G, Gutiérrez N, Atadja P, Pandiella A, San Miguel JF. The histone deacetylase inhibitor LBH589 is a potent antimyeloma agent that overcomes drug resistance. Cancer Res. 2006;66:5781–5789. doi: 10.1158/0008-5472.CAN-05-4186. [DOI] [PubMed] [Google Scholar]
- 25.Rosato RR, Grant S. Histone deacetylase inhibitors: insights into mechanisms of lethality. Expert Opin Ther Targets. 2005;9:809–824. doi: 10.1517/14728222.9.4.809. [DOI] [PubMed] [Google Scholar]
- 26.Ray PD, Huang BW, Tsuji Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell Signal. 2012;24:981–990. doi: 10.1016/j.cellsig.2012.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen AC, Arany PR, Huang YY, Tomkinson EM, Sharma SK, Kharkwal GB, Saleem T, Mooney D, Yull FE, Blackwell TS, et al. Low-level laser therapy activates NF-kB via generation of reactive oxygen species in mouse embryonic fibroblasts. PLoS One. 2011;6:e22453. doi: 10.1371/journal.pone.0022453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Corcoran A, Cotter TG. Redox regulation of protein kinases. FEBS J. 2013;280:1944–1965. doi: 10.1111/febs.12224. [DOI] [PubMed] [Google Scholar]
- 29.Simon HU, Haj-Yehia A, Levi-Schaffer F. Role of reactive oxygen species (ROS) in apoptosis induction. Apoptosis. 2000;5:415–418. doi: 10.1023/a:1009616228304. [DOI] [PubMed] [Google Scholar]
- 30.Son Y, Kim S, Chung HT, Pae HO. Reactive oxygen species in the activation of MAP kinases. Methods Enzymol. 2013;528:27–48. doi: 10.1016/B978-0-12-405881-1.00002-1. [DOI] [PubMed] [Google Scholar]
- 31.Ying YL, Balaban CD. Regional distribution of manganese superoxide dismutase 2 (Mn SOD2) expression in rodent and primate spiral ganglion cells. Hear Res. 2009;253:116–124. doi: 10.1016/j.heares.2009.04.006. [DOI] [PubMed] [Google Scholar]
- 32.Orr AL, Ashok D, Sarantos MR, Shi T, Hughes RE, Brand MD. Inhibitors of ROS production by the ubiquinone-binding site of mitochondrial complex I identified by chemical screening. Free Radic Biol Med. 2013;65:1047–1059. doi: 10.1016/j.freeradbiomed.2013.08.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Klein LE, Cui L, Gong Z, Su K, Muzumdar R. A humanin analog decreases oxidative stress and preserves mitochondrial integrity in cardiac myoblasts. Biochem Biophys Res Commun. 2013;440:197–203. doi: 10.1016/j.bbrc.2013.08.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Qiu X, Brown K, Hirschey MD, Verdin E, Chen D. Calorie restriction reduces oxidative stress by SIRT3-mediated SOD2 activation. Cell Metab. 2010;12:662–667. doi: 10.1016/j.cmet.2010.11.015. [DOI] [PubMed] [Google Scholar]
- 35.Bell EL, Guarente L. The SirT3 divining rod points to oxidative stress. Mol Cell. 2011;42:561–568. doi: 10.1016/j.molcel.2011.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Luedde T, Schwabe RF. NF-κB in the liver--linking injury, fibrosis and hepatocellular carcinoma. Nat Rev Gastroenterol Hepatol. 2011;8:108–118. doi: 10.1038/nrgastro.2010.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ji L, Xue R, Tang W, Wu W, Hu T, Liu X, Peng X, Gu J, Chen S, Zhang S. Toll like receptor 2 knock-out attenuates carbon tetrachloride (CCl4)-induced liver fibrosis by downregulating MAPK and NF-κB signaling pathways. FEBS Lett. 2014;588:2095–2100. doi: 10.1016/j.febslet.2014.04.042. [DOI] [PubMed] [Google Scholar]
- 38.Kuo MH, Allis CD. Roles of histone acetyltransferases and deacetylases in gene regulation. Bioessays. 1998;20:615–626. doi: 10.1002/(SICI)1521-1878(199808)20:8<615::AID-BIES4>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 39.Choo QY, Ho PC, Tanaka Y, Lin HS. Histone deacetylase inhibitors MS-275 and SAHA induced growth arrest and suppressed lipopolysaccharide-stimulated NF-kappaB p65 nuclear accumulation in human rheumatoid arthritis synovial fibroblastic E11 cells. Rheumatology (Oxford) 2010;49:1447–1460. doi: 10.1093/rheumatology/keq108. [DOI] [PubMed] [Google Scholar]