

Abstract
Influenza infection causes an increase in indoleamine 2, 3-dioxygenase (IDO) activity in the lung parenchyma. IDO catabolizes tryptophan into kynurenine, leading to immune dampening. Multiple cell types express IDO, and while IFN-γ upregulates IDO in dendritic cells and macrophages, it is unclear how IDO is affected in respiratory epithelial cells during influenza infection. In this study, the role of IFN-λ in IDO regulation was investigated after influenza infection of respiratory epithelial cells. IDO1 expression increased concurrently with IFN-λ expression. In differentiated NHBE cells, the IDO metabolite was released basolaterally. Recombinant IFN-λ upregulated IDO1 activity, and silencing of IFN-λ decreased IDO1 expression during influenza infection. During IFN-λ stimulation, most differentiated cell types are able to express IDO but during influenza infection, IDO is primarily expressed in uninfected cells. These studies show a role for IDO in the host response to influenza infection, and they provide insights into novel approaches for enhancing vaccine responses and therapeutic approaches.
Introduction
Influenza is a significant worldwide health concern. During respiratory infection, influenza mainly infects the respiratory epithelium that supports virus replication as well as initiates the antiviral state in response to infection (Sanders and others 2010). Virus infection of the epithelium stimulates the production of type I and type III IFNs (IFN-λ) with IFN-λ being the main IFN expressed in response to influenza infection in mice (Jewell and others 2010).
Interferon lambda consists of 3 subtypes, IFN-λ1 (IL-29), IFN-λ2 (IL-28a), and IFN-λ3 (IL-28b), with IFN-λ1 being a pseudogene in mice (Commins and others 2008). Although IFN-λ and type I IFNs have similar activities, a primary difference between them is receptor utilization (Wang and others 2009). IFN-λ binds to the IFN-λR, which consists of the IFNLR1 (IL-28Rα) and IL-10R2 (Commins and others 2008). IL-10R2 is ubiquitously expressed on the surface of most cells, while IFNLR1 is expressed on epithelial cells lining the respiratory and gastrointestinal tract (Sommereyns and others 2008), as well as by plasmacytoid dendritic cells (pDCs) in mice (Ank and others 2008). In contrast, type I IFNs utilizes IFN-αR1 and IFN-αR2 that are present in most cell types (de Weerd and others 2007). Since the complete IFN-λ receptor is primarily found in epithelial cells, its role in antiviral immunity may be unique to these cell types. After binding to their receptors, type I and type III IFNs have similar signaling pathways that act via dimerization of Stat1 and Stat2, which, in turn, recognize IFN-γ activation site sequence and IFN-stimulated response elements (Commins and others 2008).
Indoleamine 2, 3-dioxygenase (IDO) is an intracellular enzyme in the kynurenine pathway that catabolizes tryptophan (trp) into kynurenine (kyn), which leads to immune suppression and attenuation (Munn and others 2005). IDO is expressed by dendritic cells, macrophages (Fallarino and others 2002), and epithelial cells (van Wissen and others 2002). Since respiratory epithelial cells are a primary target for influenza replication, it is critical to understand the role that IDO has during influenza infections. The IDO gene contains an IFN-stimulated response element in its promoter (Popov and others 2006), is highly upregulated by stimulation with IFN-γ, and is modestly upregulated by type I IFNs (Bianchi and others 1988); however, the effect of type III IFNs has not been evaluated.
To understand the relationship between IFN and IDO during the early stages of the host response to influenza infection, mouse and differentiated human respiratory epithelial cells were investigated. The findings showed that IDO1 was upregulated after influenza virus infection and by concomitant IFN-λ expression. IFN-λ directly induced IDO activity, and removal of IFN-λ reduced IDO expression while increasing type I IFNs. Furthermore, kyn (a metabolite of IDO) was secreted basally from influenza-infected cells. These results define a role for IFN-λ during influenza infection.
Materials and Methods
Cell culture and viruses
MLE-15 cells were cultured in HITES media [RPMI 1640 media (Cellgro, Manassas, VA) with 10 nM hydrocortisone (Sigma-Aldrich, St. Louis, MO), 10 nM β-estradiol (Sigma-Aldrich), 2 mM L-glutamine (Gibco, Carlsbad, CA), and 1% ITS (insulin-transferring-selenium; Gibco)] with 4% FBS. Madin Darby Canine Kidney (MDCK) cells were cultured in DMEM (HyClone, Logan, UT) with 5% FBS. Normal human bronchial epithelial (NHBE) cells (LifeLine Cell Technology, Frederick, MD) were cultured and maintained as previously described (Maneglier and others 2007; Oshansky and others 2010). NHBE cells were maintained at an air–liquid interface at 37°C with 5% CO2 until they were fully differentiated (4 weeks once at air). Basal media was changed every 2 days, and apical surface was rinsed with PBS after 2 weeks in air. A/WSN/33 (H1N1; WSN) influenza virus was propagated in the allantoic cavity of 9-day-old embryonated chicken eggs. Titers were determined by a standard plaque assay on MDCK cells in the presence of 5% FBS at 37°C (Matrosovich and others 2006).
WSN infection of epithelial cells and stimulation with recombinant IFN-λ
MLE-15 cells were cultured on 24-well plates at 6×105 cells/well. Cells were rinsed once with PBS followed by infection with WSN in infection media (HITES media+4% FBS for MLE-15 cells) for 1 h at 37°C. Cells were subsequently rinsed thrice with PBS, and fresh infection media was added. The cells were incubated at 37°C for the indicated amount of time. Differentiated NHBE cells were cultured on a transwell plate as described earlier. Cells were rinsed thrice with PBS followed by infection (MOI=0.5) with WSN apically in BronchialLife Basal media (LifeLine Cell Technology) without supplements for 1 h at 37°C. After infection, cells were rinsed thrice with PBS, the final rinse was removed, and the cells were incubated at 37°C for the indicated times. RNA was harvested as described next, and supernatant was collected and stored at −80°C.
IFN-λ activity was blocked during infection on NHBE cells using an IL-29 or IL-28a neutralizing antibody (nAb) (R&D Systems, Minneapolis, MN) at 4 μg/mL delivered in the basal media. IFN-λ expression was silenced in MLE-15 cells with a small interfering RNA (siRNA) targeting IL28b (IFN-λ3; siIL28b) (ON-TARGETplus Il28b siRNA SMARTpool; Thermo Scientific, Pittsburgh, PA). Briefly, MLE-15 cells were transfected 16 h before infection with WSN using Dharmafect 1 following the manufacturer's protocol with the siRNA at a concentration of 100 nM. A nontargeting control (siNEG; Thermo Scientific) was included at the same concentration. IDO activity was blocked during infection using 1-methyl-D, L-tryptophan (1MT; Sigma-Aldrich) in molecular-grade water at 750 μM. Viral titers were determined from cell culture supernatant at times indicated using a TCID50 method as previously described (Smith and others 2011), with dilutions prepared in DMEM (Hyclone) with 5% FBS. The TCID50 was calculated using the Reed and Meunch method (Reed and Meunch 1938).
MLE-15 cells were stimulated with recombinant IFN-λ3 (rIFN-λ3; eBiosciences, San Diego, CA) prepared in culture media with an unstimulated control. Differentiated NHBE cells were apically stimulated with recombinant IFN-λ1 (rIFN-λ1; eBiosciences) or IFN-λ2 (rIFN-λ2; eBiosciences) at 25 nM in BronchialLife Basal media without supplements for 1 h at 37°C. After stimulation, cells were rinsed thrice with PBS, the final rinse was removed, and the cells were incubated at 37°C for indicated times.
qPCR for detection of mRNA
RNA was isolated at respective time points from samples using the RNeasy mini kit (Qiagen, Valencia, CA) following the manufacturer's protocol and was stored at −20°C. Isolated RNA was DNase treated using DNase I recombinant (Roche, Indianapolis, IN) following the manufacturer's protocol. DNase-treated RNA was quantified using the Nanodrop 1000 (Thermo Scientific, Wilmington, DE). cDNA was synthesized using Verso cDNA kits (Thermo Scientific, Lafayette, CO) following the manufacturer's protocol using equivalent concentrations of RNA for each experiment. qPCR was used to detect murine (Mm) IDO1 (Forward-GCACGACATAGCTACCAGTCT, Reverse- CCACAAAGTCACGCATCCTCTTAA, Probe-5′-6FAM-AAAGCCAAGGAAATTT-MGBNFQ-3′), Mm IDO2 (Forward-CTTCATCCTAGTGACAGTCTTGGT, Reverse-GCCTCCATTCCCTGAACCA, Probe-5′-6FAM-CACTGCTGCCTTCTC-MGBNFQ-3′). The cycling times were 95°C for 10 min, followed by 40 cycles of 95°C for 30 s, 52°C for 1 min, and 68°C for 1 min for IDO1 and IDO2 detection. All samples were normalized to a housekeeping gene, HPRT (Applied Biosystems, Foster City, CA) or GAPDH (Forward- TGTGATGGGTGTGAACCACGAGAA, Reverse-GAGCCCTTCCACAATGCCAAAGTT, Probe-5′-6FAM-AATGCATCCTGCACCACCAAC-MGBNFQ-3′). mRNA expression was determined using 2−ΔΔCt.
Evaluating IDO activity by measuring kyn concentrations
MLE-15 cells were either infected with WSN as previously described or stimulated with rIFN-λ in phenol red-free culture media. Exogenous trp (50 μM; Sigma-Aldrich) was added to each well 24 h before collection of supernatant. At 24 h post addition of trp, cellular supernatant was collected and stored at −80°C. For NHBE cells, exogenous trp (50 μM; Sigma-Aldrich) was added to the basal media 24 h before collection. At time of collection, the apical surface was rinsed with PBS and the basal media was collected and stored at −80°C. The concentration of kyn was determined using a kyn colorimetric assay and a standard curve of kyn concentrations in PBS (Sigma-Aldrich), or basal media for determining activity in NHBE cells since the media contains phenol red. Briefly, proteins were removed by addition of 30% tricholoracetic acid (TCA; VWR, Radnor, PA) and incubated at 50°C for 30 min to hydrolyze n-formylkynurenine to kyn. Samples were then centrifuged at 2400 rpm for 10 min at 4°C. Supernatants were incubated with Erlich's reagent for 10 min. Absorbance was read at 490 nm using an Epoch microplate reader (BioTek, Winooski, VT).
Quantification of IFN-λ and IFN-α/β
IFN-λ was quantified from infected MLE-15 cell supernatant using a VeriKine-DIY Mouse interferon lambda 2/3 ELISA (PBL Interferon Source, Piscataway, NJ) following the manufacturer's protocol. IFN-α was quantified using the Mouse IFN-alpha FlowCytomix Simplex Kit (eBiosciences, San Diego, CA) following the manufacturer's protocol. Samples were run on an LSRII (BD Biosciences, San Jose, CA) and analyzed using the provided software. IFN-λ, IFN-β, and IFN-α were quantified from human cells (NHBE) using the VeriKine-DIY Human Interleukin-29/Interferon Lambda ELISA, Verikine Human Interferon Beta ELISA kit, and VeriKine Human Interferon Alpha ELISA Kit (PBL Interferon Source), respectively, following the manufacturer's protocol.
Evaluation of IDO expression in specific cell types
NHBE cells were infected with WSN or stimulated with IFN-λ as previously described. Cells were rinsed with PBS and fixed with 3.7% formaldehyde on the apical and basal sides. The cells were permeabilized with 0.5% Triton-X 100 and then blocked with 3% BSA. Cells were stained apically with anti-beta-actin (Abcam, Cambridge, MA), anti-MUC5AC (Santa Cruz Biotechnology, Dalla, TX), anti-IDO1 (Santa Cruz Biotechnology), and/or anti-influenza NP (H16-L10-4R5) followed by incubation with a fluorescently conjugated antibody. The basal side remained in blocking buffer. Cells were counterstained with DAPI (Invitrogen, Carlsbad, CA) and mounted on a coverslip using SlowFade (Invitrogen). Cells were imaged using an LSM 510 Meta Confocal Microscope (Zeiss, Thornwood, NY).
Evaluation of cell death after infection
MLE-15 cells were infected with WSN as described earlier, and cell supernatants were collected at the indicated time points. Adenylate kinase release was detected using the ToxiLight kit (Lonza, Allendale, NJ) following the manufacturer's instructions. Complete lysis controls were treated with 1% triton-x 100 (Sigma-Aldrich) in PBS for 10 min. Percent cell death was calculated using the equation [(I- UI)/(C-UI)]*100, where I=infected sample, UI=uninfected samples of the same treatment as infected sample, and C=complete lysis control.
Statistical analysis
Statistics were performed using GraphPad Prism Version 5.04 (La Jolla, CA). Significance was assigned when the *P<0.05, **P<0.01, ***P<0.001, and ****P<0.0001 using either a Student's t-test or ANOVA with a Bonferroni post hoc test, as listed in the figure legends.
Results
Influenza infection upregulates expression of IDO1 compared with IDO2
A paucity of information is available regarding IDO1 or IDO2 expression in epithelial cells during influenza infection. Furthermore, both IDO1 and IDO2 function in trp catabolism but have been shown to be differentially regulated and have varied tissue expression (Ball and others 2009; Maiwald and others 2011). A few studies have differentiated between IDO1 and IDO2 during influenza infection. IDO1 and IDO2 are expressed in lung tissue (Metz and others 2007; Meininger and others 2011); thus, MLE-15 cells were infected with WSN and the levels of IDO1 and IDO2 mRNA were determined over the course of infection (Fig. 1A). At 48 h postinfection (hpi), IDO1 and IDO2 were upregulated compared with uninfected controls; however, IDO1 was preferentially expressed compared with IDO2 (Fig. 1A). To determine whether the level of IDO upregulation correlated with virus replication, MLE-15 cells were infected at varying MOI with WSN and IDO activity was evaluated. IDO activity was measured by the level of the metabolite, kyn, produced via IDO-mediated trp catabolism. As the MOI increased, there was also an increase in IDO activity, particularly at 48 and 72 hpi (Fig. 1B). These findings indicate that influenza infection induces increased IDO1 expression and activity in MLE-15 cells.
FIG. 1.

Influenza infection upregulates IDO1 expression. (A) MLE-15 cells were infected with WSN at an MOI of 0.001. At indicated time points, RNA was harvested. IDO1 and IDO2 mRNA expression was determined by qRT-PCR. The fold change over mock-infected cells is graphed, and the dotted line indicates the mock mRNA levels. Significance is indicated using a one-way ANOVA for each time point with an increase compared with mock-infected cells. (B) MLE-15 cells were infected with varying MOIs of WSN. Cell supernatant was collected at 48 and 72 hpi, and the concentration of kyn was determined. Significance is indicated using a one-way ANOVA for each time point. (C) Differentiated NHBE cells were infected with WSN at an MOI of 0.5, and IDO activity was determined in the basal media. Significance is indicated using a one-way ANOVA with an increase compared with UI controls. (D) IDO activity was determined in the apical and basal media before air–liquid interface (ALI) and in the basal media after ALI until the cells were fully differentiated (week 4). A representative graph is shown of the mean and standard deviation (SD) from at least 2 independent experiments (*P<0.05, **P<0.01, ****P<0.0001).
Since IDO1 mRNA expression and activity were determined in a mouse cell line, it was important to validate the increase in IDO activity after influenza infection by utilizing a translational relevant human bronchial epithelial cell line; thus, fully differentiated primary NHBE cells were examined. Similar to MLE-15 cells, WSN infection of NHBE cells increased IDO activity. Interestingly, kyn was only present in the basal media (Fig. 1C) with no detectable kyn present in the NHBE apical washes (data not shown). This suggests that the effect of IDO induction after influenza infection is reducing the trp concentration in the undifferentiated basal-lateral side of the lung airways. Oddly, kyn was detected in the basal media of mock-infected cells (Fig. 1D). To determine whether the presence of IDO activity was associated with the differentiation of the cells, basal media was collected at various time points pre–air–liquid interface (ALI) and post–ALI until they were fully differentiated. Increased levels of IDO activity began at 1 week at ALI compared with pre–ALI and reached mock-infected levels at week 2 of differentiation (Fig. 1D). This suggests that one or more subsets of differentiated NHBE cells produce a basal level of IDO activity. Importantly, these results indicate that IDO1 mRNA expression and activity are increased across mouse and human lung epithelial cells after influenza infection.
Peak IDO1 and IFN-λ expression coincide
During influenza, virus infection antiviral IFNs are expressed. IFN-λ is highly expressed in the mouse lung airways after influenza infection (Jewell and others 2010), but there is little known regarding the expression profile of type I, II, and III IFNs in respiratory epithelial cells during influenza virus infection. To determine the pattern of IFN expression, MLE-15 cells were infected with WSN and IFN-λ and IFN-α levels were determined at 24, 48, and 72 hpi (Fig. 2A). At 24 hpi, IFN-λ was primarily detected in the MLE-15 cell supernatant; however, at 48 hpi, the concentration of IFN-λ peaked and was higher than IFN-α. Interestingly, peak IDO1 mRNA expression also occurred at 48 hpi (Fig. 1A), a feature consistent with the hypothesis that IFN-λ drives IDO1 expression. No IFN-γ mRNA was present in the cells at any time point postinfection (data not shown), indicating that IDO1 was not upregulated by IFN-γ. A significant increase in IFN-λ production compared with IFN-α/β was also observed during WSN infection of NHBE cells (Fig. 2B). Peak IFN-λ expression also occurred at 48 hpi (Fig. 2B), which is consistent with increased IDO activity (Fig. 1C). Similar to the release of kyn in the NHBE cells, IFN-λ was initially detected in the basal media and was released at higher levels than compared with the apical wash (Fig. 2B). Together, these results suggest that IFN-λ produced during influenza infection upregulates the expression and activity of IDO and that IFN-λ may be signaling neighboring cells through the IFN-λR to induce IDO1 expression.
FIG. 2.

Influenza induces type III IFN production compared with type I IFNs. (A) MLE-15 cells were infected with WSN at an MOI of 0.01. Cell supernatant was collected at 24, 48, and 72 hpi and IFN-λ and IFN-α concentrations were determined through ELISA. (B) Differentiated NHBE cells were infected with WSN at an MOI of 0.5 and IFN-λ, and type I IFN concentrations were determined from apical and basal supernatant at indicated times points via ELISA. IFN-α and IFN-β concentrations were determined by ELISA independently, and the data were combined. The graph shows the mean and standard deviation (SD) from at least 2 independent experiments combined. Significance was determined by a 2-way ANOVA (*P<0.05, ***P<0.001). ND, not detectable.
IFN-λ upregulates IDO1 activity
It is established that type I and type II IFNs can induce IDO activity (Bianchi and others 1988). However, as IFN-λ was predominantly expressed after WSN infection, recombinant IFN-λ (rIFN-λ) was evaluated for its ability to stimulate IDO expression and activity. Stimulation of the MLE-15 cells for 24 h with rIFN-λ3 induced a substantial increase in IDO1 mRNA expression compared with unstimulated cells (Fig. 3A). This increase in IDO1 expression was dose dependent (Fig. 3A). IFN-λ-mediated upregulation of IDO1 was generally specific and had minimal effect on IDO2 expression, a finding that is consistent with influenza results (Fig. 1A). Notably, there was also a dose-response effect in IDO activity after IFN-λ stimulation (Fig. 3B), where increasing concentrations of IFN-λ were associated with increased IDO activity. Similar results were observed in the NHBE cells. Apical stimulation with rIFN-λ1 (IL-29) or rIFN-λ2 (IL-28a) was able to significantly increase IDO activity in NHBE cells compared with unstimulated cells (Fig. 3C). As previously observed after WSN infection (Fig. 1D), IFN-λ stimulation increased the kyn levels in the basal media with low concentrations detected in apical washes (Fig. 3C). These findings show that IFN-λ can directly stimulate IDO activity primarily through IDO1.
FIG. 3.

rIFN-λ upregulates the expression of IDO. (A, B) MLE-15 cells were stimulated with varying concentrations of rIL28b (IFN-λ3). (A) RNA was collected at 24 h poststimulation, and IDO1 and IDO2 expression was determined by qRT-PCR. (B) Supernatants were collected at 48 h poststimulation to assess IDO activity. (C) Differentiated NHBE cells were stimulated with rIL28a (IFN-λ2) or rIL29 (IFN-λ1) at 25 nM. Supernatants were collected at 48 h poststimulation for IDO activity analysis. (D) MLE-15 cells were transfected for 16 h with siIFN-λ3 or siNEG followed by infection with WSN at an MOI of 0.01. RNA was harvested 48 hpi. IDO1 gene expression was determined through qRT-PCR. Significance was assigned by Student's t-test compared with negative control. (E) Differentiated NHBE cells were infected with WSN at an MOI of 0.5. After infection, neutralizing antibodies against IL28b (IFN-λ3) or IL29 (IFN-λ1) (4 μg/mL) were added to the basal media. Basal media was harvested at indicated time points, and the concentration of type I IFNs was determined by ELISA. A representative graph is shown of the mean and standard deviation (SD) from combined data from at least 2 independent experiments. Unless otherwise stated, significance was determined by a one-way ANOVA (*P<0.05, **P<0.01, ***P<0.001).
Since the recombinant IFN-λ stimulated IDO1 expression, it was important to confirm that IFN-λ was mediating IDO1 stimulation during influenza infection. The role of IFN-λ in IDO induction was examined by silencing IFN-λ3 using siRNA in the MLE-15 cells. Treatment with the siRNA targeting IFN-λ3 (siIFN-λ3) substantially reduced the relative gene expression of IFN-λ compared with nontargeting control (siNEG) (Supplementary Fig. S1A; Supplementary Data are available online at www.liebertpub.com/jir). When the MLE-15 cells were infected with WSN after transfection with siIFN-λ3, there was a significant reduction in IDO1 mRNA expression compared with siNEG-treated cells (Fig. 3D). However, IDO1 mRNA was elevated compared with uninfected cells after siRNA knockdown, suggesting that another factor is affecting IDO1 expression. To determine whether suppression of type III IFNs is inducing another IDO stimulator, NHBE cells were infected with WSN in the presence or absence of a neutralizing antibody to IFN-λ1 (anti-IL29) or IFN-λ3 (anti-IL28b) and the level of type I IFNs was determined. In the presence of anti-IL28b, there was a significant increase in IFN-β in the basal media (Fig. 3F), which may be an alternative method to induce IDO1 expression in the absence of type III IFNs. These results show that IFN-λ is, to a major extent, responsible for the upregulation of IDO during influenza infection. It is likely that other factors, including type I IFNs, may be involved in IDO upregulation and it is also possible that when both type I and type III IFNs are present, they may act synergistically to increase IDO1 expression.
Cellular expression of IDO after IFN-λ stimulation and WSN infection
Unlike immortalized cell lines, NHBE cells are able to be differentiated into ciliated, nonciliated, and goblet cells. Since there was measureable IDO activity in the mock-treated differentiated NHBE cells (Fig. 1D), the expression of IDO1 was determined in specific cell types. At homeostatic conditions, IDO was expressed in goblet cells (Fig. 4A), ciliated and nonciliated cells (Fig. 4C) as determined by MUC5AC and beta actin staining, respectively. To determine whether IFN-λ stimulation altered the cellular expression of IDO, differentiated NHBE cells were treated with IFN-λ and stained for ciliated cells or goblet cells and IDO1. Interestingly, IFN-λ stimulation induced IDO expression in all the differentiated cells rather than one particular cell type (Fig. 4B, D). This suggests that the IFN-λR is not restricted to a subset of respiratory epithelial cells.
FIG. 4.
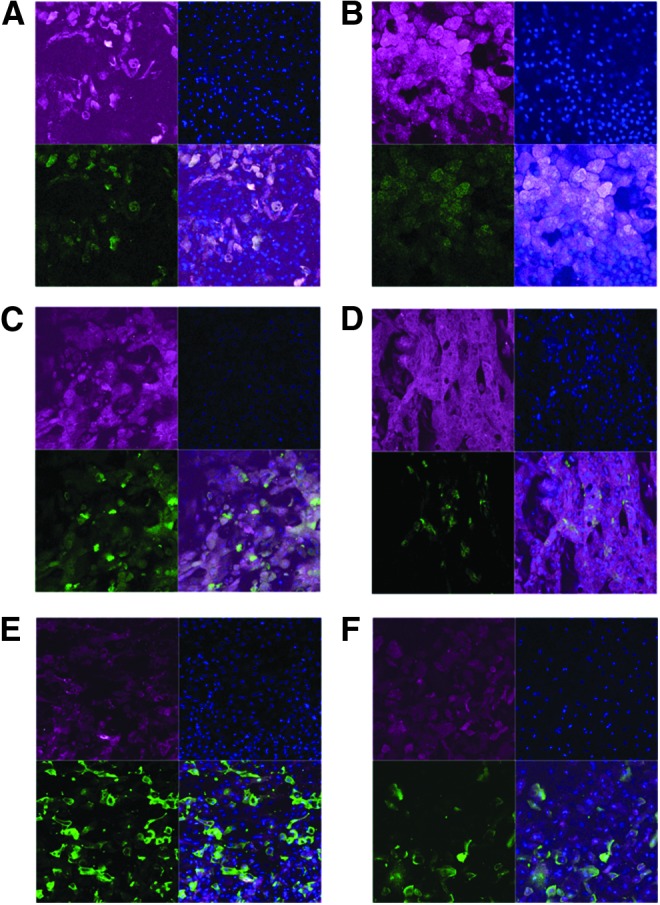
IDO expression is not differentially regulated in cell types after IFN-λ stimulation, but it is primarily expressed in uninfected cells after influenza infection. (A-D) Differentiated NHBE cells were stimulated with (B, D) 25 nM of rIL28a (IFN-λ2) or (A, C) mock stimulated. Cells were fixed and stained at 24 h poststimulation for (A, B) goblet cells or (C, D) ciliated cells (green) and IDO (red). Cells were counterstained with DAPI. (E-F) Differentiated NHBE cells were infected with WSN at an MOI of 0.5 and fixed at (E) 24 or (F) 48 hpi. Cells were stained for IDO (red) and NP (green) and counterstained with DAPI.
Human influenza viruses preferentially bind to α-2, 6 sialic acid moieties that are highly expressed on ciliated cells and goblet cells in the respiratory tract (Chan and others 2010; Oshansky and others 2011; Zeng and others 2013). These cells are the main replication site of human influenza viruses (Ibricevic and others 2006). After an influenza infection in the NHBE cells, there was a significant increase in IDO activity (Fig. 1C). To determine whether this increase was associated with infected or uninfected cells, differentiated NHBE cells were infected with WSN and stained for IDO1 and NP at 24 48 h postinfection. IDO1 was primarily expressed in uninfected cells with some IDO1-positive infected cells (Fig. 4E, F). The high IDO1 expression in uninfected cells is consistent with the expression of type III IFNs and potential paracrine signaling of IFN-λ.
Inhibition of IDO decreases viral load and is associated with increased cell death
Since IDO is upregulated during influenza infection of MLE-15 cells, a feature linked to IFN-λ expression, the relationship between IDO expression and virus load was determined. MLE-15 cells were infected with WSN in the presence or absence of 1-methyl-D, L-tryptophan (1MT). 1MT blocks IDO activity through competitive inhibition, and the racemic mixture will inhibit both IDO1 and IDO2 activity (Lob and others 2009; Qian and others 2012). The concentration of 1MT used inhibited IDO activity during influenza infection based on kyn levels (Supplementary Fig. S1B), and it caused negligible cellular cytotoxicity (data not shown). IDO inhibition during influenza infection resulted in a decrease in viral load at 24 hpi, and a slight but significant decrease in viral titers at 48 hpi, compared with untreated controls (Fig. 5A). To determine whether the decrease in viral titers was associated with increased cell death, cellular supernatants were tested for the presence of adenylate kinase and compared with uninfected and completely lysed cells. At 24 and 48 hpi, there were no substantial differences between 1MT-treated cells and control media (Fig. 5B). However, at 72 hpi, there was a significant increase in the amount of cell death with cells receiving 1MT (Fig. 5B). These results show that the absence of IDO activity decreases viral burden early during infection and reduces cellular viability at later time points postinfection.
FIG. 5.

Inhibition of IDO decreases viral titers and reduces cellular viability. MLE-15 cells were infected with WSN at an MOI of 0.01 with or without 1-methyl-D, L-tryptophan (1MT). Cellular supernatant was collected at indicated time points. (A) Viral titers were determined from supernatant using a TCID50. (B) Cell death was evaluated by adenylate kinase release. A representative graph is shown of the mean and standard deviation (SD) from at least 2 independent experiments. Significance is indicated when compared with control treated cells using a t-test at each time point (*P<0.05).
Discussion
Respiratory epithelial cells respond to influenza infection to limit virus replication through elaboration of antiviral IFNs (Ank and Paludan 2009). The results from this study show that influenza infection of respiratory epithelial cells upregulates IDO activity, specifically IDO1, which is partially driven by IFN-λ. This finding is important as IDO attenuates the immune response to virus infection, and because this is the first demonstration that IFN-λ is an inducer of IDO1 in the context of influenza infection. Recently, a second IDO enzyme, IDO2, was recognized and findings are emerging on the differential regulation between IDO1 and IDO2. IDO1 has been shown to be highly upregulated in response to IFN stimulation (Ball and others 2007), while IDO2 appears more involved in tumor evasion (Ball and others 2009). The findings reported here that influenza preferentially upregulates IDO1 compared with IDO2 are important when considering features driving the antiviral state.
Another noteworthy result from these studies was the finding that IDO is basally secreted from respiratory epithelial cells after influenza virus infection or after IFN stimulation in primary normal fully differentiated NHBE cells (Figs. 1C and 3C). The NHBE cell model closely mimics the lung airways as these cells undergo similar differentiation and are maintained at an air–liquid interface. The culture method for NHBE cells also provides the opportunity to evaluate secretions from the apical and basal surfaces of the cells. In this study, kyn was only detected in the basal media, suggesting that the effects of IDO activity are directed toward undifferentiated cell types and perhaps immune cell types that are recruited to sites of infection rather than the virus. Interestingly, there was a high concentration of kyn present in the basal media of uninfected and unstimulated NHBE cells (Fig. 1D). This suggests that a bronchial epithelial cell type constitutively supplies a low level of IDO to the environment. A recent study showed colonic goblet cells as an important source of IDO activity for epithelial cell turnover after T. muris infections (Bell and Else 2011). Our data suggest that goblet cells and ciliated cells are constitutive sources of IDO1 (Fig. 4A, C) but after IFN-λ stimulation almost all of the cells present express IDO1 protein (Fig. 4B, D). During a human influenza infection, uninfected cells are the main source of IDO activity (Fig. 4E, F). Since IFN-λ is the main IFN produced by respiratory epithelial cells after infection, the increased activity of IDO is most likely driven by IFN-λR stimulation rather than by infection. This is consistent with increasing levels of IDO activity and IFN-λ with increasing MOI (Fig 1B and data not shown). Interestingly, when IL-28b is blocked during infection, type I IFNs are increased (Fig. 3F), suggesting that type I IFNs are able to compensate for the removal of the dominant IFN.
Although IFN-λ is involved in mediating an antiviral state, IFN-λ-mediated IDO induction was linked to higher viral titers compared with cells with IDO inhibited (Fig. 5A). In the absence of IDO activity, there was a significant decrease in viral titers or load. However, there was no difference in viral titers with 1MT treatment in the C57/BL6 mouse model after X31 infection (Fox and others 2013). This suggests that a reduction in viral titers in epithelial cells may not have substantial in vivo effects. Reduced cell viability may be one possibility linking decreased viral titers in vitro when IDO activity is blocked after infection. Although there was no significant difference between 1MT administration and cell death at 48 hpi (Fig. 5B), it is possible that changes in cellular functions may reduce the capacity of the cell to replicate virus. IDO has been shown to impart antioxidant properties after influenza infection by using superoxide anion as a substrate, thus protecting the cell from oxidative damage (Hirata and Hayaishi 1975; Jacoby and Choi 1994). These cellular changes would occur at early stages of cell death, and perhaps be more distinguishable at later time points, thus providing a pro-survival response for the epithelial cells.
In addition to sharing downstream signaling pathways with type I and II IFNs for IDO induction, IFN-λ has also been shown to dampen the immune response through Treg stimulation. One report showed that dendritic cells stimulated with IFN-λ triggered the proliferation of Foxp3-expressing Treg cells (Mennechet and Uze 2006). In addition, IFN-λ in conjunction with IFN-α expression during respiratory syncytial virus infection has been shown to suppress CD4+ T cell proliferation, where the suppression could be blocked by addition of neutralizing antibodies to the IFN receptors (Chi and others 2006). These studies support a mechanism by which IFN-λ is inducing IDO expression and inducing a regulatory phenotype.
In summary, the results from this study show a mechanism of IDO upregulation through IFN-λ signaling. This finding is of significance, as IFN-λ can only act on a limited number of cells, so this response is unique to epithelial cells and potentially pDCs. These studies also show a role of IDO in increasing viral titers in epithelial cells and potential modulation recruited immune cells by basal secretion of IDO's metabolite. Thus, this study enhances the link between IDO activity and regulation during infection and supports the need for continued studies on IDO's role in vaccine development.
Supplementary Material
Acknowledgments
The authors thank Elizabeth O'Connor for her help and Dr. Wendy Watford for providing and assisting with reagents. This work was supported by the National Institutes of Health U01 grant AI083005-01 and the Georgia Research Alliance.
Author Disclosure Statement
No competing financial interests exist.
References
- Ank N, Iversen MB, Bartholdy C, Staeheli P, Hartmann R, Jensen UB, Dagnaes-Hansen F, Thomsen AR, Chen Z, Haugen H, Klucher K, Paludan SR. 2008. An important role for type III interferon (IFN-lambda/IL-28) in TLR-induced antiviral activity. J Immunol 180(4):2474–2485 [DOI] [PubMed] [Google Scholar]
- Ank N, Paludan SR. 2009. Type III IFNs: new layers of complexity in innate antiviral immunity. Biofactors 35(1):82–87 [DOI] [PubMed] [Google Scholar]
- Ball HJ, Sanchez-Perez A, Weiser S, Austin CJ, Astelbauer F, Miu J, McQuillan JA, Stocker R, Jermiin LS, Hunt NH. 2007. Characterization of an indoleamine 2,3-dioxygenase-like protein found in humans and mice. Gene 396(1):203–213 [DOI] [PubMed] [Google Scholar]
- Ball HJ, Yuasa HJ, Austin CJD, Weiser S, Hunt NH. 2009. Indoleamine 2,3-dioxygenase-2; a new enzyme in the kynurenine pathway. Int J Biochem Cell Biol 41(3):467–471 [DOI] [PubMed] [Google Scholar]
- Bell LV, Else KJ. 2011. Regulation of colonic epithelial cell turnover by IDO contributes to the innate susceptibility of SCID mice to Trichuris muris infection. Parasite Immunol 33(4):244–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchi M, Bertini R, Ghezzi P. 1988. Induction of indoleamine dioxygenase by interferon in mice: a study with different recombinant interferons and various cytokines. Biochem Biophys Res Commun 152(1):237–242 [DOI] [PubMed] [Google Scholar]
- Chan RW, Yuen KM, Yu WC, Ho CC, Nicholls JM, Peiris JS, Chan MC. 2010. Influenza H5N1 and H1N1 virus replication and innate immune responses in bronchial epithelial cells are influenced by the state of differentiation. PLoS One 5(1):e8713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi B, Dickensheets HL, Spann KM, Alston MA, Luongo C, Dumoutier L, Huang J, Renauld JC, Kotenko SV, Roederer M, Beeler JA, Donnelly RP, Collins PL, Rabin RL. 2006. Alpha and lambda interferon together mediate suppression of CD4 T cells induced by respiratory syncytial virus. J Virol 80(10):5032–5040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Commins S, Steinke JW, Borish L. 2008. The extended IL-10 superfamily: IL-10, IL-19, IL-20, IL-22, IL-24, IL-26, IL-28, and IL-29. J Allergy Clin Immunol 121(5):1108–1111 [DOI] [PubMed] [Google Scholar]
- de Weerd NA, Samarajiwa SA, Hertzog PJ. 2007. Type I interferon receptors: biochemistry and biological functions. J Biol Chem 282(28):20053–20057 [DOI] [PubMed] [Google Scholar]
- Fallarino F, Vacca C, Orabona C, Belladonna ML, Bianchi R, Marshall B, Keskin DB, Mellor AL, Fioretti MC, Grohmann U, Puccetti P. 2002. Functional expression of indoleamine 2,3-dioxygenase by murine CD8 alpha(+) dendritic cells. Int Immunol 14(1):65–68 [DOI] [PubMed] [Google Scholar]
- Fox JM, Sage LK, Huang L, Barber J, Klonowski KD, Mellor AL, Tompkins SM, Tripp RA. 2013. Inhibition of indoleamine 2,3-dioxygenase enhances the T-cell response to influenza virus infection. J Gen Virol 94(Pt 7):1451–1461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirata F, Hayaishi O. 1975. Studies on indoleamine 2,3-dioxygenase. I. Superoxide anion as substrate. J Biol Chem 250(15):5960–5966 [PubMed] [Google Scholar]
- Ibricevic A, Pekosz A, Walter MJ, Newby C, Battaile JT, Brown EG, Holtzman MJ, Brody SL. 2006. Influenza virus receptor specificity and cell tropism in mouse and human airway epithelial cells. J Virol 80(15):7469–7480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacoby DB, Choi AM. 1994. Influenza virus induces expression of antioxidant genes in human epithelial cells. Free Radic Biol Med 16(6):821–824 [DOI] [PubMed] [Google Scholar]
- Jewell NA, Cline T, Mertz SE, Smirnov SV, Flano E, Schindler C, Grieves JL, Durbin RK, Kotenko SV, Durbin JE. 2010. Lambda interferon is the predominant interferon induced by influenza A virus infection in vivo. J Virol 84(21):11515–11522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lob S, Konigsrainer A, Zieker D, Brucher BL, Rammensee HG, Opelz G, Terness P. 2009. IDO1 and IDO2 are expressed in human tumors: levo- but not dextro-1-methyl tryptophan inhibits tryptophan catabolism. Cancer Immunol Immunother 58(1):153–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiwald S, Wehner R, Schmitz M, Bornhauser M, Loeb S, Wassmuth R. 2011. IDO1 and IDO2 gene expression analysis by quantitative polymerase chain reaction. Tissue Antigens 77(2):136–142 [DOI] [PubMed] [Google Scholar]
- Maneglier B, Rogez-Kreuz C, Spreux-Varoquaux O, Malleret B, Therond P, Samah B, Drouet I, Dormont D, Advenier C, Clayette P. 2007. Comparative effects of two type I interferons, human IFN-alpha and ovine IFN-tau on indoleamine-2,3-dioxygenase in primary cultures of human macrophages. Fundam Clin Pharmacol 21(1):29–34 [DOI] [PubMed] [Google Scholar]
- Matrosovich M, Matrosovich T, Garten W, Klenk HD. 2006. New low-viscosity overlay medium for viral plaque assays. Virol J 3:63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meininger D, Zalameda L, Liu Y, Stepan LP, Borges L, McCarter JD, Sutherland CL. 2011. Purification and kinetic characterization of human indoleamine 2,3-dioxygenases 1 and 2 (IDO1 and IDO2) and discovery of selective IDO1 inhibitors. Biochim Biophys Acta 1814(12):1947–1954 [DOI] [PubMed] [Google Scholar]
- Mennechet FJ, Uze G. 2006. Interferon-lambda-treated dendritic cells specifically induce proliferation of FOXP3-expressing suppressor T cells. Blood 107(11):4417–4423 [DOI] [PubMed] [Google Scholar]
- Metz R, Duhadaway JB, Kamasani U, Laury-Kleintop L, Muller AJ, Prendergast GC. 2007. Novel tryptophan catabolic enzyme IDO2 is the preferred biochemical target of the antitumor indoleamine 2,3-dioxygenase inhibitory compound D-1-methyl-tryptophan. Cancer Res 67(15):7082–7087 [DOI] [PubMed] [Google Scholar]
- Munn DH, Sharma MD, Baban B, Harding HP, Zhang Y, Ron D, Mellor AL. 2005. GCN2 kinase in T cells mediates proliferative arrest and anergy induction in response to indoleamine 2,3-dioxygenase. Immunity 22(5):633–642 [DOI] [PubMed] [Google Scholar]
- Oshansky CM, Barber JP, Crabtree J, Tripp RA. 2010. Respiratory syncytial virus F and G proteins induce interleukin 1alpha, CC, and CXC chemokine responses by normal human bronchoepithelial cells. J Infect Dis 201(8):1201–1207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oshansky CM, Pickens JA, Bradley KC, Jones LP, Saavedra-Ebner GM, Barber JP, Crabtree JM, Steinhauer DA, Tompkins SM, Tripp RA. 2011. Avian influenza viruses infect primary human bronchial epithelial cells unconstrained by sialic acid alpha 2, 3 residues. PLoS One 6(6):e21183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popov A, Abdullah Z, Wickenhauser C, Saric T, Driesen J, Hanisch FG, Domann E, Raven EL, Dehus O, Hermann C, Eggle D, Debey S, Chakraborty T, Kronke M, Utermohlen O, Schultze JL. 2006. Indoleamine 2,3-dioxygenase-expressing dendritic cells form suppurative granulomas following Listeria monocytogenes infection. J Clin Invest 116(12):3160–3170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian F, Liao J, Villella J, Edwards R, Kalinski P, Lele S, Shrikant P, Odunsi K. 2012. Effects of 1-methyltryptophan stereoisomers on IDO2 enzyme activity and IDO2-mediated arrest of human T cell proliferation. Cancer Immunol Immunother [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed LJ, Meunch H. 1938. A simple method for estimating fifty percent endpoints. Am J Hygiene 27:493–497 [Google Scholar]
- Sanders CJ, Doherty PC, Thomas PG. 2010. Respiratory epithelial cells in innate immunity to influenza virus infection. Cell Tissue Res 343(1):13–21 [DOI] [PubMed] [Google Scholar]
- Smith JH, Brooks P, Johnson S, Tompkins SM, Custer KM, Haas DL, Mair R, Papania M, Tripp RA. 2011. Aerosol vaccination induces robust protective immunity to homologous and heterologous influenza infection in mice. Vaccine 29(14):2568–2575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommereyns C, Paul S, Staeheli P, Michiels T. 2008. IFN-lambda (IFN-lambda) is expressed in a tissue-dependent fashion and primarily acts on epithelial cells in vivo. PLoS Pathog 4(3):e1000017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Wissen M, Snoek M, Smids B, Jansen HM, Lutter R. 2002. IFN-gamma amplifies IL-6 and IL-8 responses by airway epithelial-like cells via indoleamine 2,3-dioxygenase. J Immunol 169(12):7039–7044 [DOI] [PubMed] [Google Scholar]
- Wang J, Oberley-Deegan R, Wang S, Nikrad M, Funk CJ, Hartshorn KL, Mason RJ. 2009. Differentiated human alveolar type II cells secrete antiviral IL-29 (IFN-lambda 1) in response to influenza A infection. J Immunol 182(3):1296–1304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng H, Goldsmith CS, Maines TR, Belser JA, Gustin KM, Pekosz A, Zaki SR, Katz JM, Tumpey TM. 2013. Tropism and infectivity of influenza virus, including highly pathogenic avian H5N1 virus, in ferret tracheal differentiated primary epithelial cell cultures. J Virol 87(5):2597–2607 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.