

Abstract
Background and Purpose
β2-adrenoceptor agonists are widely used in the management of obstructive airway diseases. Besides their bronchodilatory effect, several studies suggest inhibitory effects on various aspects of inflammation. The aim of our study was to determine the efficacy of the long-acting β2-adrenoceptor agonist olodaterol to inhibit pulmonary inflammation and to elucidate mechanism(s) underlying its anti-inflammatory actions.
Experimental Approach
Olodaterol was tested in murine and guinea pig models of cigarette smoke- and LPS-induced lung inflammation. Furthermore, effects of olodaterol on the LPS-induced pro-inflammatory mediator release from human parenchymal explants, CD11b adhesion molecule expression on human granulocytes TNF-α release from human whole blood and on the IL-8-induced migration of human peripheral blood neutrophils were investigated.
Key Results
Olodaterol dose-dependently attenuated cell influx and pro-inflammatory mediator release in murine and guinea pig models of pulmonary inflammation. These anti-inflammatory effects were observed at doses relevant to their bronchodilatory efficacy. Mechanistically, olodaterol attenuated pro-inflammatory mediator release from human parenchymal explants and whole blood and reduced expression of CD11b adhesion molecules on granulocytes, but without direct effects on IL-8-induced neutrophil transwell migration.
Conclusions and Implications
This is the first evidence for the anti-inflammatory efficacy of a β2-adrenoceptor agonist in models of lung inflammation induced by cigarette smoke. The long-acting β2-adrenoceptor agonist olodaterol attenuated pulmonary inflammation through mechanisms that are separate from direct inhibition of bronchoconstriction. Furthermore, the in vivo data suggest that the anti-inflammatory properties of olodaterol are maintained after repeated dosing for 4 days.
Tables of Links
| TARGETS |
|---|
| GPCRsa |
| β2-adrenoceptors |
| β1-adrenoceptors |
| CXCR2 |
| Catalytic receptorsb |
| CD11b |
| Enzymesc |
| MMP-9 |
| LIGANDS | |
|---|---|
| ACh | IL-8 |
| CCL2 | KC (mouse orthologue of CXCL1) |
| CCL4 | LPS |
| CGP-20712A | M-CSF-1 |
| CXCL9 | Olodaterol |
| GM-CSF | TNFα |
| ICI-118,551 |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (a,b,cAlexander et al., 2013a,b,c,,).
Introduction
β2-adrenoceptor agonists are widely used in the treatment of obstructive lung diseases. According to the GOLD guidelines, inhaled long-acting β2-adrenoceptor agonist (LABAs) are recommended for treatment of patients with chronic obstructive pulmonary disease (COPD) who are symptomatic despite regular use of short-acting β2-adrenoceptor agonists (Vestbo et al., 2013). Recently, olodaterol, a new LABA with true once-daily dosing, has been developed (Bouyssou et al., 2010a,b,). Olodaterol is a potent, enantiomerically pure LABA that induces bronchodilation up to 24 h after dosing in patients with COPD (van Noord et al., 2011).
β2-adrenoceptor agonist improve airway function mainly by a direct relaxant effect on airway smooth muscle (ASM). However, the expression of β2-adrenoceptors is not restricted to ASM and these receptors are widely distributed throughout the lung being expressed on epithelial, endothelial and vascular smooth muscle cells and pneumocytes (Barnes et al., 1982; Carstairs et al., 1984; 1985,). Inflammatory disorders, like COPD, are characterized by the over-expression of adhesion molecules leading to the recruitment of leukocytes to the site of inflammation. Stimulation of β2-adrenoceptors attenuated neutrophil adhesion to the vascular endothelium (Derian et al., 1995; Bolton et al., 1997). Furthermore, the IFN-γ-induced increase in ICAM-1 expression on bronchial epithelial cells was inhibited by fenoterol (Oddera et al., 1998) and olodaterol was shown to inhibit neutrophil adhesion to bronchial epithelial cells (Profita et al., 2012).
Moreover, β2-adrenoceptors are also expressed on inflammatory cells, such as neutrophils, monocytes and lymphocytes (Barnes, 1999). Several studies demonstrated that stimulation of β2-adrenoceptors on these immunocompetent cells results in a direct inhibitory effect on the release of pro-inflammatory cytokines. Clenbuterol, formoterol and salmeterol suppressed the LPS-induced TNF-α and IL-6 expression in monocyte-derived macrophages (Izeboud et al., 1999; Donnelly et al., 2010). Furthermore, β2-adrenoceptor agonists reduced the LPS-stimulated TNF-α and IL-1β release from human peripheral blood mononuclear cells (Yoshimura et al., 1997a,b,). Salmeterol reduced IL-8 release from human macrophages, induced by exposure to extracts of cigarette smoke (CS) (Sarir et al., 2007) and fenoterol reduced the CS extract-induced IL-8 release from primary human ASM cells (Oldenburger et al., 2012).
Preclinical in vivo experiments and clinical data further add evidence to the anti-inflammatory capacity of β2-adrenoceptor agonists. In murine models of LPS-induced pulmonary inflammation, formoterol and salmeterol reduced the recruitment of neutrophils to the lung and inhibited the release of pro-inflammatory mediators (Maris et al., 2004; Bosmann et al., 2012). In a clinical setting, salmeterol reduced the neutrophil influx, neutrophil degranulation and TNF-α release after LPS inhalation in healthy volunteers at a clinically relevant dose (Maris et al., 2005). Furthermore, a meta-analysis revealed that LABAs reduced the frequency of COPD exacerbations (Wang et al., 2012).
Taken together, all this evidence suggests that β2-adrenoceptor agonists exert inhibitory effects on various aspects of the inflammatory processes that characterize obstructive lung diseases. Therefore, the efficacy of olodaterol to inhibit cell influx in animal models of LPS- and CS-induced pulmonary inflammation was tested. To our knowledge, this is the first published study testing the efficacy of a β2-adrenoceptor agonist in models of CS-induced lung inflammation. Furthermore, the anti-inflammatory action of olodaterol was also assessed on human peripheral blood cells and pulmonary primary cellular preparations. Our results showed that stimulation of β2-adrenoceptors with olodaterol prevented airway inflammation induced by different stimuli relevant to human lung disease and that this anti-inflammatory effect was mediated through mechanisms that were distinct from direct inhibition of bronchoconstriction.
Methods
Animal welfare and ethical statement
For mice, animal care and experimental procedures complied with German national guidelines and legal regulations and were approved by the ethical committee Regierungspräsidium Tübingen (Germany) (Permit Nos. 08-005, 12-014 and 12-020). C57BL/6 mice were obtained from Charles River Laboratories (Germany), housed in isolated ventilated cages under a 12 h light-dark cycle and received food and water ad libitum. Experiments using guinea pigs complied with the conditions of the UK Home Office Project Licence No. 30-2072 and were performed at Argenta Discovery 2009 Ltd (Harlow, UK). Dunkin–Hartley guinea pigs were obtained from B&K Universal Limited (UK) and were housed in polycarbonate solid-bottomed cages. Food and water were provided ad libitum. All procedures used were as humane as possible and the description of studies involving animals complied with the recommendations of ARRIVE (Kilkenny et al., 2010; McGrath et al., 2010). A total of 186 animals (136 mice; 50 guinea pigs) were used in the experiments described here.
Mouse model of CS-induced lung inflammation
Female C57BL/6 mice were exposed to the mainstream smoke of six cigarettes (Roth-Händle without filters; Badische Tabakmanufaktur Roth-Händle®, Lahr, Germany) on days 1 and 2, and to five cigarettes on days 3 and 4. One hour prior to each CS exposure, animals were exposed for 5 min to an aqueous solution of olodaterol (Boehringer Ingelheim Pharma GmbH & Co. KG, Biberach, Germany) aerosolized with a jet nebulizer (Pari Master; Pari GmbH, Starnberg, Germany). Eighteen hours after the last CS exposure, animals were killed and lungs were lavaged. CS exposure, drug inhalation and lavage were performed as detailed elsewhere (Wollin and Pieper, 2012).
Mouse model of LPS-induced lung inflammation
Female C57BL/6 mice were exposed to 1 mg·mL−1 of LPS from Escherichia coli serotype O111:B4 (Sigma-Aldrich, St. Louis, MO, USA) in PBS by inhalation for 30 min in a cylindrical 32 L Perspex box using a jet nebulizer. One hour prior to LPS inhalation, animals were treated with olodaterol as described above. Four hours after the LPS exposure, animals were killed and lungs were lavaged as detailed elsewhere (Wollin and Pieper, 2012).
Guinea pig model of CS-induced lung inflammation
Female Dunkin–Hartley guinea pigs were exposed individually to the mainstream smoke of nine cigarettes (Marlboro 100) daily for 4 consecutive days. Exposure to the smoke lasted 45 min (flow rate of 100 mL·min−1). Animals received olodaterol intranasally under light anaesthesia, 1 h prior to each CS exposure. Twenty-four hours after the final CS exposure, animals were killed by barbiturate anaesthetic overdose and lungs were lavaged with 4 mL PBS. Total and differential cell counts in the lavage fluid were determined using light microscopy.
Lung function measurement and acetylcholine-induced bronchoconstriction
Lung function in female C57BL/6 mice was measured as detailed previously (Wollin and Pieper, 2012). Briefly, olodaterol was administered as an aerosol of aqueous solutions at different doses for 5 min. One hour later, mice were anaesthetized by i.p. injection of 60 mg·kg−1 pentobarbital (Narcoren; Merial GmbH, Hallbergmoos, Germany) and 2.5 mg·kg−1 xylazine hydrochloride (Rompun; Bayer Vital GmbH, Leverkusen, Germany) in a volume of 10 mL·kg−1. A tracheal cannula was inserted and connected to an integrated ventilation and lung function measurement device (Scireq flexiVent; EmkaTechnologies, Paris, France). To prevent spontaneous breathing, the animals received 0.8 mg·kg−1 pancuronium bromide (Pancuronium Inresa, Inresa Arzneimittel GmbH, Freiburg, Germany) by i.v. administration at a volume of 5 mL·kg−1. Ventilation was conducted with 150 breaths min−1, a tidal volume of 6.5 mL·kg−1 and 3 cmH2O end-expiratory pressure. After ventilating to total lung capacity (30 cmH2O), baseline airway resistance and compliance were determined. A bolus of acetylcholine (50 μL) was administered intravenously as an aqueous solution at increasing concentrations (50, 100, 200 and 400 μg·mL−1) resulting in doses of 125–1000 μg·kg−1 body weight. Maximal bronchoconstriction within 3 min was determined after each bolus and lungs were ventilated to total lung capacity before applying the next bolus. The AUC of resistance increase between 50 and 400 μg·mL−1 acetylcholine of the placebo group was defined as 100%. Concentration–response curves were calculated using log-linear regression analysis with variable slope.
Mediator measurement in bronchoalveolar lavage (BAL) supernatant
Pro-inflammatory mediators in BAL supernatant were measured at Myriad RBM (Austin, TX, USA) using a multiplex immunoassay (RodentMAP v3.0).
LPS-induced mediator release from human parenchymal explants
The use of lung tissues resected from patients was approved by the regional investigational review board (CPP Ile de France VIII, France) and written informed consent was obtained from each patient. Human lung parenchyma was obtained from 14 patients (age: 66 ± 3 years; males/females, 7/7; non-smokers: 2; smokers and ex-smokers: 8 and 4; pack years: 44 ± 6) undergoing surgical resection for lung carcinoma and who had not received recent chemo/steroid therapy. The procedure for preparation of lung explants has been described elsewhere (Hackett et al., 2008; Buenestado et al., 2013). Explants (5 fragments of 3–5 mm3, total weight ∼50–100 mg) were maintained overnight at 4°C in RPMI supplemented with 2 mM L-glutamine, 100 μg·mL−1 streptomycin and 100 U·mL−1 penicillin. The fragments were distributed into six-well plates and incubated with olodaterol or vehicle (DMSO) for 1 h before LPS stimulation (1 μg·mL−1). The maximal DMSO concentration applied to explants in culture did not exceed 0.01% and did not alter the production of cytokines or TNF-α. After 24 h, supernatants were collected and stored at −80°C. Production of cytokines was measured in medium supernatants with elisa techniques (R&D Systems, Minneapolis, MN, USA).
CD11b expression and TNF-α release assay using human whole blood
Human whole blood was obtained from healthy adult donors who did not take any medication for at least 10 days before venepuncture. Blood samples were pre-incubated with olodaterol for 30 min at 37°C in the CO2 incubator. Then samples were stimulated with 400 pg·mL−1 LPS at 37°C in the presence of the drug. CD11b expression on granulocytes was determined by FACS using a monoclonal mouse anti-human PE-labelled CD11b/Mac-1 antibody (BD Biosciences, San Jose, CA, USA) after 60 min of LPS stimulation. TNF-α in the supernatant was determined after 4 h of LPS stimulation using a commercially available TNF-α elisa kit (BD Biosciences, San Diego, CA, USA). To test the efficacy of the β-receptor antagonists, human whole blood was pre-incubated with either 30 nM ICI-118,551 or 100 nM CGP-20712A for 30 min at 37°C before the addition of olodaterol.
Transwell migration experiments
Primary human neutrophils were isolated from peripheral blood, obtained as above (healthy adult donors who did not take any medication for at least 10 days before venipuncture). Different concentrations of IL-8 (Biotrend Chemicals, Destin, FL, USA) were tested to stimulate transwell migration ChemoTx® System; Neuro Probe, Inc., Gaithersburg, MD, USA). Cells were pre-incubated with olodaterol for 30 min at room temperature. Migration was stimulated by addition of 3 nM IL-8 to the lower chamber of the transwell. The number of migrated cells was determined using the CellTiter-Glo Luminescent Cell Viability Assay (Promega Corporation, Madison, WI, USA) after 1 h incubation at 37°C.
Data analysis
All data are presented as mean ± SEM. Statistical differences between groups were analysed by one-way anova, followed by Dunnett's multiple comparison test. The mediator release from human parenchymal explants was analysed by Friedman test (non-parametric repeated-measure test), followed by Dunn's multiple comparisons test. Statistical significance was accepted at P < 0.05. The tests were performed using GraphPad Prism version 6.01 for Windows (GraphPad Software, La Jolla, California, USA; www.graphpad.com).
Materials
ICI-118,551 and CGP-20712A were supplied by Sigma-Aldrich (St. Louis, MO, USA) and SCH-527123 was from SynphaBase AG (Pratteln, Switzerland).
Results
Olodaterol attenuates pulmonary neutrophilia and pro-inflammatory mediator release in mouse models of CS- and LPS-induced lung inflammation
To determine the anti-inflammatory potential of olodaterol in a mouse model of CS-induced lung inflammation, mice were exposed to CS on 4 consecutive days and treated with olodaterol aerosol before every smoke exposure. CS exposure led to a prominent neutrophilic pulmonary inflammation (Figure 1). Preventive olodaterol treatment dose dependently reduced cell counts with a maximal effect at the highest dose of 1.0 mg·mL−1 attenuating the total cell count by 60% and the neutrophilic cell count by 69% (Figure 1A and B).
Figure 1.
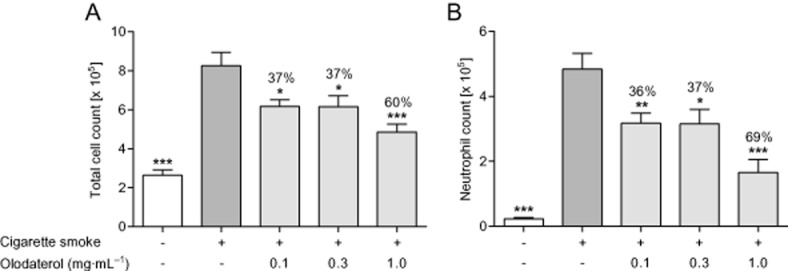
Effect of olodaterol on CS-induced lung inflammation in C57BL/6 mice. (A) Total and (B) neutrophilic cell counts in the BAL fluid of mice after CS exposure on 4 consecutive days. Mice were exposed to olodaterol aerosol for 5 min, 1 h before every CS exposure. Data shown are means ± SEM (n = 6–8 animals per group). *P < 0.05, **P < 0.01, ***P < 0.001; significantly different from CS-exposed, vehicle-treated animals.
In the LPS model, mice were first exposed to different doses of aerosolised olodaterol and 1 h later to aerosolised LPS for 30 min. As seen in the CS model, 4 h after LPS inhalation, a prominent neutrophilic inflammatory response was measured in the BAL (Figure 2). Olodaterol dose-dependently reduced the neutrophil influx into the lung with a maximal efficacy of 82% at the highest dose of 1 mg·mL−1 (Figure 2B). To test the hypothesis that olodaterol might reduce the release of pro-inflammatory mediators, thereby attenuating pulmonary cell influx, mediators in the BAL supernatant were determined. The highest dose of 1 mg·mL−1 olodaterol significantly reduced TNF-α, myeloperoxidase (MPO), MMP-9, M-CSF-1, GM-CSF and the chemokine CCL4 by 83, 69, 76, 59, 86 and 86%, respectively, but had no significant effects on the release of IL-1 β or of KC, the mouse orthologue of the chemokine CXCL1 (Table 1).
Figure 2.
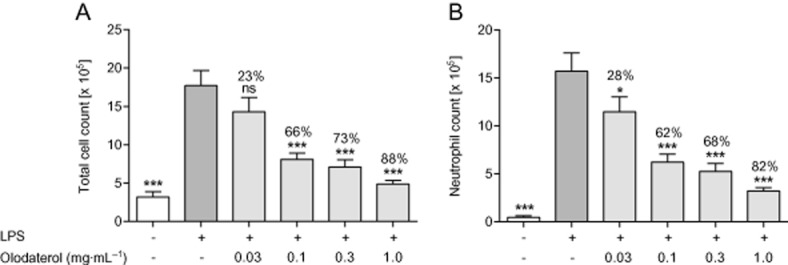
Effect of olodaterol on LPS-induced pulmonary inflammation in C57BL/6 mice. (A) Total and (B) neutrophilic cell counts in the BAL fluid of mice 4 h after inhalation of 1 mg·mL−1 LPS for 30 min. Mice were exposed to olodaterol aerosol for 5 min 1 h prior to LPS exposure. Data shown are means ± SEM (n = 8 animals per group). *P < 0.05, ***P < 0.001: significantly different from LPS-exposed, vehicle-treated animals.
Table 1.
Pro-inflammatory mediators measured in the BAL fluid of C57BL/6 mice 4 h after inhalation of LPS
| PBS inhalation | LPS inhalation | LPS inhalation + 1 mg·mL−1 olodaterol | Inhibition by olodaterol (%) | |
|---|---|---|---|---|
| TNF-α (ng·mL−1) | 0.0 ± 0.0*** | 0.63 ± 0.03 | 0.11 ± 0.01*** | 82.9 ± 1.3 |
| MPO (ng·mL−1) | 3.51 ± 1.37*** | 53.75 ± 3.71 | 18.88 ± 1.39*** | 69.4 ± 2.8 |
| MMP-9 (ng·mL−1) | 1.09 ± 0.44*** | 87.00 ± 6.99 | 21.75 ± 2.29*** | 76.0 ± 2.7 |
| M-CSF-1 (ng·mL−1) | 0.08 ± 0.005*** | 0.25 ± 0.01 | 0.15 ± 0.01*** | 58.5 ± 3.9 |
| GM-CSF (ng·mL−1) | 0.004 ± 0.0*** | 0.23 ± 0.02 | 0.04 ± 0.004*** | 85.8 ± 1.8 |
| CCL4 (ng·mL−1) | 0.05 ± 0.01*** | 14.51 ± 1.54 | 2.03 ± 0.17*** | 86.3 ± 1.2 |
| IL-1β (ng·mL–1) | 0.60 ± 0.0** | 1.30 ± 0.06 | 1.35 ± 0.12 | – |
| KC (ng·mL–1) | 0.0 ± 0.0*** | 0.46 ± 0.04 | 0.39 ± 0.03 | 15.4 ± 5.9 |
Data are presented as mean ± SEM (n = 8 animals per group).
P < 0.01
P < 0.001 represent significant differences compared with LPS-exposed, vehicle-treated animals. KC: murine form of CXCL1.
Olodaterol diminishes pulmonary cell influx in a guinea pig model of CS-induced lung inflammation
The anti-inflammatory efficacy of olodaterol was also tested in a guinea pig model of CS-induced pulmonary inflammation to exclude the possibility that the anti-inflammatory mechanism was species-specific. Guinea pigs were exposed to CS on 4 consecutive days and olodaterol was administered intranasally 1 h prior to each CS exposure. CS induced a significant increase in neutrophilic and monocytic cell counts in the BAL of CS-exposed animals (Figure 3). CS increased the total BAL cell count, to about three-fold that from air-exposed animals (Figure 3A). CS induced the most prominent increase in BAL neutrophils (Figure 3B). Treatment with 10 μg·kg−1 olodaterol markedly reduced the total (Figure 3A) and neutrophil (Figure 3B) cell count. In contrast to the mouse model, monocytic cells were also significantly increased and all three doses of olodaterol reduced the monocytic cell accumulation with a maximal effect at 1 μg·kg−1 olodaterol (Figure 3C).
Figure 3.
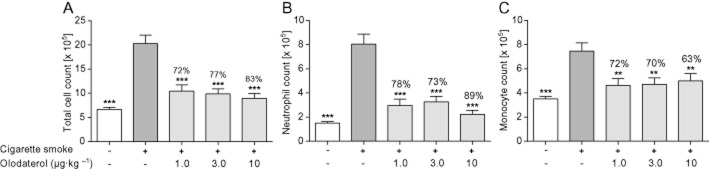
Effect of olodaterol on CS-induced lung inflammation in Dunkin–Hartley guinea pigs. (A) Total, (B) neutrophilic and (C) monocytic cell counts in the BAL fluid of guinea pigs after CS exposure on 4 consecutive days. Olodaterol was administered intranasally 1 h before every CS exposure. Data shown are means ± SEM (n = 9–10 animals per group). **P < 0.01, ***P < 0.001; significantly different from CS-exposed, vehicle-treated animals.
Olodaterol inhibits acetylcholine-induced bronchoconstriction in mice
The pharmacologically active dose of olodaterol to induce bronchodilation was determined in a model of acetylcholine-induced bronchoconstriction in mice. Olodaterol dose-dependently induced bronchodilation (Figure 4). Analysis of the data suggested biphasic behaviour with a first plateau at 25% bronchoprotection at a dose of 0.03 mg·mL−1 and a half-maximal effective concentration (EC50) of 0.0044 mg·mL−1 and a maximal bronchoprotection of 71% at a dose of 10 mg·mL−1 and an EC50 of 0.21 mg·mL−1.
Figure 4.
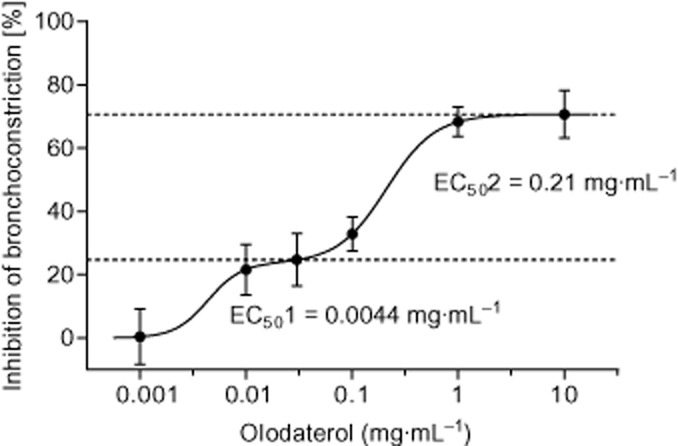
Effect of olodaterol on acetylcholine-induced bronchoconstriction in C57BL/6 mice. One hour after exposure of the animals to an olodaterol aerosol (0.001–10 mg·mL−1) for 5 min, a transient bronchoconstriction was elicited by i.v. injection of acetylcholine (125–1000 μg·kg−1 body weight). The AUC of resistance increase between 125 and 1000 μg·kg−1 acetylcholine of the placebo group was defined as 100%. Data shown are means ± SEM (n = 4–7 animals per group).
Olodaterol attenuates pro-inflammatory mediator release from unstimulated and LPS-stimulated parenchymal explants
To translate our findings to human tissue, the efficacy of olodaterol to inhibit the LPS-induced release of pro-inflammatory mediators from human parenchymal explants was tested. At nanomolar concentrations, olodaterol significantly attenuated the LPS-induced release of TNF-α, CCL4, CCL2 and CXCL9 with a maximal efficacy of 45, 32, 43 and 54% (Figure 5) and significantly reduced the mediator release from non-stimulated lung explants with a maximal efficacy of 46% for TNF-α, 42% for CCL4, 37% for CCL2 and 35% for CXCL9 (data not shown).
Figure 5.

Effect of olodaterol on LPS-induced cytokine release from human lung parenchymal explants. (A) Levels of TNF-α, (B) CCL4, (C) CCL2 and (D) CXCL9 measured in the supernatant of human lung parenchymal explants stimulated with 1 μg·mL−1 LPS for 24 h. Tissue was incubated with olodaterol for 1 h before and during LPS stimulation. Data shown are means ± SEM (n = 9–14 donors). *P < 0.05, **P < 0.01, ***P < 0.001; significantly different from LPS-stimulated, vehicle-treated tissue.
Olodaterol reduces CD11b adhesion molecule expression and TNF-α release but has no effect on the transwell migration of primary human neutrophils
Stimulation of human whole blood with LPS concentration dependently induced the expression of CD11b on granulocytes and the release of TNF-α into the medium (data not shown). Olodaterol significantly attenuated the LPS-induced CD11b expression with a maximal inhibition of 48% at 10−8 M and a half-maximal inhibitory concentration (IC50) value of 1.31 × 10−10 M (Figure 6A) and almost completely blocked the TNF-α release with an IC50 value of 1.99 × 10−11 M (Figure 6B). In both assays, addition of the selective β2-adrenoceptor antagonist ICI-118,551 shifted the olodaterol curve to the right (133- and 168-fold respectively), whereas the selective β1-adrenoceptor antagonist CGP-20712A had no effect on neither the potency nor the maximal efficacy of olodaterol in both assays. In a transwell migration assay, IL-8 induced the migration of primary human neutrophils isolated from peripheral blood with a bell-shaped concentration dependence and a maximum at 3 nM IL-8 (Figure 6C). Olodaterol up to the highest concentration of 10−7 M showed no effect on the migratory capacity induced by 3 nM IL-8 (Figure 6D). To make sure that the lack of efficacy of olodaterol was not due to the lack of expression of functional β2-adrenoceptors on human neutrophils, a cAMP assay was performed, demonstrating a concentration-dependent increase in cAMP with olodaterol treatment (data not shown). Furthermore, to validate the transwell migration assay, the efficacy of the CXCR2 antagonist SCH-527123 was tested. SCH-527123 exhibited a potent and almost full inhibition of the IL-8-induced neutrophil migration (Figure 6D).
Figure 6.
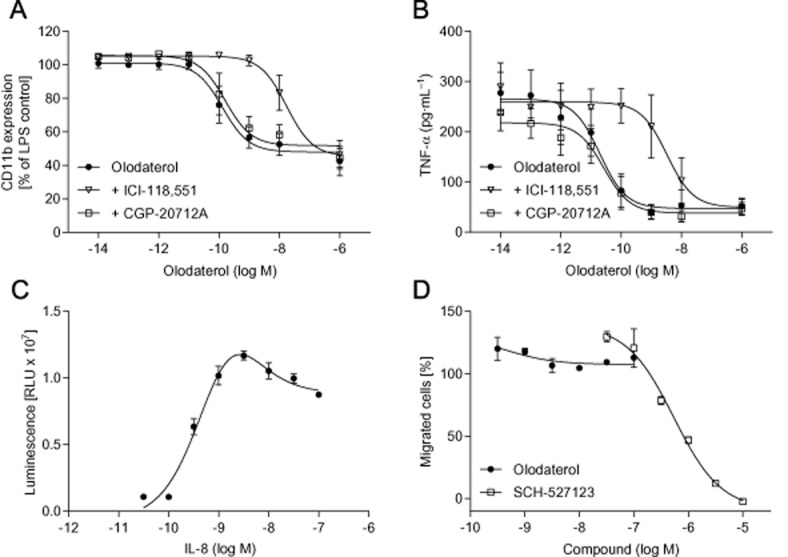
Effect of olodaterol on CD11b expression, TNF-α release and neutrophil migration. (A) CD11b expression on granulocytes was measured after incubation of human peripheral whole blood with olodaterol for 30 min and subsequently stimulated with 400 pg·mL−1 LPS for 1 h in the presence of the drug. CD11b expression was determined by FACS using a monoclonal mouse anti-human PE-labelled CD11b/Mac-1 antibody. (B) TNF-α release was measured after incubation of blood with olodaterol for 30 min and subsequently stimulated with 400 pg·mL−1 LPS for 4 h in the presence of the drug. To test the antagonist effects, whole blood was pre-incubated with either ICI-118,551 (open triangles) or CGP-20712A (open squares) for 30 min before the addition of olodaterol (filled circles). (C) Transwell migration of isolated human peripheral blood neutrophils stimulated by different concentrations of IL-8. The number of migrated cells was determined using the CellTiter-Glo luminescent cell viability assay. Generation of the luminescent signal is proportional to the amount of ATP present, which signals the presence of metabolically active cells in the lower compartment. (D) Cells were pre-incubated with olodaterol (filled circles) or the CXCR2 antagonist SCH-527123 (open squares) for 30 min. Migration was stimulated by addition of 3 nM IL-8 to the lower chamber of the transwell. Data shown are means ± SEM (n = 3–4 donors).
Discussion and conclusions
β2-adrenoceptor agonists are widely used in the management of obstructive lung diseases, such as COPD. Besides their bronchodilatory effect, several studies suggest an inhibitory effect on various aspects of inflammation that characterize pulmonary diseases. The aim of our study was to test the novel long-acting β2-adrenoceptor agonist olodaterol in models of pulmonary inflammation and to shed light on its potential anti-inflammatory mechanism of action to better understand its mode of action, beyond bronchodilation, in clinical use.
In a first set of experiments, olodaterol was tested in acute models of CS-induced pulmonary inflammation in mice and guinea pigs. To our knowledge, this is the first published study testing a β2-adrenoceptor agonist in in vivo models of CS-induced lung inflammation. In both species, olodaterol dose-dependently and highly significantly inhibited the influx of neutrophils into the lung. Furthermore, olodaterol attenuated the influx of inflammatory cells into the lung in a murine model of LPS-induced pulmonary inflammation. To determine whether the doses used in the inflammation models were relevant with regard to their bronchodilatory efficacy, olodaterol was tested in models of acetylcholine-induced bronchoconstriction in both species. In guinea pigs, olodaterol intratracheally applied, using the Respimat soft mist inhaler, showed full bronchoprotection of 100% at a dose of 3 μg·kg−1 (Bouyssou et al., 2010a). With regard to its anti-inflammatory efficacy, olodaterol highly significantly reduced cell influx into the lung already at the lowest tested dose of 1 μg·kg−1 when applied intranasally, which for neutrophils was numerically, however not significantly, further attenuated up to the highest dose of 10 μg·kg−1. Because the Respimat soft mist inhaler is a device which ensures optimal delivery of the drug to the lung and intranasal dosing probably leads to lower deposition rates in the lung, these data reflect the fact that, in guinea pigs, the anti-inflammatory effect of olodaterol occurs at (or even below) a dose which is relevant in terms of its bronchodilatory capacity.
In mice, β-adrenoceptor-mediated relaxation is attributed predominantly to activation of β1-adrenoceptors and to be only partially mediated via the β2-subtype (Henry and Goldie, 1990; Raemdonck et al., 2012). This might explain the biphasic dose–response curve that was observed in our model of acetylcholine-induced bronchoconstriction in mice. Bronchodilation induced at low doses of olodaterol is probably mediated via β2-adrenoceptors, whereas at higher doses bronchodilation might be induced by β1-adrenoceptors. This would fit the selectivity profile of olodaterol determined in in vitro experiments which showed a 241 times higher affinity for the human β2-adrenoceptor compared with the β1-adrenoceptor (Bouyssou et al., 2010a). Our data suggest that in the mouse, the β2-adrenoceptor-mediated bronchodilation accounts for ∼25% of bronchodilation and that the maximal bronchodilation of ∼70% achieved with olodaterol might be the accumulated response of the activation of both β1- and β2-adrenoceptors. However, to further dissect the precise role of β1- and β2-adrenoceptor activation on smooth muscle relaxation and inflammation in mice, additional experiments would be necessary.
To further investigate the anti-inflammatory efficacy of olodaterol, pro-inflammatory mediators were measured in the BAL supernatant obtained in the LPS experiment. At 1 mg·mL−1, a dose which inhibits the acetylcholine-induced bronchoconstriction by 68% in mice, olodaterol significantly attenuated the levels of TNF-α, MPO, MMP-9, M-CSF-1, GM-CSF and CCL4, adding evidence for anti-inflammatory mechanisms, other than bronchodilation, for olodaterol.
Our in vivo data are in accordance with many in vivo studies demonstrating that short-term treatment with different β2-adrenoceptor agonists reduced the cell recruitment and inhibited the release of pro-inflammatory mediators in LPS-induced inflammation models in mice and rats (Izeboud et al., 1999; Shinkai et al., 2003; Maris et al., 2004; Miyamoto et al., 2004; Bosmann et al., 2012). Furthermore, in two clinical settings in healthy volunteers, a single dose of salmeterol or salbutamol reduced the neutrophil influx and TNF-α release after LPS inhalation at a clinically relevant dose (Masclans et al., 1996) and prevented platelet-activating factor-induced pulmonary sequestration of radio-labelled neutrophils (Maris et al., 2005). However, although diverse preclinical and clinical studies have clearly demonstrated the anti-inflammatory efficacy of β2-adrenoceptor activation, rapid desensitization of β2-adrenoceptors on airway inflammatory cells is often considered a potential pitfall in translating preclinical findings obtained in short-term models into clinical situations (Barnes, 1999). In this regard, our data clearly demonstrate that the anti-inflammatory efficacy of olodaterol was shown not only after single dosing (e.g. in the LPS experiment), but was maintained both in mice and guinea pigs, after repetitive dosing for 4 consecutive days in the CS experiments. Thus, our data confirm previous observations and also extend our knowledge by showing that the anti-inflammatory properties of the LABA olodaterol were maintained after repeated dosing. Furthermore, in patients with mild asthma, a 4 week treatment with formoterol significantly reduced sputum IL-8 levels and neutrophil numbers (Maneechotesuwan et al., 2005). Additionally, treatment with salmeterol over a period of 6 weeks decreased the number of neutrophils in bronchial biopsies and MPO levels in serum, adding more evidence for the persistence of the anti-inflammatory efficacy of β2-adrenoceptor agonists, after repeated dosing (Jeffery et al., 2002; Maneechotesuwan et al., 2005).
To test whether our findings from the LPS and CS models did translate to human tisues, the efficacy of olodaterol to inhibit pro-inflammatory mechanisms in human parenchymal explants was determined. Comparable to the data obtained in the BAL fluid from the murine LPS model, olodaterol reduced the LPS-induced release of TNF-α, CCL4, CCL2 and CXCL9 from human parenchymal explants. These data are in agreement with a recent report by Buenestado et al. (2013), demonstrating that increasing cAMP levels in human parenchymal explants using the PDE4 inhibitor roflumilast attenuated LPS-induced TNF-α and chemokine production. Furthermore, we demonstrate that olodaterol almost completely blocks the LPS-induced TNF-α release from human peripheral whole blood.
Because inflammatory disorders, such as COPD, are characterized by the over-expression of adhesion molecules which lead to the recruitment of leukocytes to the site of inflammation, human peripheral whole blood was stimulated with LPS and the expression of the adhesion molecule CD11b on granulocytes was measured. As expected, LPS induced a concentration-dependent increase in CD11b expression, and olodaterol partly inhibited this increased expression by up to 48% at a concentration of 10−8 M. These data are in agreement with a study by Profita et al. (2012), demonstrating that olodaterol significantly reduced CD11b expression on human neutrophils which was induced by the incubation of cells with induced sputum supernatants from patients with COPD. Furthermore, other in vitro studies showed that stimulation of β2-adrenoceptors attenuated neutrophil adhesion to the vascular endothelium (Derian et al., 1995; Bolton et al., 1997) and IFN-γ-induced increase in ICAM-1 expression on bronchial epithelial cells (Oddera et al., 1998). However, whether the reduced CD11b expression was due to a direct effect on CD11b synthesis and release, or indirectly through reducing TNF-α expression (which induces CD11b up-regulation) remains to be determined (Diez-Fraile et al., 2000).
Furthermore, in the CD11b and TNF-α assays, we demonstrated that the anti-inflammatory effects of olodaterol were indeed mediated through the β2-receptor because the selective β2-adrenoceptor antagonist ICI-118,551 reduced the potency of olodaterol in inhibiting both the TNF-α release and the CD11b expression. However, the selective β1-adrenoceptor antagonist CGP-20712A had no effect on the potency and maximal efficacy of olodaterol. Because there are conflicting data on whether β2-agonists directly inhibit neutrophil migration, we isolated the primary human neutrophils from peripheral blood, confirmed the expression of functional β2-adrenoceptors by performing cAMP assays, and stimulated transwell migration with human IL-8. Olodaterol had no effect on the transwell migration of human neutrophils up to a concentration of 10−7 M, which is compatible with the results of Strandberg et al. (2010) who showed that formoterol did not affect IL-8-induced migration of human peripheral blood neutrophils.
In conclusion, in the present study, we have demonstrated the anti-inflammatory activity of the novel once-daily LABA olodaterol in different preclinical models of COPD in mice and guinea pigs. This provides the first evidence for anti-inflammatory activity of a β2-adrenoceptor agonist in in vivo models of CS-induced lung inflammation. Furthermore, our study demonstrated that the anti-inflammatory effect was mediated through additional mechanisms that were separate from direct inhibition of bronchoconstriction and that even repetitive treatment with olodaterol on 4 consecutive days inhibited cell influx into the lung. Mechanistically, olodaterol inhibited release of pro-inflammatory cytokines from lung parenchyma and whole blood and attenuated adhesion molecule expression on granulocytes, thereby attenuating the influx of inflammatory cells into the lung. As chronic inflammation in the respiratory tract is a characteristic of obstructive lung diseases, such as COPD, knowledge of the effects of olodaterol, apart from bronchodilation, is important for a full understanding of the mechanisms underlying its clinical efficacy.
Acknowledgments
The research of EN and PD was supported by a grant from the Boehringer Ingelheim Pharma GmbH & Co. KG.
Glossary
- ASM
airway smooth muscle
- BAL
bronchoalveolar lavage
- COPD
chronic obstructive pulmonary disease
- CS
cigarette smoke
- LABA
long-acting β2-adrenoceptor agonist
Author contributions
E. W., I. K. and E. N. performed the research. E. W., L. W., E. N. and P. D. designed the research study. E. W., I. K., M. J. D., E. N., L. W. and P. D. analysed the data. E. W., L. W. and P. D. wrote the paper.
Conflict of interest
E. W., I. K., M. J. D. and L. W. are employees of Boehringer Ingelheim Pharma GmbH & Co. KG.
References
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: G Protein-Coupled Receptors. Br J Pharmacol. 2013a;170:1459–1581. doi: 10.1111/bph.12445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Catalytic Receptors. Br J Pharmacol. 2013b;170:1676–1705. doi: 10.1111/bph.12449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Enzymes. Br J Pharmacol. 2013c;170:1797–1867. doi: 10.1111/bph.12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes PJ. Effect of beta-agonists on inflammatory cells. J Allergy Clin Immunol. 1999;104:S10–S17. doi: 10.1016/s0091-6749(99)70269-1. [DOI] [PubMed] [Google Scholar]
- Barnes PJ, Basbaum CB, Nadel JA, Roberts JM. Localization of beta-adrenoreceptors in mammalian lung by light microscopic autoradiography. Nature. 1982;299:444–447. doi: 10.1038/299444a0. [DOI] [PubMed] [Google Scholar]
- Bolton PB, Lefevre P, McDonald DM. Salmeterol reduces early- and late-phase plasma leakage and leukocyte adhesion in rat airways. Am J Respir Crit Care Med. 1997;155:1428–1435. doi: 10.1164/ajrccm.155.4.9105089. [DOI] [PubMed] [Google Scholar]
- Bosmann M, Grailer JJ, Zhu K, Matthay MA, Sarma JV, Zetoune FS, et al. Anti-inflammatory effects of beta2 adrenergic receptor agonists in experimental acute lung injury. FASEB J. 2012;26:2137–2144. doi: 10.1096/fj.11-201640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouyssou T, Casarosa P, Naline E, Pestel S, Konetzki I, Devillier P, et al. Pharmacological characterization of olodaterol, a novel inhaled beta2-adrenoceptor agonist exerting a 24-hour-long duration of action in preclinical models. J Pharmacol Exp Ther. 2010a;334:53–62. doi: 10.1124/jpet.110.167007. [DOI] [PubMed] [Google Scholar]
- Bouyssou T, Hoenke C, Rudolf K, Lustenberger P, Pestel S, Sieger P, et al. Discovery of olodaterol, a novel inhaled beta2-adrenoceptor agonist with a 24 h bronchodilatory efficacy. Bioorg Med Chem Lett. 2010b;20:1410–1414. doi: 10.1016/j.bmcl.2009.12.087. [DOI] [PubMed] [Google Scholar]
- Buenestado A, Chaumais MC, Grassin-Delyle S, Risse PA, Naline E, Longchampt E, et al. Roflumilast inhibits lipopolysaccharide-induced tumor necrosis factor-alpha and chemokine production by human lung parenchyma. PLoS ONE. 2013;8:e74640. doi: 10.1371/journal.pone.0074640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carstairs JR, Nimmo AJ, Barnes PJ. Autoradiographic localisation of beta-adrenoceptors in human lung. Eur J Pharmacol. 1984;103:189–190. doi: 10.1016/0014-2999(84)90211-5. [DOI] [PubMed] [Google Scholar]
- Carstairs JR, Nimmo AJ, Barnes PJ. Autoradiographic visualization of beta-adrenoceptor subtypes in human lung. Am Rev Respir Dis. 1985;132:541–547. doi: 10.1164/arrd.1985.132.3.541. [DOI] [PubMed] [Google Scholar]
- Derian CK, Santulli RJ, Rao PE, Solomon HF, Barrett JA. Inhibition of chemotactic peptide-induced neutrophil adhesion to vascular endothelium by cAMP modulators. J Immunol. 1995;154:308–317. [PubMed] [Google Scholar]
- Diez-Fraile A, Meyer E, Massart-Leen AM, Burvenich C. Effect of isoproterenol and dexamethasone on the lipopolysaccharide induced expression of CD11b on bovine neutrophils. Vet Immunol Immunopathol. 2000;76:151–156. doi: 10.1016/s0165-2427(00)00199-9. [DOI] [PubMed] [Google Scholar]
- Donnelly LE, Tudhope SJ, Fenwick PS, Barnes PJ. Effects of formoterol and salmeterol on cytokine release from monocyte-derived macrophages. Eur Respir J. 2010;36:178–186. doi: 10.1183/09031936.00158008. [DOI] [PubMed] [Google Scholar]
- Hackett TL, Holloway R, Holgate ST, Warner JA. Dynamics of pro-inflammatory and anti-inflammatory cytokine release during acute inflammation in chronic obstructive pulmonary disease: an ex vivo study. Respir Res. 2008;9:47. doi: 10.1186/1465-9921-9-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry PJ, Goldie RG. Beta 1-adrenoceptors mediate smooth muscle relaxation in mouse isolated trachea. Br J Pharmacol. 1990;99:131–135. doi: 10.1111/j.1476-5381.1990.tb14666.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izeboud CA, Monshouwer M, van Miert AS, Witkamp RF. The beta-adrenoceptor agonist clenbuterol is a potent inhibitor of the LPS-induced production of TNF-alpha and IL-6 in vitro and in vivo. Inflamm Res. 1999;48:497–502. doi: 10.1007/s000110050493. [DOI] [PubMed] [Google Scholar]
- Jeffery PK, Venge P, Gizycki MJ, Egerod I, Dahl R, Faurschou P. Effects of salmeterol on mucosal inflammation in asthma: a placebo-controlled study. Eur Respir J. 2002;20:1378–1385. doi: 10.1183/09031936.02.00542001. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. Animal research: Reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maneechotesuwan K, Essilfie-Quaye S, Meah S, Kelly C, Kharitonov SA, Adcock IM, et al. Formoterol attenuates neutrophilic airway inflammation in asthma. Chest. 2005;128:1936–1942. doi: 10.1378/chest.128.4.1936. [DOI] [PubMed] [Google Scholar]
- Maris NA, van der Sluijs KF, Florquin S, de Vos AF, Pater JM, Jansen HM, et al. Salmeterol, a beta2-receptor agonist, attenuates lipopolysaccharide-induced lung inflammation in mice. Am J Physiol Lung Cell Mol Physiol. 2004;286:L1122–L1128. doi: 10.1152/ajplung.00125.2003. [DOI] [PubMed] [Google Scholar]
- Maris NA, de Vos AF, Dessing MC, Spek CA, Lutter R, Jansen HM, et al. Antiinflammatory effects of salmeterol after inhalation of lipopolysaccharide by healthy volunteers. Am J Respir Crit Care Med. 2005;172:878–884. doi: 10.1164/rccm.200503-451OC. [DOI] [PubMed] [Google Scholar]
- Masclans JR, Barbera JA, MacNee W, Pavia J, Piera C, Lomena F, et al. Salbutamol reduces pulmonary neutrophil sequestration of platelet-activating factor in humans. Am J Respir Crit Care Med. 1996;154:529–532. doi: 10.1164/ajrccm.154.2.8756833. [DOI] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamoto M, Tomaki M, Lotvall J, Linden A. Beta-adrenoceptor stimulation and neutrophil accumulation in mouse airways. Eur Respir J. 2004;24:231–237. doi: 10.1183/09031936.04.00035204. [DOI] [PubMed] [Google Scholar]
- van Noord JA, Smeets JJ, Drenth BM, Rascher J, Pivovarova A, Hamilton AL, et al. 24-hour bronchodilation following a single dose of the novel beta(2)-agonist olodaterol in COPD. Pulm Pharmacol Ther. 2011;24:666–672. doi: 10.1016/j.pupt.2011.07.006. [DOI] [PubMed] [Google Scholar]
- Oddera S, Silvestri M, Lantero S, Sacco O, Rossi GA. Downregulation of the expression of intercellular adhesion molecule (ICAM)-1 on bronchial epithelial cells by fenoterol, a beta2-adrenoceptor agonist. J Asthma. 1998;35:401–408. doi: 10.3109/02770909809048948. [DOI] [PubMed] [Google Scholar]
- Oldenburger A, Roscioni SS, Jansen E, Menzen MH, Halayko AJ, Timens W, et al. Anti-inflammatory role of the cAMP effectors Epac and PKA: implications in chronic obstructive pulmonary disease. PLoS ONE. 2012;7:e31574. doi: 10.1371/journal.pone.0031574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, et al. NC-IUPHAR. The IUPHAR/BPS Guide to PHARMACOLOGY: an expert-driven knowledge base of drug targets and their ligands. Nucl Acids Res. 2014;42(Database Issue):D1098–D1106. doi: 10.1093/nar/gkt1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Profita M, Bonanno A, Montalbano AM, Albano GD, Riccobono L, Siena L, et al. beta(2) long-acting and anticholinergic drugs control TGF-beta1-mediated neutrophilic inflammation in COPD. Biochim Biophys Acta. 2012;1822:1079–1089. doi: 10.1016/j.bbadis.2012.03.002. [DOI] [PubMed] [Google Scholar]
- Raemdonck K, de Alba J, Birrell MA, Grace M, Maher SA, Irvin CG, et al. A role for sensory nerves in the late asthmatic response. Thorax. 2012;67:19–25. doi: 10.1136/thoraxjnl-2011-200365. [DOI] [PubMed] [Google Scholar]
- Sarir H, Mortaz E, Karimi K, Johnson M, Nijkamp FP, Folkerts G. Combination of fluticasone propionate and salmeterol potentiates the suppression of cigarette smoke-induced IL-8 production by macrophages. Eur J Pharmacol. 2007;571:55–61. doi: 10.1016/j.ejphar.2007.05.034. [DOI] [PubMed] [Google Scholar]
- Shinkai N, Takasuna K, Takayama S. Inhibitory effects of formoterol on lipopolysaccharide-induced premature delivery through modulation of proinflammatory cytokine production in mice. Reproduction. 2003;125:199–203. doi: 10.1530/rep.0.1250199. [DOI] [PubMed] [Google Scholar]
- Strandberg K, Blidberg K, Sahlander K, Palmberg L, Larsson K. Effect of formoterol and budesonide on chemokine release, chemokine receptor expression and chemotaxis in human neutrophils. Pulm Pharmacol Ther. 2010;23:316–323. doi: 10.1016/j.pupt.2010.03.004. [DOI] [PubMed] [Google Scholar]
- Vestbo J, Hurd SS, Agusti AG, Jones PW, Vogelmeier C, Anzueto A, et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: GOLD executive summary. Am J Respir Crit Care Med. 2013;187:347–365. doi: 10.1164/rccm.201204-0596PP. [DOI] [PubMed] [Google Scholar]
- Wang J, Nie B, Xiong W, Xu Y. Effect of long-acting beta-agonists on the frequency of COPD exacerbations: a meta-analysis. J Clin Pharm Ther. 2012;37:204–211. doi: 10.1111/j.1365-2710.2011.01285.x. [DOI] [PubMed] [Google Scholar]
- Wollin L, Pieper MP. Tiotropium bromide exerts anti-inflammatory activity in a cigarette smoke mouse model of COPD. Pulm Pharmacol Ther. 2010;23:345–354. doi: 10.1016/j.pupt.2010.03.008. [DOI] [PubMed] [Google Scholar]
- Yoshimura T, Kurita C, Nagao T, Usami E, Nakao T, Watanabe S, et al. Inhibition of tumor necrosis factor-alpha and interleukin-1-beta production by beta-adrenoceptor agonists from lipopolysaccharide-stimulated human peripheral blood mononuclear cells. Pharmacology. 1997a;54:144–152. doi: 10.1159/000139481. [DOI] [PubMed] [Google Scholar]
- Yoshimura T, Kurita C, Nagao T, Usami E, Nakao T, Watanabe S, et al. Effects of cAMP-phosphodiesterase isozyme inhibitor on cytokine production by lipopolysaccharide-stimulated human peripheral blood mononuclear cells. Gen Pharmacol. 1997b;29:633–638. doi: 10.1016/s0306-3623(96)00580-0. [DOI] [PubMed] [Google Scholar]