

Abstract
Background and Purpose
Most forms of human obesity are characterized by impaired leptin sensitivity and, therefore, the effectiveness of anti-obesity leptin therapy in these leptin-resistant obese patients is marginal. Hence, the development of strategies to increase leptin sensitivity is of high priority in the field of obesity research.
Experimental Approach
We first examined the effects of co-administration of leptin and meta-chlorophenylpiperazine (mCPP), an agonist of 5-HT2C and 5-HT1B receptors, on energy balance in leptin-resistant diet-induced obese (DIO) mice. We further assessed leptin-induced phosphorylation of the STAT-3 (pSTAT3) in various brain regions of DIO mice pretreated with mCPP or in mice genetically lacking 5-HT2C receptors.
Results
Co-administration of mCPP with leptin had an additive effect on reducing body weight in DIO mice. Furthermore, mCPP pretreatment in DIO mice enhanced leptin-induced pSTAT3 in the arcuate nucleus, the ventromedial hypothalamic nucleus, and the ventral premammillary nucleus. Finally, deletion of 5-HT2C receptors significantly blunted leptin-induced pSTAT3 in these same hypothalamic regions.
Conclusions and Implications
Our study provides evidence that drugs, which activate 5-HT2C receptors, could function as leptin sensitizers and be used in combination with leptin to provide additional weight loss in DIO.
Tables of Links
| TARGETS | |
|---|---|
| GPCRsa | Catalytic receptorsb |
| 5-HT1B receptor | Leptin receptor |
| 5-HT2C receptor | Enzymesc |
| Melanocortin 4 receptor | JAK2 |
| LIGANDS | |
|---|---|
| 5-HT | Leptin |
| Agouti-related peptide | Lorcaserin |
| Amylin | Meta-chlorophenylpiperazine |
| Glycine | Neuropeptide Y |
| H2O2 | POMC |
| Isoflurane | Sibutramine |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (a,b,cAlexander et al., 2013a, b, c).
Introduction
Obese individuals are at an increased risk of developing type II diabetes, cardiovascular disease and cancer. Obesity is associated with 300 000 premature deaths every year. These figures highlight the urgent need to develop more effective therapies to combat the disease.
Leptin, an adipocyte-derived hormone that mirrors the body's fat stores, plays an essential role in preventing obesity (Halaas et al., 1995). Leptin initiates multiple intracellular signalling cascades, including the JAK2/STAT-3 pathway (Tartaglia, 1997). STAT3 has a critical role in mediating the anti-obesity effects of leptin (Bates et al., 2003), and the phosphorylation of STAT3 (pSTAT3) after leptin administration has been widely used to assess leptin sensitivity in animals (Munzberg et al., 2003; 2004).
Leptin acts primarily through the long form of the leptin receptor (LepRb) to suppress food intake, increase energy expenditure and, therefore, decrease body weight (Tartaglia et al., 1995). Various brain regions that express LepRb have been identified as important sites for the ability of leptin to produce anti-obesity effects. These include neurons in the arcuate nucleus (ARC) expressing pro-opiomelanocortin (POMC; Balthasar et al., 2004; Hill et al., 2010) or co-expressing neuropeptide Y (NPY)/agouti-related peptide (AgRP; van de Wall et al., 2008), neurons in the ventromedial hypothalamic nucleus (VMH, also known as VMN; Dhillon et al., 2006; Bingham et al., 2008), neurons in the ventral premammillary nucleus (PMV; Leshan et al., 2012), and neurons in the medial nucleus of solitary tract (NTS) and the area postrema (Hayes et al., 2010).
Obesity caused by congenital leptin deficiency can be completely cured by leptin replacement therapy (Farooqi et al., 1999). However, most forms of human obesity are characterized by hyperleptinaemia with leptin being ineffective, a state referred to as leptin resistance (Myers et al., 2010). The anti-obesity effects of leptin in these leptin-resistant obese patients are minimal even at high doses. Thus, developing strategies to increase leptin sensitivity has been a high priority in the field of obesity research. A few ‘leptin sensitizers’ have been identified, which can increase leptin sensitivity and produce additive anti-obesity effects when combined with leptin. For example, while leptin alone fails to reduce body weight in rats with diet-induced obesity (DIO, an animal model most commonly used to replicate human obesity), the combination of leptin and the pancreatic hormone amylin produces additional weight loss in DIO rats (Roth et al., 2008). Importantly, the leptin-sensitizing effects of amylin were also observed in obese patients (Roth et al., 2008). Similarly, sibutramine also increases leptin sensitivity in DIO rats, demonstrated by the additive effects on body weight loss induced by the two reagents combined (Boozer et al., 2001). Notably, sibutramine acts as a re-uptake inhibitor to increase the levels of 5-HT, as well as noradrenaline and dopamine (Heal et al., 1998). This raises the possibility that drugs targeting the 5-HT system may serve as leptin sensitizers.
In the current study, we first tested if co-administration of leptin and meta-chlorophenylpiperazine (mCPP), an agonist for both 5-HT2C and 5-HT1B receptors, can induce additional body weight loss in mice with DIO. We then examined the effects of mCPP on leptin-induced pSTAT3 in the brain of DIO mice. Finally, we determined whether leptin-induced pSTAT3 was altered in 2C-null mice, which genetically lack 5-HT2C receptors.
Methods
Mice
We have previously generated a 2C-null (loxTB-5-HT2C receptor) mouse line (Xu et al., 2008). We verified that 2C-null mice lack 5-HT2C receptors globally and phenotypically resemble the conventional 5-HT2C receptor-knockout mice (Tecott et al., 1995). We crossed male C57Bl6 mice with female mice that were heterozygous for the 2C-null allele (Xu et al., 2008). This cross-generated male wild type (WT) and 2C-null littermates. All the breeders had been backcrossed to the C57Bl6 background for more than 12 generations. In addition, some C57Bl6 mice were purchased from the mouse facility of University of Texas Southwestern Medical Center or of Baylor College of Medicine.
Mice were housed in a temperature-controlled environment at 22–24°C using a 12 h light/12 h dark cycle. The mice were fed standard chow (6.5% fat, #2920, Harlan-Teklad, Madison, WI, USA) or a high-fat diet (HFD, 40% fat; TD.95217, Harlan) as specified below. Water was provided ad libitum. A total of 57 mice was used in this study. All mice experiments were approved by the Institutional Animal Care and Use Committee of University of Texas Southwestern Medical Center at Dallas or of Baylor College of Medicine at Houston. All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010).
Additive effects of leptin and mCPP
Experiment 1
This experiment was performed at the University of Texas Southwestern Medical Center at Dallas. In order to determine if leptin and mCPP can additively regulate energy and glucose homeostasis in DIO mice, male C57Bl6 mice (at 8 weeks of age) were fed on HFD for 6 weeks to induce DIO and leptin resistance (minimum body weight of 45 g). These DIO mice were then anaesthetized with inhaled 2% isoflurane and depth of anaesthesia was assessed by the absence of a toe-pinch response. Alzet osmotic minipumps (#2002, DURECT Corporation, ALZET Osmotic Pumps, Cupertino, CA, USA) containing saline, leptin (0.5 mg·kg−1·day−1), mCPP (1.5 mg·kg−1·day−1) or leptin (0.5 mg·kg−1·day−1) plus mCPP (1.5 mg·kg−1·day−1) were implanted s.c. Eight mice were included in each treatment group. Leptin (purchased from Dr E. Parlow, National Institute of Diabetes and Digestive and Kidney Diseases and The National Hormone and Pituitary Program), mCPP (#125180, Sigma, St. Louis, MO, USA), or their combination was dissolved in saline. The dose of leptin (0.5 mg·kg−1·day−1) was chosen based on a previous report that this dose of leptin alone does not reduce body weight in DIO rats (Boozer et al., 2001), and we confirmed the lack of leptin response in DIO mice. mCPP was administered at the dose of 1.5 mg·kg−1·day−1, because our pilot studies showed that this dose alone did not affect body weight and food intake in DIO mice.
Body weight, whole-body fat mass and lean mass [measured by use of quantitative magnetic resonance (QMR)] were recorded before implantation of the minipumps, and these data were used to ensure that mice subjected to different treatments had comparable baseline body weight, fat mass and lean mass. These mice were then housed singly and continued to be fed a HFD. Daily body weight and food intake were monitored for the entire 2 week treatment period. Whole body fat mass and lean mass were measured again at the end of the 2 week treatment, and changes in fat mass and lean mass in each mouse were calculated. Cumulative food intake was calculated based on daily food intake, and feeding efficiency was calculated as the ratio between the changes in body weight and cumulative food intake over the same period.
At the end of the 2 week treatment, mice were briefly deprived of food (fasted) from 08:00 h to 10:00 h to ensure their stomachs were empty, and blood glucose was measured using tail blood with the OneTouch Ultra glucometer (CVS Pharmacy, Houston, TX, USA). Mice were then deeply anaesthetized and quickly killed. Trunk blood was collected and processed to obtain serum. Serum insulin was measured using the elisa kit (#90080; Crystal Chem Inc., Owners Grove, IL, USA), as described previously (Xu et al., 2010). In parallel, hypothalami were quickly collected and stored at −80°C. Total mRNAs from the hypothalami were extracted and reverse-transcribed to cDNAs using SuperScript III First-Strand cDNA Synthesis kit (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's instructions. Quantitative real-time PCR assays were performed according to published protocols (Bookout and Mangelsdorf, 2003). Tissue mRNA levels were measured with an ABI 7900HT Sequence Detection System (Grand Island, NY, USA). We used pre-developed Taqman assays purchased from Applied Biosystems for all genes. Normalized mRNA levels were expressed in arbitrary units, obtained by dividing the averaged, efficiency-corrected values for sample mRNA expression by that for 18S RNA expression for each sample. The results are expressed as fold change above average control levels.
Leptin-induced pSTAT3
The following experiments were performed at the Baylor College of Medicine at Houston.
Experiment 2
Male C57Bl6 mice (at 4 weeks of age) were fed a HFD for 8 weeks to induce DIO and leptin resistance. During the last 2 weeks of HFD feeding, mice were divided into two groups to receive daily pretreatment of saline or mCPP (1.5 mg·kg−1·day−1, i.p.). At the end of the pretreatment, mice were deprived of food (fasted) overnight and then received a single bolus injection of saline or leptin (0.5 mg·kg−1, i.p.). Two hours after the bolus injections, mice were anaesthetized with inhaled isoflurane, and quickly perfused with 10% formalin, and brain sections were cut at 25 μm. The brain sections were subjected to pSTAT3 immunohistochemical staining as described below. Three or four mice were included in each treatment group.
Experiment 3
Male WT and 2C-null littermates were weaned on chow. At 12 weeks of age, WT and 2C-null littermates showed comparable body weight as reported previously (Xu et al., 2008). These mice were deprived of food (fasted) overnight and then received a single bolus injection of saline or leptin (0.5 mg·kg−1, i.p.). Two hours after the bolus injections, mice were anaesthetized with inhaled isoflurane, and quickly perfused with 10% formalin, and brain sections were cut at 25 μm. The brain sections were subjected to pSTAT3 immunohistochemical staining as described below. Three mice were included in each treatment group.
pSTAT3 immunohistochemistry
The brain sections were pretreated (1% H2O2, 1% NaOH, 0.3% glycine, 0.03% SDS), blocked (3% goat-anti-rabbit serum for 1 h), incubated with rabbit anti-pSTAT3 antibody (1:2000; #9145, Cell Signaling, Danvers, MA, USA) on shaker at room temperature for 24 h and then put in 4°C for 48 h, followed by biotinylated anti-rabbit secondary antibody (1:1000; Vector Laboratories, Burlingame, CA, USA) for 2 h. Sections were then incubated in the avidin-biotin complex (1:500, ABC; Vector Elite Kit) and incubated in 0.04% 3, 3′-diaminobenzidine and 0.01% hydrogen peroxide. After dehydration through graded ethanol, the slides were then immersed in xylene and cover-slipped. Images were analysed using a brightfield Leica microscope (Buffalo Grove, IL, USA).
Quantifications
The numbers of pSTAT3-positive neurons in multiple brain regions were counted by blinded investigators. For each brain region [e.g. ARC, VMH, dorsomedial hypothalamic nucleus (DMH), PMV or NTS], pSTAT3-positive neurons were counted in 3–5 consecutive brain sections containing that region from the same mouse, and the average was treated as the value for that region of the mouse. Three to four mice were included in each treatment group for statistical analyses. Importantly, data from the WT mice (in experiment 3) were also used as lean controls and compared to data from WT DIO mice (in experiment 2), in order to determine if mCPP pretreatment in DIO mice can restore leptin-induced pSTAT3 to the levels seen in lean mice. In the initial study, we found that leptin-induced pSTAT3 levels in the DMH and NTS were comparable between lean WT mice and WT DIO mice (after the 8 week HFD feeding). These are consistent with previous findings from DIO mice fed with HFD for 16 weeks (Munzberg et al., 2004). Therefore, pSTAT3 levels in these two regions were not quantified in later experiments.
Statistics
The data are presented as mean ± SEM. Statistical analyses were performed using GraphPad Prism. Data were compared by two-way anova, followed by post hoc Newman–Keuls Student's tests. The number of animals in each group is specified in the figure legends. P < 0.05 was considered to be statistically significant.
Results
Co-administration of sub-threshold doses of leptin and mCPP additively decreased body weight in DIO mice
We first examined if co-administration of leptin and mCPP can produce additive benefits on energy and glucose balance in leptin-resistant DIO mice. To this end, WT mice were first fed a HFD to induce DIO and leptin resistance (minimum body weight of 45 g). These DIO mice were implanted with Alzet minipumps containing saline, leptin (0.5 mg·kg−1·day−1), mCPP (1.5 mg·kg−1·day−1) or leptin (0.5 mg·kg−1·day−1) plus mCPP (1.5 mg·kg−1·day−1). Body weight changes and food intake were monitored daily for 2 weeks (Figure 1A). Consistent with a leptin-resistant state of these DIO mice, we found that leptin alone failed to significantly alter body weight compared with saline treatment (Figure 1B). Similarly, mice treated with mCPP alone showed comparable body weight changes to mice treated with saline or leptin alone (Figure 1B). Interestingly, co-administration of leptin and mCPP (at the same sub-threshold doses) significantly reduced body weight compared with the saline-treated mice at many time points (Figure 1B). In addition, body weight-lowering effects after co-administration of the two compounds were significantly greater than those of leptin alone or mCPP alone, respectively, at the various time points (Figure 1B). At the end of the 2 week treatment, mice co-administered leptin and mCPP showed significantly greater reductions in fat mass compared to mice treated with saline, leptin alone or mCPP alone, respectively, while the latter three groups had comparable fat mass (Figure 1C). No significant difference in lean mass was observed among the four groups (Figure 1C).
Figure 1.
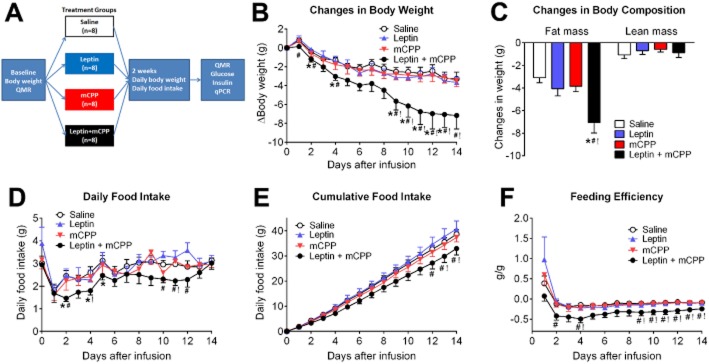
Effects of mCPP and leptin on energy balance in DIO mice. (A) Diagram of the schedule used for the experiments in Figures 3. Body weight-matched WT DIO mice were implanted with minipumps containing saline, leptin (0.5 mg·kg−1·day−1), mCPP (1.5 mg·kg−1·day−1) or leptin (0.5 mg·kg−1·day−1) plus mCPP (1.5 mg·kg−1·day−1). Baseline body weight and QMR were measured before minipump implantation; body weight and food intake were monitored daily for 2 weeks after minipump implantation; QMR, serum glucose/insulin and qPCR were measured at the end of the 2 week treatment. (B) Changes in body weight. (C) Changes in fat mass and lean mass. (D) Daily food intake. (E) Cumulative food intake. (F) Feeding efficiency. Data are presented as mean ± SEM. n = 8 in each group. *P < 0.05 between saline versus leptin + mCPP; #P < 0.05 between leptin versus leptin + mCPP; !P < 0.05 between mCPP versus leptin + mCPP; two-way anova followed by the post hoc Newman–Keuls Student's tests.
Co-administration of leptin and mCPP significantly suppressed daily food intake as well as cumulative food intake compared with saline, leptin alone or mCPP alone, at the various time points (Figure 1D and 1E). Furthermore, feeding efficiency (calculated as ratio of body weight changes over cumulative food intake) was significantly reduced in mice co-administered leptin and mCPP, when compared with mice treated with leptin or mCPP alone (Figure 1F).
We also measured blood glucose and insulin levels at the end of the 2 week treatment. Mice in all four groups showed comparable blood glucose levels (Figure 2A ). While leptin alone did not alter insulin levels, mCPP alone significantly reduced insulin levels compared to the saline group (Figure 2B). The co-administration of leptin and mCPP trended to decrease insulin levels compared to saline group, although these responses did not reach statistical significance (Figure 2B).
Figure 2.
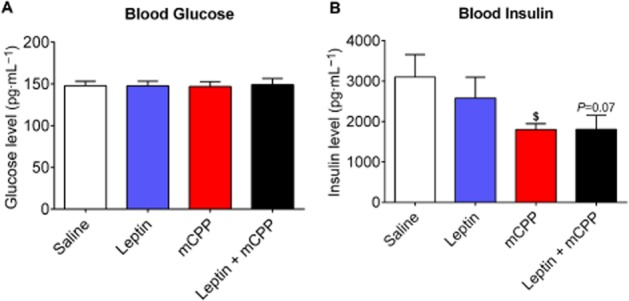
Effects of mCPP and leptin on glucose balance in DIO mice. (A) Blood glucose measured 2 weeks after treatment. (B) Blood insulin measured 2 weeks after treatment. Data are presented as mean ± SEM. n = 8 in each group. $P < 0.05 between saline versus mCPP; two way anova followed by the post hoc Newman–Keuls Student's tests.
We further examined the expression of relevant genes in the hypothalamus after the 2 week treatment. We found that mCPP alone tended to increase POMC mRNAs compared with the saline group, while the co-administration of leptin and mCPP significantly elevated POMC levels (Figure 3A ). In contrast, both mCPP alone and co-administration significantly reduced AgRP mRNAs compared to the saline group (Figure 3B). No significant changes in NPY expression was observed among the four groups (Figure 3C). Interestingly, we found that STAT3 mRNAs were significantly increased by either mCPP alone or by the co-administration of leptin and mCPP (Figure 3D).
Figure 3.
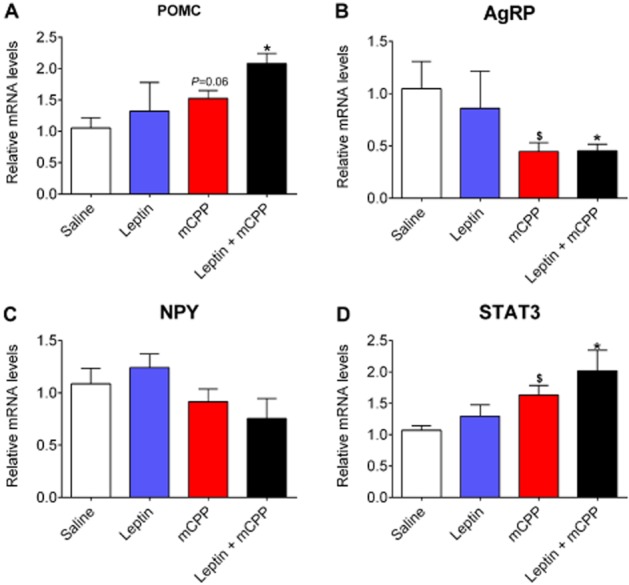
Effects of mCPP and leptin on hypothalamic gene expression. Relative mRNA levels for POMC (A), AgRP (B), NPY (C) and STAT3 (D) in the hypothalamus 2 weeks after various treatments. Data are presented as mean ± SEM. n = 4 in each group. $P < 0.05 between saline versus mCPP; *P < 0.05 between leptin + mCPP versus saline; two-way anova followed by the post hoc Newman–Keuls Student's tests.
mCPP enhanced leptin-induced pSTAT3 in many hypothalamic regions in DIO mice
Since leptin and mCPP showed additive effects on body weight balance in leptin-resistant DIO mice, we sought to examine if mCPP can enhance leptin sensitivity (as measured by leptin-induced pSTAT3) in the brain (Figure 4A). To this end, WT mice were first fed a HFD to induce DIO and leptin resistance. These DIO mice were then randomly divided into groups that received daily pretreatment of either saline or mCPP (1.5 mg·kg−1·day−1, i.p.) for 2 weeks. Similar to our earlier observations, pretreatment with mCPP for 2 weeks did not significantly alter body weight in DIO mice compared with saline-treated mice (data not shown). These mice were further divided into groups that received a single bolus injection of either saline or leptin (0.5 mg·kg−1, i.p.), and pSTAT3 in the brain was examined by use of immunohistochemistry. As a positive control, pSTAT3 was also examined in a group of chow-fed lean mice that received a single bolus injection of either saline or leptin (0.5 mg·kg−1, i.p.).
Figure 4.
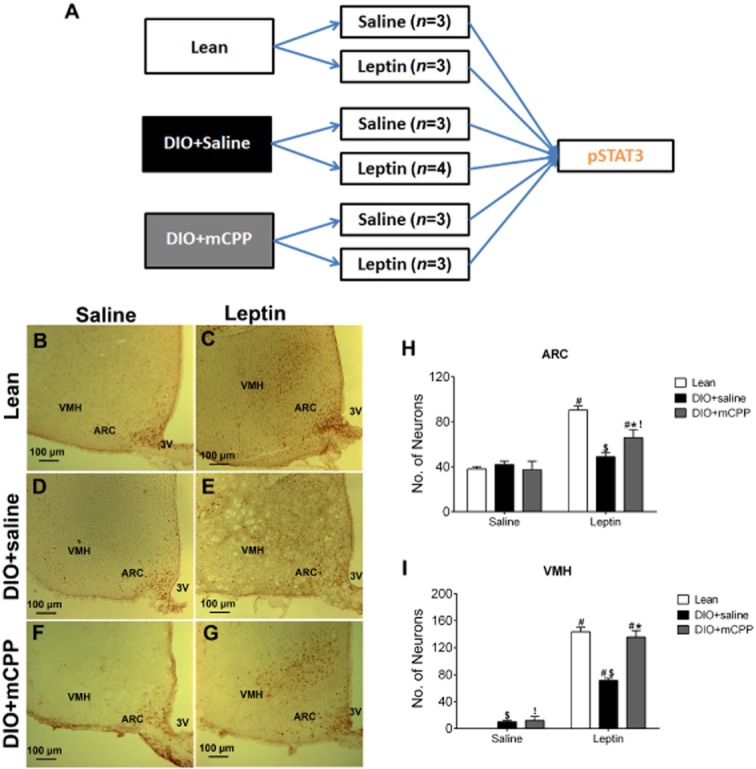
Effects of mCPP pretreatment on leptin-induced pSTAT3 in the ARC and VMH of DIO mice. (A) Diagram for the schedule used for experiments in Figures 4 and 5. (B–C) Representative pSTAT3 immunohistochemical staining in the ARC and VMH of chow-fed lean WT mice receiving a single bolus i.p. injection of saline (B) or 0.5 mg·kg−1 leptin (C). (D–E) Representative pSTAT3 immunohistochemical staining in the ARC and VMH of DIO + saline mice (8 weeks on a HFD and in the last 2 weeks given daily i.p. injections of saline), after receiving a single bolus i.p. injection of saline (D) or 0.5 mg·kg−1 leptin (E). (F–G) Representative pSTAT3 immunohistochemical staining in the ARC and VMH of DIO + mCPP mice (8 weeks on a HFD feeding and in the last 2 weeks given daily i.p. injections of 1.5 mg·kg−1·day−1 mCPP), after receiving a single bolus i.p. injection of saline (F) or 0.5 mg·kg−1 leptin (G). (H–I) Number of pSTAT3-positive neurons in the ARC (H) and VMH (I) of the various groups. Data are presented as mean ± SEM. n = 3–4 in each group. #P < 0.05 between saline and leptin within the same animal group; $P < 0.05 between Lean and DIO + saline within the same injection group; *P < 0.05 between DIO + saline and DIO + mCPP within the same injection group; !P < 0.05 between Lean and DIO + mCPP within the same injection group; two-way anova followed by the post hoc Newman–Keuls Student's tests.
We first quantified pSTAT3-postive neurons in the ARC. After the single bolus injection of saline, a baseline level of pSTAT3 staining in the ARC was observed in lean mice, DIO + saline mice and DIO + mCPP mice, and there was no significant difference among these three groups (Figure 4B, D, F and H). As expected, leptin significantly increased pSTAT3 levels in the ARC of lean mice (Figure 4B, C and H); in contrast, leptin failed to increase pSTAT3 levels in DIO + saline mice (Figure 4D, E and H). Importantly, leptin significantly increased pSTAT3 levels in DIO + mCPP mice (Figure 4F, 4G and 4H). In particular, the leptin-induced pSTAT3 in DIO + mCPP mice was significantly higher than that in DIO + saline mice (Figure 4E, 4G and 4H), but was still significantly lower than that in lean mice (Figure 4C, 4G and 4H).
We also quantified pSTAT3 levels in the VMH. After a single injection of saline, no pSTAT3 staining was observed in the VMH of lean mice; minimal pSTAT3 staining was detected in the VMH of DIO + saline mice and DIO + mCPP mice (Figure 4B, D, F and I). Leptin significantly increased pSTAT3 levels in the VMH of lean, DIO + saline or DIO + mCPP mice respectively (Figure 4B–G and I). However, leptin-induced pSTAT3 in DIO + saline mice was significantly lower than those in lean mice and in DIO + mCPP mice (Figure 4C, E, G and I). No significant difference was observed in leptin-induced pSTAT3 in lean mice and in DIO + mCPP mice (Figure 4C, G and I).
We then quantified pSTAT3 staining in the PMV. After a single injection of saline, no pSTAT3 staining was observed in the PMV of lean mice; minimal pSTAT3 staining was detected in the PMV of DIO + saline mice and DIO + mCPP mice (Figure 5A, C, E and G). Leptin significantly increased pSTAT3 levels in the PMV of lean, DIO + saline or DIO + mCPP mice respectively (Figure 5A–F and G). In particular, leptin-induced pSTAT3 in the PMV was significantly attenuated in DIO + saline mice compared with those in lean mice and in DIO + mCPP mice (Figure 5B, D, F and G). Interestingly, leptin-induced pSTAT3 in DIO + mCPP mice was significantly higher than that in lean mice (Figure 5B, F and G).
Figure 5.
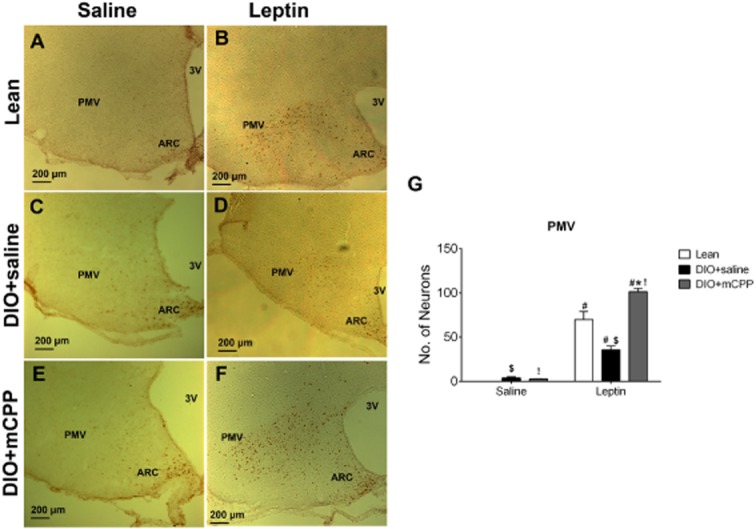
Effects of mCPP pretreatment on leptin-induced pSTAT3 in the PMV of DIO mice. (A–B) Representative pSTAT3 immunohistochemical staining in the PMV of chow-fed lean WT mice receiving a single bolus i.p. injection of saline (A) or 0.5 mg·kg−1 leptin (B). (C–D) Representative pSTAT3 immunohistochemical staining in the PMV of DIO + saline mice (8 weeks on a HFD and in the last 2 weeks given daily i.p. injections of saline), after receiving a single bolus i.p. injection of saline (C) or 0.5 mg·kg−1 leptin (D). (E–F) Representative pSTAT3 immunohistochemical staining in the PMV of DIO + mCPP mice (8 weeks on a HFD and in the last 2 weeks given daily i.p. injections of 1.5 mg·kg−1·day−1 mCPP), after receiving a single bolus i.p. injection of saline (E) or 0.5 mg·kg−1 leptin (F). (G) Number of pSTAT3-positive neurons in the PMV of the various groups. Data are presented as mean ± SEM. n = 3–4 in each group. #P < 0.05 between saline and leptin within the same animal group; $P < 0.05 between Lean and DIO + saline within the same injection group; *P < 0.05 between DIO + saline and DIO + mCPP within the same injection group; !P < 0.05 between Lean and DIO + mCPP within the same injection group; two-way anova followed by the post hoc Newman–Keuls Student's tests.
Deletion of 5-HT2C receptors blunts leptin-induced pSTAT3 in many hypothalamic regions
Given that mCPP co-administration can enhance leptin sensitivity and that mCPP has been shown to reduce food intake at least partly through acting upon 5-HT2C receptors (Xu et al., 2008; Berglund et al., 2013), we sought to test if 5-HT2C receptors are involved in leptin's actions. To this end, we compared the leptin-induced pSTAT3 in the ARC, VMH and PMV of lean WT mice and 2C-null mice, which genetically lack 5-HT2C receptors (Xu et al., 2008; 2010; Figure 6A). In the ARC, leptin significantly increased pSTAT3 levels in 2C-null mice, but this leptin-induced pSTAT3 in 2C-null mice was significantly lower than that in WT mice (Figure 6B–D). Similarly, in the VMH, while leptin significantly increased pSTAT3 levels in 2C-null mice, this leptin-induced pSTAT3 in 2C-null mice was significantly lower than that in WT mice (Figure 6B, C and E). Most strikingly, while leptin strongly increased pSTAT3 levels in the PMV of WT mice (Figure 5A, B and G), this leptin-induced pSTAT3 was diminished in 2C-null mice (Figure 7A–C). Interestingly, we also observed that the baseline pSTAT3 level in the ARC of 2C-null mice (after saline injection) was significantly lower than that of WT mice (Figure 6B and D), whereas the baseline pSTAT3 levels in the VMH and PMV were modestly increased in 2C-null mice compared with WT mice (Figures 6B, E, 7A and C).
Figure 6.
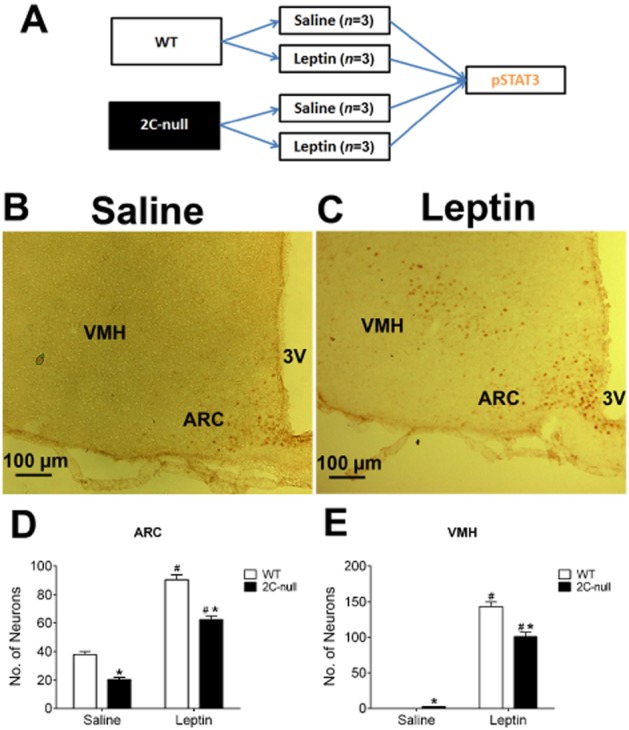
Leptin-induced pSTAT3 in the ARC and VMH of 2C-null mice. (A) Diagram of the schedule used for the experiments in Figures 6 and 7. (B–C) Representative pSTAT3 immunohistochemical staining in the ARC and VMH of chow-fed 2C-null mice receiving a single bolus i.p. injection of saline (B) or 0.5 mg·kg−1 leptin (C). Note that representative images from WT mice have been included as the ‘lean’ group (from Figure 3). (D–E) Quantification of number of pSTAT3-positive neurons in the ARC (D) and VMH (E) of WT or 2C-null littermates. Data are presented as mean ± SEM. n = 3 in each group. #P < 0.05 between saline and leptin within the same genotype; *P < 0.05 between WT and 2C-null within the same injection groups; two-way anova followed by the post hoc Newman–Keuls Student's tests.
Figure 7.
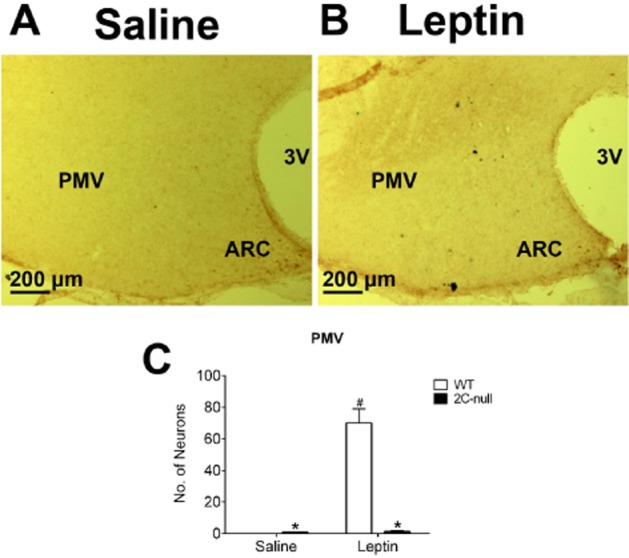
Leptin-induced pSTAT3 in the PMV of 2C-null mice. (A–B) pSTAT3 immunohistochemical staining in the PMV of chow-fed 2C-null mice receiving a single bolus i.p. injection of saline (A) or 0.5 mg·kg−1 leptin (B). Note that representative images from WT mice have been included as the ‘lean’ group (from Figure 4). (C) Quantification of number of pSTAT3-positive neurons in the PMV of WT or 2C-null littermates. Data are presented as mean ± SEM. n = 3 in each group. #P < 0.05 between saline and leptin within the same genotype; *P < 0.05 between WT and 2C-null within the same injection groups; two way anova followed by the post hoc Newman–Keuls Student's tests.
Discussion
Since leptin resistance is the hallmark of common forms of human obesity, developing strategies to enhance leptin sensitivity and/or overcome leptin resistance has been prioritized in the field of obesity research. One important finding of our study is that mCPP, an agonist of 5-HT2C and 5-HT1B receptors, can enhance leptin sensitivity in leptin-resistant DIO mice. Supporting this notion, we showed that co-administration of leptin and mCPP, both at sub-threshold doses, lowered body weight and body fat in DIO mice. Importantly, leptin or mCPP alone, at the same doses, failed to reduce body weight and body fat in DIO mice compared with saline treatment, indicating that the anti-obesity benefits of co-administration stem from the additive effects of these two regimens. The anti-obesity effects of leptin and mCPP co-administration were accompanied by reductions both in food intake and in feeding efficiency. Thus, we suggest that both decreased food intake and increased energy expenditure (as demonstrated by decreased feeding efficiency) may contribute to the overall anti-obesity effects of leptin and mCPP co-administration.
The additive effects of leptin and mCPP on body weight balance could partly result from a sum of two independent actions of leptin and mCPP. For example, both leptin (Duan et al., 2007) and 5-HT2C receptor agonists (Zhou et al., 2007; Lam et al., 2008) have been shown to stimulate the expression of the anorexigenic gene, POMC, in the hypothalamus. Here we found that leptin alone failed to stimulate POMC expression in DIO mice, presumably because of leptin resistance; mCPP alone only tended to increase POMC expression probably due to the sub-threshold dose we used. However, the co-administration of leptin and mCPP significantly elevated POMC expression by about twofold (compared with the saline group). Given the well-characterized anorexigenic effects of POMC gene products (Yaswen et al., 1999), it is conceivable that the combined effects of leptin and mCPP on POMC gene expression at least partly contribute to the decreased body weight in mice co-treated with these two compounds. Notably, mCPP significantly reduced AgRP mRNAs in the hypothalamus, which may also contribute to the reduction in body weight of DIO mice.
Another possibility is that mCPP may function as a ‘leptin sensitizer’ to enhance the effects of leptin in otherwise leptin-resistant DIO mice. Supporting this notion, we observed that mCPP alone significantly increased STAT3 levels in the hypothalamus of DIO mice. There is compelling evidence indicating that the STAT3-associated pathway mediates most of the anti-obesity effects of leptin (Bates et al., 2003; Myers et al., 2010). For example, a point mutation on the leptin receptor (1138 tyrosine→serine) specifically disrupts the leptin-induced STAT3 pathway and mice with this mutation reproduce the severe obesity seen in db/db mice (lacking the leptin receptor; Bates et al., 2003). Similar massive obesity is caused by deletion of STAT3 in the entire brain (Gao et al., 2004) or only in leptin receptor-expressing neurons (Piper et al., 2008). Thus, elevated STAT3 levels upon mCPP treatment may provide more ‘substrates’ for leptin's actions and, therefore, lead to enhanced leptin sensitivity.
Further supporting the leptin-sensitizing effects of mCPP, we showed that leptin-induced pSTAT3 immunoreactivity was enhanced in many brain regions in mCPP-pretreated DIO mice. Neurons in the ARC, including POMC and NPY/AgRP neurons, have been shown to lose their ability to respond to leptin-induced pSTAT3 as early as 2 days–2 weeks after being fed a HFD (Olofsson et al., 2013). Consistently, we found that the leptin-induced pSTAT3 was completely blocked in the ARC of DIO mice (after 8 weeks of a HFD). Interestingly, pretreatment of DIO mice with mCPP enhanced leptin-induced pSTAT3 in the ARC, although this enhancement did not bring the pSTAT3 level to that seen in lean mice.
Compared with the ARC, other brain regions have been shown to be more protected from leptin resistance in DIO mice. For example, in DIO mice fed a HFD for 16 weeks a high dose of leptin (5 mg·kg−1, i.p.) fails to induce pSTAT3 in the ARC, whereas in the VMH, PMV, DMH and NTS leptin is still able to increase pSTAT3 (Munzberg et al., 2004). Consistent with this, we found in our DIO mice (fed a HFD for 8 weeks), that leptin at a very low dose (0.5 mg·kg−1, i.p.) still significantly induced pSTAT3 in the VMH and PMV, although the ARC did not respond to leptin at all. However, we also found that leptin-induced pSTAT3 in the VMH and PMV was significantly weaker in DIO mice than that in lean mice, suggesting the presence of modest leptin resistance in these two regions. We speculate that the low leptin dose we used may have allowed us to unmask a subtle leptin resistance in these non-ARC regions. More importantly, we showed that pretreatment of DIO mice with mCPP enhanced leptin-induced pSTAT3 in the VMH to the level seen in lean mice; the most robust enhancement was observed in the PMV, which even exceeded the normal physiological leptin sensitivity (observed in lean mice). Together, our results indicate that DIO develop leptin resistance most severely in the ARC, and to a lesser extent in the VMH and the PMV; importantly, mCPP can partially restore leptin sensitivity in the ARC, and fully restore leptin sensitivity in the VMH and PMV. Thus, the enhanced leptin sensitivity in these hypothalamic regions may have at least partly contributed to the additive effects of leptin and mCPP on body weight balance in DIO mice.
Since mCPP is known to act partly though 5-HT2C receptors to inhibit food intake (Xu et al., 2008; Berglund et al., 2013), we further examined leptin sensitivity in 2C-null mice, which generally lack 5-HT2C receptors. Interestingly, in 2C-null mice, we found that leptin-induced pSTAT3 was partially suppressed in the ARC and VMH, and completely abolished in the PMV. Importantly, it has been reported that mice lacking 5-HT2C receptors are more sensitivity to HFD-induced body weight gain and hyperleptinaemia (Nonogaki et al., 1998; Xu et al., 2008). Furthermore, chow-fed 5-HT2C receptor knockout mice demonstrate a partial resistance to leptin-induced anorexia and body weight loss, although this resistance can be overcome by higher doses of leptin (Nonogaki et al., 1998). Together, these results indicate that 5-HT2C receptors are required to maintain the full strength of leptin signalling in the hypothalamus as well as leptin-induced anti-obesity effects.
It is worth noting that deletion of 5-HT2C receptors leads to an impairment in leptin-induced pSTAT3 primarily in the PMV, and to a lesser extent in the ARC and VMH. However, mCPP improved leptin-induced pSTAT3 most robustly in the PMV and to a lesser extent in the ARC and VMH. This pattern suggests that PMV neurons may be an important target where both leptin and 5-HT2C receptor signals converge to regulate energy homeostasis. Supporting this possibility, it has been shown that deletion of LepRb in the PVM causes massive obesity in male mice, which almost reproduces the severe obesity seen in db/db mice (Leshan et al., 2012). However, loss of LepRb in ARC neurons, including POMC neurons (Balthasar et al., 2004; Hill et al., 2010) and/or NPY/AgRP neurons (van de Wall et al., 2008), only produced modest obesity in mice. Similarly, deletion of LepRb from VMH neurons leads to modest obesity (Dhillon et al., 2006; Bingham et al., 2008). 5-HT2C receptors are abundantly expressed by various hypothalamic nuclei, including the ARC, VMH and PVM (Hoffman and Mezey, 1989; Pasqualetti et al., 1999). In particular, we have shown that 5-HT2C receptors expressed by POMC neurons in the ARC play an important role in regulating food intake and body weight (Xu et al., 2008; Berglund et al., 2013). However, the physiological importance of 5-HT2C receptors in PMV neurons as well as VMH neurons remains unclear, and this warrants future investigations.
Notably, a highly selective 5-HT2C receptor agonist, namely lorcaserin, was approved by the FDA in 2012 for the treatment of obesity. However, 1 year treatment with lorcaserin, in combination with diet and exercise, only achieved 4.5–5.8% weight loss in obese patients (Smith et al., 2010; Fidler et al., 2011; O'Neil et al., 2012). Thus, our results raise a possibility that lorcaserin could be used as a leptin sensitizer and be combined with leptin therapy to produce more robust body weight loss in obese patients. While the potential additive efficacy of leptin and 5-HT2C receptor agonists (e.g. lorcaserin) warrants further investigation, we cannot rule out the possibility that the leptin-sensitizing effects of mCPP could be attributed to other 5-HT receptors, including 5-HT1B receptors. Indeed, it has been shown that mCPP acts partly upon 5-HT1B receptors expressed by NPY/AgRP neurons to suppress food intake (Heisler et al., 2006).
Another interesting finding is that mCPP alone significantly decreased blood insulin levels in DIO mice, while it had no effect on blood glucose, body weight, body fat and food intake. These findings suggest that mCPP treatment can also enhance the whole-body insulin sensitivity, independently of its effects to suppress food intake and body weight. Indeed, it has been reported that a slightly lower dose of mCPP (1 mg·kg−1·day−1 for 2 weeks) decreases blood insulin, increases glucose tolerance and improves insulin sensitivity in DIO mice, effects that require brain melanocortin 4 receptors (Zhou et al., 2007), the receptors for POMC gene products. Furthermore, we have shown that the ability of mCPP to improve insulin sensitivity is blocked in mice lacking 5-HT2C receptors only in POMC neurons (Berglund et al., 2013), but can be restored in mice expressing 5-HT2C receptors only in POMC neurons (Xu et al., 2010). Together, these findings indicate that drugs acting upon 5-HT2C receptors on POMC neurons can improve the whole-body insulin sensitivity and, therefore, suggest that 5-HT2C receptor agonists, including lorcaserin, could also be used to treat diabetic patients with insulin resistance.
In summary, we showed that co-administration of leptin and mCPP (an agonist of 5-HT2C and 5-HT1B receptors) can provide an additive effect to reduce body weight in leptin-resistant DIO mice. We demonstrated that mCPP enhances leptin sensitivity in the ARC, VMH and PMV and that endogenous 5-HT2C receptor-mediated signals are required to maintain the full strength of leptin sensitivity in the same hypothalamic regions. Thus, our study provides evidence that 5-HT2C receptor agonists (e.g. lorcaserin) could function as leptin sensitizers and be used in combination with leptin to provide additional weight loss in obese patients.
Acknowledgments
This work was supported by grants from the National Institutes of Health (R01DK093587 and R01DK101379 to Y. X.; R01DK092605 to Q. T.; R25GM56929 to A. H. J.) and by an American Heart Association postdoctoral fellowship (P. X.).
Glossary
- AgRP
agouti-related peptide
- ARC
arcuate nucleus
- DIO
diet-induced obesity
- DMH
dorsomedial hypothalamic nucleus
- LepRb
the long form of leptin receptors
- mCPP
meta-chlorophenylpiperazine
- NPY
neuropeptide Y
- NTS
nucleus of solitary tract
- PMV
ventral premammillary nucleus
- POMC
pro-opiomelanocortin
- pSTAT3
phosphorylated STAT-3
- VMH
ventromedial hypothalamic nucleus
Author contributions
C. Y. performed most of the experiments; M. F. and Y. X. designed the study and performed the minipump experiments; C. Y., M. F. and Y. X. wrote the manuscript; J. K. E. generated and validated the 2C-null mice and provided expertise in mouse genetics; Y. Y., K. S., P. X. provided technical assistance in injections, perfusions and immunohistochemistry; C. W., A. O. H. and X. Y. helped to produce the mice used in this study; Q. W. and Q. T. helped in study design and manuscript editing.
Conflict of interest
The authors declare that they have no conflicts of interest.
References
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: G protein-coupled receptors. Br J Pharmacol. 2013a;170:1459–1581. doi: 10.1111/bph.12445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: catalytic receptors. Br J Pharmacol. 2013b;170:1676–1705. doi: 10.1111/bph.12449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: enzymes. Br J Pharmacol. 2013c;170:1797–1867. doi: 10.1111/bph.12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balthasar N, Coppari R, McMinn J, Liu SM, Lee CE, Tang V, et al. Leptin receptor signaling in POMC neurons is required for normal body weight homeostasis. Neuron. 2004;42:983–991. doi: 10.1016/j.neuron.2004.06.004. [DOI] [PubMed] [Google Scholar]
- Bates SH, Stearns WH, Dundon TA, Schubert M, Tso AW, Wang Y, et al. STAT3 signalling is required for leptin regulation of energy balance but not reproduction. Nature. 2003;421:856–859. doi: 10.1038/nature01388. [DOI] [PubMed] [Google Scholar]
- Berglund ED, Liu C, Sohn JW, Liu T, Kim MH, Lee CE, et al. Serotonin 2C receptors in pro-opiomelanocortin neurons regulate energy and glucose homeostasis. J Clin Invest. 2013;123:5061–5070. doi: 10.1172/JCI70338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bingham NC, Anderson KK, Reuter AL, Stallings NR, Parker KL. Selective loss of leptin receptors in the ventromedial hypothalamic nucleus results in increased adiposity and a metabolic syndrome. Endocrinology. 2008;149:2138–2148. doi: 10.1210/en.2007-1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bookout AF, Mangelsdorf DJ. Quantitative real-time PCR protocol for analysis of nuclear receptor signaling pathways. Nucl Recept Signal. 2003;1:e012. doi: 10.1621/nrs.01012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boozer CN, Leibel RL, Love RJ, Cha MC, Aronne LJ. Synergy of sibutramine and low-dose leptin in treatment of diet-induced obesity in rats. Metabolism. 2001;50:889–893. doi: 10.1053/meta.2001.24917. [DOI] [PubMed] [Google Scholar]
- Dhillon H, Zigman JM, Ye C, Lee CE, McGovern RA, Tang V, et al. Leptin directly activates SF1 neurons in the VMH, and this action by leptin is required for normal body-weight homeostasis. Neuron. 2006;49:191–203. doi: 10.1016/j.neuron.2005.12.021. [DOI] [PubMed] [Google Scholar]
- Duan J, Choi YH, Hartzell D, Della-Fera MA, Hamrick M, Baile CA. Effects of subcutaneous leptin injections on hypothalamic gene profiles in lean and ob/ob mice. Obesity (Silver Spring) 2007;15:2624–2633. doi: 10.1038/oby.2007.314. [DOI] [PubMed] [Google Scholar]
- Farooqi IS, Jebb SA, Langmack G, Lawrence E, Cheetham CH, Prentice AM, et al. Effects of recombinant leptin therapy in a child with congenital leptin deficiency. N Engl J Med. 1999;341:879–884. doi: 10.1056/NEJM199909163411204. [DOI] [PubMed] [Google Scholar]
- Fidler MC, Sanchez M, Raether B, Weissman NJ, Smith SR, Shanahan WR, et al. A one-year randomized trial of lorcaserin for weight loss in obese and overweight adults: the BLOSSOM trial. J Clin Endocrinol Metab. 2011;96:3067–3077. doi: 10.1210/jc.2011-1256. [DOI] [PubMed] [Google Scholar]
- Gao Q, Wolfgang MJ, Neschen S, Morino K, Horvath TL, Shulman GI, et al. Disruption of neural signal transducer and activator of transcription 3 causes obesity, diabetes, infertility, and thermal dysregulation. Proc Natl Acad Sci U S A. 2004;101:4661–4666. doi: 10.1073/pnas.0303992101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halaas JL, Gajiwala KS, Maffei M, Cohen SL, Chait BT, Rabinowitz D, et al. Weight-reducing effects of the plasma protein encoded by the obese gene. Science. 1995;269:543–546. doi: 10.1126/science.7624777. [DOI] [PubMed] [Google Scholar]
- Hayes MR, Skibicka KP, Leichner TM, Guarnieri DJ, DiLeone RJ, Bence KK, et al. Endogenous leptin signaling in the caudal nucleus tractus solitarius and area postrema is required for energy balance regulation. Cell Metab. 2010;11:77–83. doi: 10.1016/j.cmet.2009.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heal DJ, Aspley S, Prow MR, Jackson HC, Martin KF, Cheetham SC. Sibutramine: a novel anti-obesity drug. A review of the pharmacological evidence to differentiate it from d-amphetamine and d-fenfluramine. Int J Obes Relat Metab Disord. 1998;22(Suppl. 1):S18–S28. discussion S29. [PubMed] [Google Scholar]
- Heisler LK, Jobst EE, Sutton GM, Zhou L, Borok E, Thornton-Jones Z, et al. Serotonin reciprocally regulates melanocortin neurons to modulate food intake. Neuron. 2006;51:239–249. doi: 10.1016/j.neuron.2006.06.004. [DOI] [PubMed] [Google Scholar]
- Hill JW, Elias CF, Fukuda M, Williams KW, Berglund ED, Holland WL, et al. Direct insulin and leptin action on pro-opiomelanocortin neurons is required for normal glucose homeostasis and fertility. Cell Metab. 2010;11:286–297. doi: 10.1016/j.cmet.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman BJ, Mezey E. Distribution of serotonin 5-HT1C receptor mRNA in adult rat brain. FEBS Lett. 1989;247:453–462. doi: 10.1016/0014-5793(89)81390-0. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. Animal research: Reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam DD, Przydzial MJ, Ridley SH, Yeo GS, Rochford JJ, O'Rahilly S, et al. Serotonin 5-HT2C receptor agonist promotes hypophagia via downstream activation of melanocortin 4 receptors. Endocrinology. 2008;149:1323–1328. doi: 10.1210/en.2007-1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leshan RL, Greenwald-Yarnell M, Patterson CM, Gonzalez IE, Myers MG., Jr Leptin action through hypothalamic nitric oxide synthase-1-expressing neurons controls energy balance. Nat Med. 2012;18:820–823. doi: 10.1038/nm.2724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munzberg H, Huo L, Nillni EA, Hollenberg AN, Bjorbaek C. Role of signal transducer and activator of transcription 3 in regulation of hypothalamic proopiomelanocortin gene expression by leptin. Endocrinology. 2003;144:2121–2131. doi: 10.1210/en.2002-221037. [DOI] [PubMed] [Google Scholar]
- Munzberg H, Flier JS, Bjorbaek C. Region-specific leptin resistance within the hypothalamus of diet-induced obese mice. Endocrinology. 2004;145:4880–4889. doi: 10.1210/en.2004-0726. [DOI] [PubMed] [Google Scholar]
- Myers MG, Jr, Leibel RL, Seeley RJ, Schwartz MW. Obesity and leptin resistance: distinguishing cause from effect. Trends Endocrinol Metab. 2010;21:643–651. doi: 10.1016/j.tem.2010.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nonogaki K, Strack AM, Dallman MF, Tecott LH. Leptin-independent hyperphagia and type 2 diabetes in mice with a mutated serotonin 5-HT2C receptor gene. Nat Med. 1998;4:1152–1156. doi: 10.1038/2647. [DOI] [PubMed] [Google Scholar]
- Olofsson LE, Unger EK, Cheung CC, Xu AW. Modulation of AgRP-neuronal function by SOCS3 as an initiating event in diet-induced hypothalamic leptin resistance. Proc Natl Acad Sci U S A. 2013;110:E697–E706. doi: 10.1073/pnas.1218284110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Neil PM, Smith SR, Weissman NJ, Fidler MC, Sanchez M, Zhang J, et al. Randomized placebo-controlled clinical trial of lorcaserin for weight loss in type 2 diabetes mellitus: the BLOOM-DM study. Obesity (Silver Spring) 2012;20:1426–1436. doi: 10.1038/oby.2012.66. [DOI] [PubMed] [Google Scholar]
- Pasqualetti M, Ori M, Castagna M, Marazziti D, Cassano GB, Nardi I. Distribution and cellular localization of the serotonin type 2C receptor messenger RNA in human brain. Neuroscience. 1999;92:601–611. doi: 10.1016/s0306-4522(99)00011-1. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, et al. NC-IUPHAR. The IUPHAR/BPS Guide to PHARMACOLOGY: an expert-driven knowledgebase of drug targets and their ligands. Nucleic Acids Res. 2014;42(Database Issue):D1098–D1106. doi: 10.1093/nar/gkt1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piper ML, Unger EK, Myers MG, Jr, Xu AW. Specific physiological roles for signal transducer and activator of transcription 3 in leptin receptor-expressing neurons. Mol Endocrinol. 2008;22:751–759. doi: 10.1210/me.2007-0389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth JD, Roland BL, Cole RL, Trevaskis JL, Weyer C, Koda JE, et al. Leptin responsiveness restored by amylin agonism in diet-induced obesity: evidence from nonclinical and clinical studies. Proc Natl Acad Sci U S A. 2008;105:7257–7262. doi: 10.1073/pnas.0706473105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith SR, Weissman NJ, Anderson CM, Sanchez M, Chuang E, Stubbe S, et al. Multicenter, placebo-controlled trial of lorcaserin for weight management. N Engl J Med. 2010;363:245–256. doi: 10.1056/NEJMoa0909809. [DOI] [PubMed] [Google Scholar]
- Tartaglia LA. The leptin receptor. J Biol Chem. 1997;272:6093–6096. doi: 10.1074/jbc.272.10.6093. [DOI] [PubMed] [Google Scholar]
- Tartaglia LA, Dembski M, Weng X, Deng N, Culpepper J, Devos R, et al. Identification and expression cloning of a leptin receptor, OB-R. Cell. 1995;83:1263–1271. doi: 10.1016/0092-8674(95)90151-5. [DOI] [PubMed] [Google Scholar]
- Tecott LH, Sun LM, Akana SF, Strack AM, Lowenstein DH, Dallman MF, et al. Eating disorder and epilepsy in mice lacking 5-HT2c serotonin receptors. Nature. 1995;374:542–546. doi: 10.1038/374542a0. [DOI] [PubMed] [Google Scholar]
- van de Wall E, Leshan R, Xu AW, Balthasar N, Coppari R, Liu SM, et al. Collective and individual functions of leptin receptor modulated neurons controlling metabolism and ingestion. Endocrinology. 2008;149:1773–1785. doi: 10.1210/en.2007-1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Jones JE, Kohno D, Williams KW, Lee CE, Choi MJ, et al. 5-HT2CRs expressed by pro-opiomelanocortin neurons regulate energy homeostasis. Neuron. 2008;60:582–589. doi: 10.1016/j.neuron.2008.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Berglund ED, Sohn JW, Holland WL, Chuang JC, Fukuda M, et al. 5-HT2CRs expressed by pro-opiomelanocortin neurons regulate insulin sensitivity in liver. Nat Neurosci. 2010;13:1457–1459. doi: 10.1038/nn.2664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaswen L, Diehl N, Brennan MB, Hochgeschwender U. Obesity in the mouse model of pro-opiomelanocortin deficiency responds to peripheral melanocortin. Nat Med. 1999;5:1066–1070. doi: 10.1038/12506. [DOI] [PubMed] [Google Scholar]
- Zhou L, Sutton GM, Rochford JJ, Semple RK, Lam DD, Oksanen LJ, et al. Serotonin 2C receptor agonists improve type 2 diabetes via melanocortin-4 receptor signaling pathways. Cell Metab. 2007;6:398–405. doi: 10.1016/j.cmet.2007.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]