

Abstract
Background and Purpose
There are conflicting data regarding whether netrin-1 retards or accelerates atherosclerosis progression, as it can lead either to monocyte repulsion from or retention within plaques depending on its cellular source. We investigated the effect of aspirin, which is widely used in cardiovascular prophylaxis, on the synthesis of different isoforms of netrin-1 by endothelial cells under pro-inflammatory conditions, and defined the net effect of aspirin-dependent systemic modulation of netrin-1 on atherosclerosis progression.
Experimental Approach
Netrin-1 synthesis was studied in vitro using human endothelial cells stimulated with TNF-α, with or without aspirin treatment. In vivo experiments were conducted in ApoE−/− mice fed with a high-fat diet (HFD), receiving either aspirin or clopidogrel.
Key Results
TNF-α-induced NF-κB activation up-regulated the nuclear isoform of netrin-1, while simultaneously reducing secreted netrin-1. Down-regulation of the secreted isoform compromised the chemorepellent action of the endothelium against monocyte chemotaxis. Aspirin counteracted TNF-α-mediated effects on netrin-1 synthesis by endothelial cells through COX-dependent inhibition of NF-κB and concomitant histone hyperacetylation. Administration of aspirin to ApoE−/− mice on HFD increased blood and arterial wall levels of netrin-1 independently of its effects on platelets, accompanied by reduced plaque size and content of monocytes/macrophages, compared with untreated or clopidogrel-treated mice. In vivo blockade of netrin-1 enhanced monocyte plaque infiltration in aspirin-treated ApoE−/− mice.
Conclusions and Implications
Aspirin counteracts down-regulation of secreted netrin-1 induced by pro-inflammatory stimuli in endothelial cells. The aspirin-dependent increase of netrin-1 in ApoE−/− mice exerts anti-atherogenic effects by preventing arterial accumulation of monocytes.
Tables of Links
| TARGETS | |
|---|---|
| Enzymes | |
| COX-1 | |
| COX-2 | |
| HDAC, histone deacetylase | |
| HAT, histone acetyltransferase |
| LIGANDS | |
|---|---|
| Aspirin | |
| CCL2 | |
| Clopidogrel | |
| Indomethacin | |
| Salicylic acid | |
| TNF-α | |
| TSA, trichostatin A |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (Alexander et al., 2013).
Introduction
The neuroimmune guidance cue netrin-1 is known to affect atheroma development through an inhibitory action on the motility of monocytes/macrophages. The overall effect depends on the tissue expression and cellular localization of netrin-1 (Gerszten and Tager, 2012). Endothelial-derived netrin-1, which is down-regulated under pro-atherogenic conditions, prevents monocyte vascular infiltration (Ly et al., 2005; Mirakaj et al., 2010; van Gils et al., 2013), whereas haematopoietic netrin-1, which is enhanced by pro-inflammatory stimuli (van Gils et al., 2012; Ramkhelawon et al., 2014), favours the arterial accumulation of monocytes. From a therapeutic perspective, such a divergence of effect translates into the balance between ingress and egress of monocytes into plaques requiring a cell-targeted approach, because systemic modulation of netrin-1 would exert an indiscriminate action on different cell types, resulting in opposing effects on atherosclerosis progression.
As well as the cellular source of netrin-1, it may also be important to distinguish between the different isoforms of netrin-1 synthesized by cells. In tumour cells, both full-length and truncated proteins have been found, displaying different cellular compartmentalization and biological functions. The full-length isoform is secreted, exerting its actions in the micro-environment of the cell, whereas the truncated isoform localizes within the nucleoplasm and promotes cell proliferation (Delloye-Bourgeois et al., 2012; Harter et al., 2014). Therefore, assessing both the existence and regulation of secreted and nuclear isoforms of netrin-1 in cell types that are crucial players in atheroma development might be of prime importance in evaluating the net effect of therapeutic approaches targeting netrin-1 on plaque progression.
We hypothesized that the reduction in netrin-1 observed in endothelial cells under pro-inflammatory or pro-atherogenic conditions, specifically relates to the secreted isoform of the protein. We therefore aimed to define: (i) the subcellular localization of netrin-1 in endothelial cells in response to the pro-inflammatory cytokine TNF-α; (ii) whether aspirin, a COX inhibitor which is widely used as an anti-platelet agent but also exerts anti-inflammatory properties, differentially modulates endothelial synthesis of the two distinct isoforms of netrin-1 under pro-inflammatory conditions; and (iii) the net effect of aspirin treatment on netrin-1 production and atherosclerosis progression in vivo.
Methods
Detailed methods are available in the Supporting Information.
In vitro experiments
Cells
HUVECs were isolated from fresh umbilical cords obtained from uncomplicated deliveries following healthy pregnancies; these were obtained from the labour ward at St Thomas’ Hospital (London, UK), with written informed consent from the mothers. The study was approved by the St Thomas’ Hospital Research Ethics Committee. Human aortic endothelial cells (HAoECs) were purchased from PromoCell, UK.
Experiments with HUVECs
HUVECs were incubated overnight with the following compounds: TNF-α (10 ng·mL−1, Invitrogen, Paisley, UK); Bay 11-7085 (an NF-κB inhibitor; 5 μM, Calbiochem, Nottingham, UK); aspirin or salicylic acid (0.5 mM for each; Sigma, Poole, UK); SC-560 (30 nM; Calbiochem); NS-398 (10 μM; Calbiochem); indomethacin (100 μM; Sigma); and trichostatin A (TSA; 400 nM; Invitrogen).
Immunofluorescence staining was performed using rat anti-netrin-1 (1:200; R&D System, Abingdon, UK), rabbit anti-acetylated histone (1:400; Cell Signalling, Hitchin, UK) and rabbit anti-p65 (1:100; Abcam, Cambridge, UK).
Transcript levels of nuclear and total netrin-1 were assessed by real-time PCR (RT-PCR) and quantified using the comparative Ct method (ΔΔCt).
Histone acetylation was quantified by Western blotting using rabbit anti-acetylated H3 (acH3) or total H3 (both 1:1000; Cell Signalling) primary antibodies.
Netrin-1 in cell supernatants was measured by elisa. Western blotting of both cell lysates and cell supernatants was performed with both rat anti-netrin-1 and goat anti-netrin-1 (both 1:100; R&D System) primary antibodies.
Histone deacetylase (HDAC) and histone acetyltransferase (HAT) activities were measured by commercially available colorimetric assay kits (Epigentek, Caltag MedSystem, Buckingham, UK).
Human CD14+CD16- monocytes were isolated by immunomagnetic negative isolation (kit from Invitrogen, UK) of peripheral blood mononuclear cells from healthy volunteers (n = 3; 2 female and 1 male; 18–40 years old) and on no regular medication; particularly, they denied taking any medication for at last 2 weeks before study. The study was approved by the St Thomas’ Hospital Research Ethics Committee. Briefly, the whole blood (30 ml) was collected in EDTA tubes and underwent Lymphoprep gradient centrifugation. Mononuclear cells were then isolated and underwent immunomagnetic negative selection for CD14+CD16- monocytes (kit from Invitrogen) according to the manufacturer's instructions. Purity of isolation, as determined by antigen expression (CD14+, CD16−, CD3−, CD4−, CD8−, CD20−) using flow cytometry, was 85–90% in all experiments. Viability of monocytes was assessed by Trypan Blue dye exclusion and was >95% immediately after their isolation. Human monocyte migration was assessed using a 24-multi-well Boyden chamber system (5 μm pores; Corning, Birmingham, UK).
In vivo experiments
All animal care and experimental procedures complied with the guidelines of the United Kingdom Home Office (PPL 70/6646; PIL 70/20776) and were approved by the local Animal Ethical Committee. Studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010). A total of 62 animals were used in the experiments described here.
Male ApoE−/− mice (B6.129P2–apoEtm1Unc/J; 8 weeks old; Charles River Laboratories, Edinburgh, UK) were studied before (baseline), and at 4 and 8 weeks after commencement of a high-fat diet (HFD; Special Diets Services, Witham, UK). Wild-type C57BL/6J mice (males; Charles River Laboratories, Edinburgh, UK) were used as controls. An additional group of ApoE−/− mice was kept on 8 week HFD and concomitant anti-platelet therapy with aspirin (5 mg·kg−1·day−1) or clopidogrel (25 mg·kg−1·day−1). Drugs were administered in drinking water.
Plasma netrin-1 levels were measured at each time point by elisa; for this peripheral blood was drawn by cardiac puncture using syringes containing heparin (10U/l) as anticoagulant, from mice deeply anaesthetized with isoflurane (terminal sample collection). The brachiocephalic artery was collected at death. The following variables were also measured at each time point; expression of netrin-1 in the arterial wall by immunofluorescence; phenotype of circulating monocytes by flow cytometry; plaque burden and lipid content by Oil Red O staining; and phenotype of monocytes/macrophages infiltrating plaques by histological staining for CD68 and tissue flow cytometry.
Monocyte trafficking into the brachiocephalic artery was studied by autoradiography (Phosphorimager Screen, Molecular Dynamics, GE HealthCare) (Swirski et al., 2007; Kircher et al., 2008) following transfer of [111In]-labelled exogenous monocytes in ApoE−/− mice kept on 8 week HFD with concomitant aspirin treatment (5 mg·kg−1·day−1). Experiments were performed in the presence of netrin-1 blocking obtained by i.v. administration of Unc5b-Fc, a netrin-1 receptor, but is fused to the Fc portion of IgG and because of this Unc5b-Fc is referred to as chimera antibody (800 μg·kg−1 body weight; R&D Systems) (Tadagavadi et al., 2010).
Data analysis
Data are shown as means ±SEM. In vitro and in vivo results were analysed by anova, with or without repeated measures as appropriate, with Bonferroni's multiple comparisons test. P < 0.05 (two tailed) was taken to indicate statistical significance.
Results
The nuclear and secreted isoforms of netrin-1 are differentially regulated in endothelial cells under pro-inflammatory conditions
TNF-α stimulation of HUVECs did not modify the total intracellular content of netrin-1 compared with untreated cells (Figure 1A). However, differences in cellular distribution of netrin-1 were observed, in that TNF-α-treated HUVECs displayed increased netrin-1 expression within the nuclei (Figure 1B), with a concomitant reduction in its cytoplasmic level as demonstrated by a decrease in ratio of total to nuclear immunofluorescence for netrin-1 (Figure 1C). These data were corroborated by staining of non-permeabilized HUVECs, which showed TNF-α-dependent down-regulation of netrin-1 on the cell membrane (Figure 1D). elisa of cell supernatants confirmed a decrease in netrin-1 secretion by HUVECs in response to TNF-α (Figure 1E).
Figure 1.
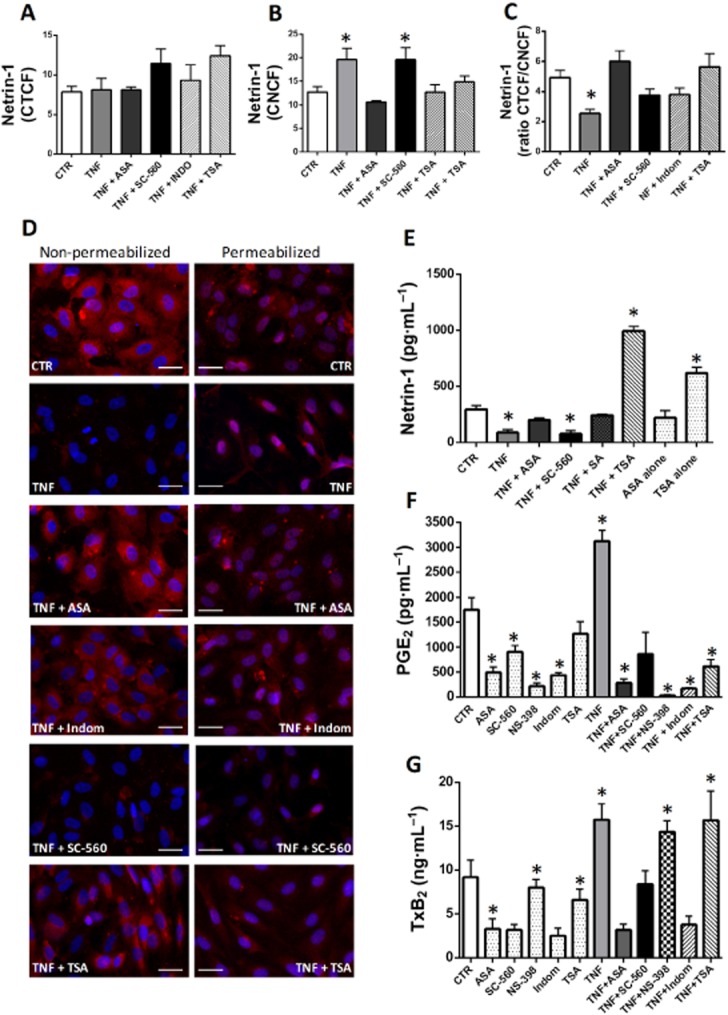
Cellular distribution of netrin-1 in HUVECs. Corrected total (A) and nuclear (B) cell fluorescence of netrin-1 [corrected total cell fluorescence (CTCF) and corrected nuclear cell fluorescence (CNCF), respectively] in HUVECs following different treatments as indicated. The ratio of CTCF to CNCF is also displayed (C). Panel (D) shows representative micrographs (20× magnification fields) of netrin-1 staining (in red) in non-permeabilized (left) and permeabilized (right) HUVECs under the different experimental conditions as specified (scale bars = 10 μm). Netrin-1 fluorescent images were merged with corresponding fluorescent nuclear staining with Hoechst (in blue). Pink colour within the nuclei in permeabilized cells derives from the overlay of netrin-1 signal (red) with nuclear staining (blue), and is indicative of nuclear netrin-1 localization. Concentration of the full-length/secreted isoform of netrin-1 was measured in cell supernatants and is displayed in (E). (F) and (G) report the levels of PGE2 and TxB2, respectively, in HUVEC supernatant following different treatments. n = 3–7. ASA: aspirin, 0.5 mM; SC-560, 30 nM; Indom: indomethacin, 100 μM; NS-398, 10 μM; SA: salicylic acid, 0.5 mM; TSA, 400 nM; TNF-α, 10 ng·mL–1. *P < 0.05 versus CTR (control HUVECs not treated with TNF-α).
Aspirin abolished both the over-expression of nuclear netrin-1 and the down-regulation of cytoplasmic netrin-1 induced by TNF-α (Figure 1A–C). Immunofluorescence staining of non-permeabilized HUVECs similarly showed that aspirin restored the expression of netrin-1 on the extracellular surface of TNF-α-treated cells (Figure 1D); and elisa of HUVEC supernatants confirmed that aspirin counteracted the decrease in secretion of netrin-1 induced by TNF-α (Figure 1E). We also examined the effect of other COX inhibitors such as the selective COX-1 inhibitor SC-560, the selective COX-2 inhibitor NS-398 and the non-selective COX inhibitor indomethacin. Only indomethacin treatment reproduced the effects of aspirin on netrin-1 synthesis (Figure 1). The inhibitory action exerted by these different compounds on COX enzymic activity was measured by evaluation of prostanoid release into HUVEC supernatants. elisa assays for TxB2 (the stable metabolite of TxA2) and PGE2 showed an increase in both prostanoids in response to TNF-α (Figure 1F–G). Aspirin, and indomethacin to a similar extent, reduced the synthesis of both PGE2 and TxB2 in both untreated and TNF-α-treated cells. NS-398 suppressed PGE2 production only, either in the absence or presence of TNF-α; while SC-560 was as effective as aspirin in suppressing TxB2 synthesis in the absence of TNF-α, but had only a minor inhibitory action on the release of both TxB2 and PGE2 in the presence of TNF-α (Figure 1F–G). These data demonstrated that aspirin restored netrin-1 production in cells stimulated with TNF-α, to baseline levels, and that this effect was COX-dependent. However, only the combined inhibition of COX-1 and COX-2, as obtained in response to aspirin and indomethacin, was able to counteract the effect of TNF-α on levels of secreted and nuclear netrin-1.
We also found that the HDAC inhibitor TSA similarly counteracted the effect of TNF-α on netrin-1 production (Figure 1), although through a COX-independent mechanism as discussed below.
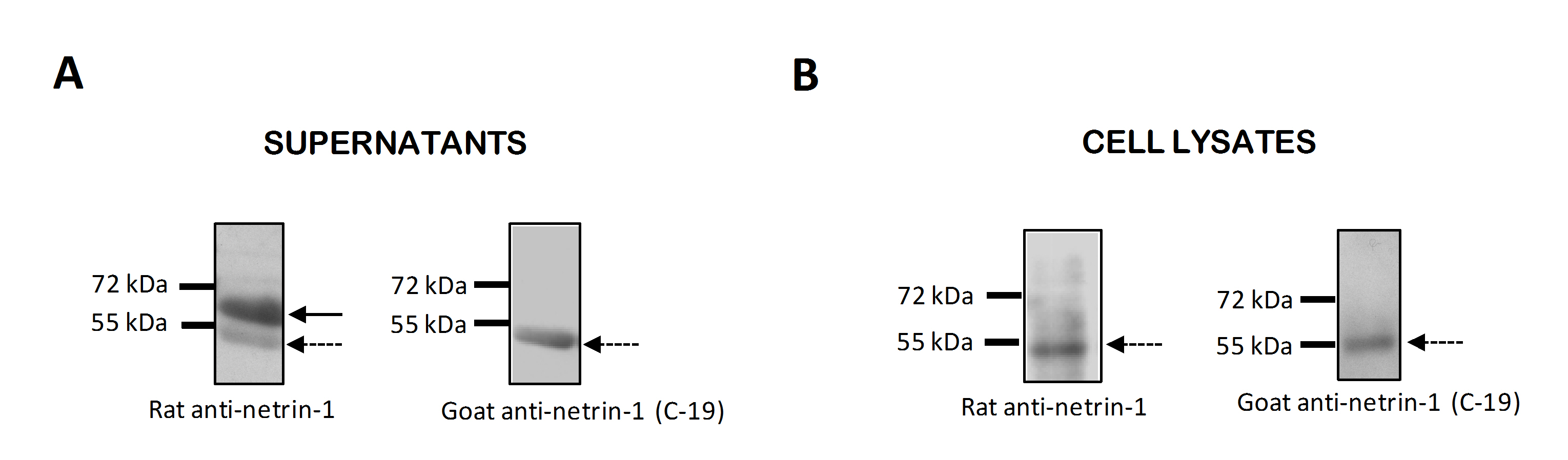
The expression of netrin-1 was next characterized by Western blotting of cell lysates and supernatants in order to investigate whether different isoforms of netrin-1, with distinct cellular distributions, were synthesized by HUVECs, as has previously been reported for tumour cells (Delloye-Bourgeois et al., 2012). Using the rat anti-netrin-1 antibody that was used for the elisa assays and immunofluorescence staining described above, two isoforms were detected in cell supernatants, at 55 kDa and around 70 kDa (Supporting Information Fig. S1a). The same antibody only detected a 55 kDa protein in cell lysates (Supporting Information Fig. S1b). A goat anti-netrin-1 antibody specifically targeting the C-terminal of the protein, which was previously shown to contain the nuclear localization signal of netrin-1 (Delloye-Bourgeois et al., 2012), only detected a 55 kDa protein in both cell lysates and cell supernatants (Supporting Information Fig. S1a,b). These data suggest that the HUVECs were able to synthesize a truncated isoform of netrin-1 that was predominantly contained within cells and, given the presence of the C-terminal nuclear signal, could localize within the nuclei; whereas the full-length isoform of netrin-1, which lacks the C-terminal nuclear signal, was secreted, as did the truncated isoform to a much lesser degree.
Differential gene regulation of secreted and nuclear netrin-1 in endothelial cells
Because gene induction of netrin-1 has been demonstrated to be under transcriptional control by NF-κB (Paradisi et al., 2008), we sought to evaluate whether activation of NF-κB in TNF-α-treated HUVECs gives rise to differential gene induction of the two isoforms of netrin-1. As expected, TNF-α treatment induced p65 nuclear translocation, consistent with NF-κB activation (Figure 2A, B). By quantitative RT-PCR using primers that nonselectively amplify both the nuclear (truncated) and secreted (full-length) isoforms of netrin-1, TNF-α was found to induce total netrin-1 gene transcription (Figure 2C). However, amplification of netrin-1 mRNA using primers that selectively recognize the full-length (secreted) isoform of the protein showed that TNF-α does not significantly modify its gene induction (Figure 2D), and in fact the ratio of secreted to total netrin-1 transcripts (Figure 2E) exhibited a trend towards reduction in response to TNF-α. These data demonstrate that TNF-α-dependent NF-κB activation gives rise to up-regulation specifically of the nuclear (truncated) isoform of netrin-1. Consistent with this, inhibition of NF-κB in TNF-α-treated HUVECs, by co-incubation with Bay 11-7085, also reduced the TNF-α-induced over-expression of netrin-1, as revealed by using primers that amplify both isoforms (Figure 2C), while it did not affect the gene transcription of full-length netrin-1 nor the ratio of secreted to total netrin-1 compared with cells treated with TNF-α only (Figure 2D, E). At the protein level, Bay 11-7085 restored the generation of secreted netrin-1 to baseline levels in TNF-α-stimulated HUVECs, as evaluated by cellular immunofluorescence staining and elisa of cell supernatants, while at the same time suppressing the expression of nuclear netrin-1 induced by TNF-α (Figure 2F–G).
Figure 2.

NF-κB activation and gene induction of truncated (nuclear) and full-length (secreted) isoforms of netrin-1. (A) Representative micrographs (20× magnification fields) of p65 staining under the different experimental conditions as indicated. p65 fluorescent images (in green) were merged with corresponding fluorescent nuclear staining with Hoechst (in blue). Yellow colour within the nuclei derives from the overlay of p65 signal (green) with nuclear staining (blue), and is indicative of nuclear p65 localization. p65 nuclear translocation was taken as an index of NF-κB activation and is reported in (B) as percentage of nuclei expressing p65 under the different experimental conditions. Panels (C) and (D) show accumulated data for quantitative RT-PCR for netrin-1 (NTN-1) using primers for (C) total netrin-1 (comprising both the full-length and truncated isoforms) or (D) the full-length/secreted isoform specifically, both normalized to the housekeeping gene GAPDH. The ratio of the two isoforms was obtained by normalizing the full-length isoform of netrin-1 to total netrin-1 (E). Data are reported as fold changes compared with control (untreated cells, dotted blue line). n = 3–5. Panels (F) and (G) show representative micrographs (20× magnification fields) of netrin-1 staining (in red) in permeabilized (F) and non-permeabilized (G) HUVECs either untreated (control) or TNF-α treated, either in the presence or absence of the NF-κB inhibitor Bay 11-7085 (BAY), as specified. Netrin-1 fluorescent images were merged with corresponding fluorescent nuclear staining with Hoechst (in blue). Pink colour within the nuclei in permeabilized cells (F) derives from the overlay of netrin-1 signal (red) with nuclear staining (blue), and is indicative of nuclear netrin-1 localization. In permeabilized cells (F), corrected total and nuclear cell fluorescence of netrin-1 (CTCF and CNCF, respectively) following different treatments was calculated and reported in the corresponding graphs. The ratio of CTCF to CNCF is also displayed. Secreted netrin-1 was measured by elisa in cell supernatants and results are reported in (G). n = 3. Scale bars = 10 μm. ASA: aspirin, 0.5 mM; SC-560, 30 nM; Indom: indomethacin, 100 μM; NS-398, 10 μM; TSA, 400 nM; TNF-α, 10 ng·mL−1; Bay 11-7085, 5 μM. *P < 0.05 versus CTR (control HUVECs not treated with TNF-α).
Quantitative RT-PCR demonstrated that none of the COX inhibitors tested in our study affected TNF-α-induced gene transcription of total netrin-1 in HUVECs (Figure 2C). However, aspirin and indomethacin specifically enhanced the mRNA level of the secreted isoform of netrin-1 (Figure 2D), as well as the ratio of secreted to total netrin-1 (Figure 2E). By immunofluorescence staining of HUVECs, we found that both aspirin and indomethacin abolished (and SC-560 significantly reduced, although to a much lesser extent than aspirin or indomethacin) the TNF-α-dependent p65 nuclear translocation (Figure 2A, B). Selective inhibition of either COX isoform alone was less effective than aspirin in abolishing NF-κB activation. We also found that inhibition of p65 nuclear translocation in response to TNF-α was strongly inhibited by TSA (Figure 2A, B), which also specifically enhanced the mRNA synthesis of secreted netrin-1 as demonstrated by quantitative RT-PCR (Figure 2C–E).
Aspirin regulation of secreted netrin-1 occurs through epigenetic modification
The above experiments demonstrate that, although NF-κB activation gives rise to gene induction and consequent over-expression of nuclear netrin-1, the synthesis of secreted netrin-1 is regulated in a different way. In accordance with this hypothesis, it has previously been reported that the two isoforms have different promoters (Delloye-Bourgeois et al., 2012). Because epigenetic modification of chromatin and DNA is considered a crucial regulator of gene expression in the TNF-α-mediated inflammatory response (Barnes et al., 2009; Pons et al., 2009; McKinsey, 2012), we postulated that modulation of histone H3 acetylation, which has a key role in vascular inflammation and atherosclerosis (Zampetaki et al., 2010), could be involved in the differential gene induction of secreted and nuclear netrin-1 under the different experimental conditions. Western blotting and immunofluorescence staining demonstrated that treatment of HUVECs with TNF-α did not alter the level of acetylated H3 (Figure 3A–C). Co-stimulation of HUVECs with TNF-α and the NF-κB inhibitor Bay 11-7085 also did not modify the level of acetylated H3 compared with untreated cells (Figure 3B). However, in the presence of aspirin, TNF-α-treated cells displayed higher expression of acetylated H3 compared with control cells (Figure 3A–C). These results were reproduced by co-incubating HUVECs with TNF-α and salicylic acid. Aspirin alone did not affect acetylated H3 (Figure 3A).
Figure 3.

Histone 3 acetylation and netrin-1 expression in HUVECs. Western blotting (A, B) and immunofluorescence (C) studies showing degree of histone acetylation in HUVECs in response to different treatments as specified. In the Western blots, densitometric analysis of acetylated H3 (acH3) was normalized to total H3. In immunofluorescence experiments, the corrected nuclear cell fluorescence (CNCF) was calculated (C). Panels (D)–(H) show the effect of aspirin (ASA) on HAT and HDAC activities in HUVECs. HAT (D) and HDAC (E) activities, and HDAC/HAT ratio (F), were measured in nuclear extracts isolated from HUVECs following treatments as specified. Nuclear extracts were isolated from HUVECs not stimulated with TNF-α, and HAT (G) and HDAC (H) activities were tested in the presence of either ASA or salicylic acid. n = 3. ASA, 0.5 mM; SC-560, 30 nM; SA: salicylic acid, 0.5 mM; Indom: indomethacin, 100 μM; TSA, 400 nM; TNF-α, 10 ng·mL−1; Bay 11-7085 (BAY), 5 μM. *P < 0.05 versus CTR (control HUVECs not treated with TNF-α).
The HDAC inhibitor TSA, which was used as a positive control to increase H3 acetylation and which increased acetylated H3 up to five times more than the level induced by aspirin or salicylic acid (Figure 3A–C), was also able not only to restore but even increase the secretion of netrin-1 into the supernatant of cells co-treated with TNF-α (Figure 1E). Moreover, TSA alone enhanced the secretion of netrin-1 by HUVECs even in the absence of TNF-α (Figure 1E). TSA treatment also suppressed TNF-α-dependent NF-κB activation (Figure 2A, B), thus reducing the level of netrin-1 within the nuclei (Figure 1A), and induced gene transcription of the secreted isoform of netrin-1 (Figure 2C–E).
In order to further investigate the biomolecular mechanism underlying aspirin/salicylate-mediated increase in acetylated H3 in HUVECs stimulated with TNF-α, we assessed the enzymic activity of HAT and HDAC in nuclear extracts. We found that aspirin, and salicylic acid to a similar extent, increased HAT (Figure 3D) and partly inhibited HDAC (Figure 3E) activities in TNF-α-treated cells, thus resulting in a marked reduction of the HDAC/HAT ratio compared with control or TNF-α-stimulated HUVECs (Figure 3F). These effects appeared to be partly mediated by a direct action of these compounds on the enzymic activity of HAT; indeed, addition of either salicylic acid or aspirin to nuclear extracts isolated from untreated cells led to enhanced HAT activity (Figure 3G). On the other hand, inhibition of HDAC activity is likely to be indirectly mediated, as neither aspirin nor salicylic acid was able to directly modify the HDAC activity of isolated nuclear extracts (Figure 3H).
Aspirin-dependent increase of endothelial-derived netrin-1 reduces monocyte chemotaxis
We next explored the hypothesis that the aspirin-dependent increase in endothelium-derived netrin-1 in TNF-α-stimulated cells would result in a chemorepellent action on arterial infiltration by monocytes. Our migration assays showed that TNF-α enhanced the monocyte chemotaxis towards activated human aortic endothelial cells (HAoECs) compared with unstimulated cells. Aspirin pretreatment of HAoECs significantly reduced monocyte migration towards TNF-α-stimulated cells, this effect being abolished by co-incubation with UNC5b-Fc (Figure 4A–C), which neutralizes soluble netrin-1 and reduces its bioactivity, as previously described (van Gils et al., 2013). Similar to the effect observed in HUVECs, aspirin counteracted the up-regulation of nuclear netrin-1 and the reduction in the secreted isoform of netrin-1 in response to TNF-α in HAoECs (Supporting Information Fig. S2). Co-staining of HAoECs for netrin-1 and p65 under the different experimental conditions showed that nuclear netrin-1 up-regulation in TNF-α-treated cells was paralleled by p65 nuclear translocation, indicative of NF-κB activation. As observed in HUVECs, aspirin inhibited TNF-α-dependent NF-κB activation in HAoECs (Supporting Information Fig. S2a,b).
Figure 4.

Effect of aspirin (ASA) on migration of human CD14+CD16− monocytes. Migration of human CD14+CD16− monocytes towards a monolayer of HAoECs (A–C) or towards an CCL2 (100 ng·mL−1) gradient (D–F) is shown under different experimental conditions, as indicated in the graphs. When tested against an HAoEC monolayer, the endothelial cells, but not monocytes, were pretreated with ASA, and migration was conducted in the presence or absence of Unc5b-Fc. When tested towards a CCL2 gradient, monocytes were pretreated with ASA and migration was performed either in the presence or absence of Unc5b-Fc. (A) and (D) show cell count under light microscopy in five microscopic fields (20× magnification) on the lower surface of the membranes of the Boyden chambers, following removal of non-migrated cells on the upper surface. (B) and (E) show count, as determined by flow cytometry, of monocytes (CD14+ cells, gated) which have migrated to the lower well of the Boyden chamber system, with representative dot plots also displayed (C and F). n = 3. CTR: TNF-α-stimulated HAoECs (C) or untreated monocytes (F); ASA: ASA-treated HAoECs (C) or monocytes (F); ASA + Unc5b-Fc: following ASA pretreatment of either HAoECs (C) or monocytes (F). Unc5b-Fc was added during monocyte migration. Histograms show count of migrated monocytes in CTR (grey), ASA (blue) and ASA + Unc5b-Fc (red). *P < 0.05 versus ASA; †P < 0.01 versus netrin-1.
Because monocytes are known to be capable of synthesizing netrin-1 (van Gils et al., 2012), we tested the possibility that aspirin exerts a direct effect on monocyte motility by enhancing their own production of secreted netrin-1. We found that monocytes pretreated with aspirin displayed reduced motility towards the chemokine CCL2 compared with untreated cells, this effect being similar to that exerted by recombinant human netrin-1, and again this was abolished by UNC5b-Fc (Figure 4D–F).
Aspirin enhances systemic and vascular expression of netrin-1 in ApoE−/− mice
The in vitro experiments decribed above suggest that aspirin might be able to enhance the expression of netrin-1 at a systemic level by acting on both the vascular endothelium and monocytes. In order to test whether such an effect is observable in vivo, as well as the resultant net effect on atherosclerosis progression, we treated ApoE−/− mice fed with a HFD with aspirin. Considering that platelet inhibition itself might have important anti-inflammatory effects, a separate group of animals was treated with a different anti-platelet drug, clopidogrel, working through an unrelated mechanism (P2Y12 receptor inhibition).
Administration of aspirin to ApoE−/− mice given 8 weeks of HFD induced a significant increase in plasma netrin-1 compared with levels measured in both untreated and clopidogrel-treated animals (Figure 5A). The up-regulation of netrin-1 in the aspirin-treated group was also evident within the arterial wall, as demonstrated by immunofluorescence of the abdominal aorta (Figure 5B, C). No atherosclerotic lesions were detectable in the abdominal aortas of HFD-fed ApoE−/− mice, either with or without anti-platelet treatment, in contrast to the abundance of plaque observed in the brachiocephalic arteries of these same animals (Figure 6). Nevertheless, netrin-1 was expressed in the abdominal aortas and co-localized with the endothelial cell marker CD31 (Figure 5C). Aspirin, but not clopidogrel, increased the expression of endothelial netrin-1 within the arterial wall, compared with that in untreated animals (Figure 5B, C). HDAC activity, as measured in nuclear extracts, was lower in the aortic wall of both aspirin- and clopidogrel-treated animals than control animals (Figure 5D), while aortic HAT activity was higher in the aspirin-treated mice only (Figure 5E). As a consequence, both anti-platelet drugs reduced the HDAC/HAT ratio, although aspirin treatment did so to a greater extent than clopidogrel (Figure 5F).
Figure 5.

Aspirin (ASA) increases systemic and vascular expression of netrin-1 in ApoE−/− mice. (A) elisa of netrin-1 in plasma, from ApoE−/− mice on 8 week HFD, either untreated (8 weeks; n = 8) or treated with ASA (5 mg·kg−1·day−1; n = 8) or clopidogrel (Clop; 25 mg·kg−1·day−1; n = 8). Also shown in (A) are plasma netrin-1 levels in ApoE−/− mice after 4 weeks of HFD (4 weeks; n = 4) for comparison. (B–C) The micrographs show immunofluorescence staining for netrin-1 (red), the endothelial marker CD31 (green) and nuclei by DAPI (blue) of aortas from ApoE−/− mice after 8 week HFD either untreated (left) or ASA treated (right, ASA 5 mg·kg−1·day−1); magnification = 20× (scale bars = 50 μm). Bottom panels show merging of the three stainings: the white arrows indicate the luminal localization of netrin-1 (red) within the arterial wall and its co-localization with the endothelial cell marker CD31 (green), which is evident in the ASA-treated group but not in either clopidogrel-treated or untreated mice. The graph reports quantification of mean fluorescence intensity for netrin-1 and CD31 in the different groups as specified, and it is reported as fold change versus control (untreated) animals. (D) and (E) report HDAC and HAT activities measured in nuclear extracts isolated from the aortic arterial wall of ApoE−/− mice at the end of the 8 week HFD period, either untreated (8 weeks) or treated with ASA (5 mg·kg−1·day−1) or clopidogrel (25 mg·kg−1·day−1). Also shown in (F) is the HDAC/HAT ratio in the different groups. Data for (A) are shown as median ± interquartile ranges. *P < 0.05 versus 8 weeks.
Figure 6.

Effect of aspirin (ASA) and clopidogrel on monocyte/macrophage plaque content. (A, B) Representative pictures of Oil Red O(ORO) (A) and CD68 (B) staining of cryosections of the brachiocephalic artery (BA) of ApoE−/− mice at the end of 8 week HFD in clopidogrel (25 mg·kg−1·day−1) or ASA-treated animals (ASA: 5 mg·kg−1·day−1) as indicated (magnification 20× and 40× on the left and right sides of each panel respectively). Counterstaining was with haematoxylin. Black arrows indicate the atherosclerotic lesion detected on histological section. Total and ORO-positive area of plaques under the different experimental conditions is reported in (C) (scale bars = 150 μm lower power; 50 μm higher power). (D) Total myeloid cell count [F4/80+ CD115+ Lin- (B220, CD90, Ly6G, NK1.1)], number of monocytes [F4/80low CD115+ Lin- (B220, CD90, Ly6G, NK1.1)] and macrophages [F4/80high CD115+ Lin- (B220, CD90, Ly6G, NK1.1)] within the BA in untreated animals (8 weeks; n = 6) and those treated with either clopidogrel (25 mg·kg−1·day−1; n = 6) or ASA (5 mg·kg−1·day−1; n = 5). Values are indicated as cell number/mg tissue. Relative prevalence of monocytes (F4/80low cells, dark grey) and macrophages (F4/80low cells, light grey) is reported in the pie charts and expressed as percentage of total myeloid cells [F4/80+ CD115+ Lin- (B220, CD90, Ly6G, NK1.1)]. (E) Representative flow cytometry plots showing gating strategy for monocyte/macrophage characterization in the BA of each group of animals. Briefly, total myeloid cells were identified within the Lin− gate as CD115+ F4/80+ cells. Within this population, monocytes (F4/80low; left quadrants in the dot plots indicating Ly6C vs. F4/80) and macrophages (F4/80high and Ly6Clow cells; lower right quadrant in the dot plots indicating Ly6C vs. F4/80) were differentiated on the basis of intensity of fluorescence for F4/80 staining. Total monocytic population was further distinguished into Ly6Chigh and Ly6Clow cells (bottom plots). The percentage shown in the dot plots refers to the frequency of gated cells within the parent population. In the contour plots, the relative prevalence of either monocytes (Lin− CD115+ F4/80low) and macrophages (Lin− CD115+ F4/80low Ly6Clow) or Ly6Chigh and Ly6Clow monocytes is shown. (F) Ly6Chigh and Ly6Clow monocyte count within the BA in the different animal groups as indicated. Values are indicated as cell number/mg tissue. Pie charts show accumulated data (8 weeks: n = 4; clopidogrel: n = 4; ASA: n = 5) reporting percentage of Ly6Chigh (dark grey) and Ly6Clow (light grey) cells over total monocytes [F4/80low CD115+ Lin- (B220, CD90, Ly6G, NK1.1)] measured in the BA of each group of animals. *P < 0.05 versus 8 weeks.
ApoE−/− mice on HFD, either untreated or treated with these anti-platelet drugs, developed atherosclerotic plaques in the brachiocephalic arteries. However, treatment with aspirin reduced the total plaque area compared with control animals (Figure 6A–C). By contrast, clopidogrel had no detectable effect on plaque extent (Figure 6A–C). Moreover, plaques in mice receiving aspirin, but not clopidogrel, displayed a reduction in lipid content, as evaluated by the area stained with Oil-Red O (Figure 6A–C), as well as in myeloid cell content, as determined by CD68 staining (Figure 6B). Suppression of infiltration of atherosclerotic lesions by inflammatory cells in the aspirin group was confirmed by tissue flow cytometry to specifically assess the content of different subsets of monocytes/macrophages. Indeed, the brachiocephalic arteries obtained from ApoE−/− mice treated with aspirin, but not clopidogrel, exhibited less total myeloid cell infiltration compared with untreated animals, with a reduction in both the monocyte (F4/80low) and macrophage (F4/80high) components (Figure 6D). Within the monocytic population, aspirin was able to suppress vascular infiltration by both Ly6Chigh and Ly6Clow subtypes (Figure 6F). Clopidogrel exerted a weaker action than aspirin on monocyte arterial infiltration (Figure 6D), specifically reducing the content of Ly6Clow cells only (Figure 6F), thus leading to a slight modulation of the prevalence between the two monocyte subpopulations within the arterial wall compared with untreated animals, with predominance of Ly6Chigh over Ly6Clow cells in the clopidogrel group (Figure 6F). Plaques from untreated or clopidogrel-treated mice exhibited a similar content of both Ly6Chigh monocytes (Figure 6F) and macrophages (Figure 6D).
Anti-platelet therapy abolishes atherosclerosis-related blood monocytosis
We next evaluated whether the reduced monocyte/macrophage plaque content in aspirin-treated animals was related to a systemic anti-inflammatory action of the drug on peripheral monocyte count and phenotype. In accordance with previous reports, we found a time-dependent expansion of total monocytes in the peripheral blood of ApoE−/− mice fed with a HFD over the 8 week period of study (Figure 7A, C). The predominant subtype in the blood was Ly6Chigh throughout the study period (Figure 7A, F), although the count and percentage of Ly6Clow monocytes also increased over time (Figure 7A, F). The increase in Ly6Chigh monocytes was attributable to the HFD regime, because a similar expansion of this particular subset of monocytes was observed in C57BL/6J animals fed with a HFD (Supporting Information Fig. S3a), even in the absence of atherosclerosis development. ApoE−/− mice, but not the wild-type strain, developed a concomitant progressive increase in circulating Ly6Clow monocytes (Supporting Information Fig. S3b) and, in agreement with previous findings (Potteaux et al., 2011), their percentage was strongly and directly related to plaque size in the brachiocephalic artery. Conversely, the level of Ly6Chigh was inversely related to lesion area, and no relationship was found between total monocytes and plaque area (Supporting Information Fig. S3c–e).
Figure 7.

Effect of aspirin (ASA) on blood monocytosis and monocyte trafficking into atherosclerotic lesions. (A) and (B) display monocyte count (SSClow B220− CD115+ F4/80+ cells) and number of both Ly6Chigh and Ly6Clow subsets in the peripheral blood of ApoE−/− mice on HFD at different time points as indicated, either with or without concomitant treatment with ASA (5 mg·kg−1·day−1) or clopidogrel (25 mg·kg−1·day−1). n = 4–8. (C) and (D) show the prevalence of total monocytes (reported as percentage over SSClow B220− mononuclear cells) in the blood of the different animal groups as indicated, and the prevalence of circulating Ly6Chigh (light grey) and Ly6Clow cells (dark grey) is reported in the pie charts in (F) (calculated as percentage of total monocytes). (E) Degree of platelet activation as measured by percentage of P-selectin-expressing platelets in the circulation under the different experimental conditions. *P < 0.05 versus 8 weeks. (G) Autoradiographic exposure of the brachiocephalic arteries from ApoE−/− mice on HFD and ASA treatment (5 mg·kg−1·day−1) for 8 weeks, excised 36 h after adoptive transfer of 111In-labelled monocytes, either injected with Un5cb-Fc antibody (left) or isotype control (right) 1 h before receiving labelled monocytes. Exposure of tissue plates was for 12 h. Dark spots in the grey-scale images (left) and red spots in the colour images (right), as indicated by the black arrows, indicate cell accumulation and localization within the brachiocephalic artery. The corresponding signal was quantified and reported as relative recruitment of cells (labelled monocytes injected into animals receiving antibody isotype control scaled to 100%). n = 3 per group. *P < 0.05 versus isotype control. (H) Representative flow cytometry dot plots indicating the gating strategy and analysis for blood monocyte characterization in the different animal groups as indicated. Briefly, B220 expression was evaluated within the mononuclear blood population only. B220+ cells were excluded and further analysis was performed on the population of B220− cells (prevalence of these within the mononuclear cell population is indicated). Within the SSClow B220− mononuclear cell gate, the expression of CD115 and F4/80 was studied to identify CD115+F4/80+ events representative of the total monocytic population. Expression of Ly6C within the SSClow B220− CD115+ F4/80+ cells was carried out to calculate the percentage and number of Ly6Chigh and Ly6Clow monocytes (upper and lower right quadrants, respectively, in the bottom plots).
Atherosclerosis-related total monocytosis in ApoE−/− mice at the end of the 8 week HFD regimen was significantly reduced by concomitant treatment with either clopidogrel or aspirin (Figure 7B–D, H). The distribution pattern of the different monocytic subsets was also modulated by anti-platelet treatment, as the progressive increase in percentage and count of Ly6Clow over Ly6Chigh cells was attenuated by both anti-platelet therapies, although not to baseline values (Figure 7F). Measurement of P-selectin-expressing platelets, as a marker of in vivo platelet activation, showed that both aspirin and clopidogrel inhibited the platelet P-selectin expression in ApoE−/− mice on HFD, clopidogrel doing so to a greater extent than aspirin (Figure 7E).
Aspirin reduces monocyte trafficking within atherosclerotic lesions through modulation of systemic netrin-1
The results described above suggested that the greater action of aspirin over clopidogrel on atherosclerotic plaque size and composition in ApoE−/− mice fed with a HFD was independent of its inhibitory effect on platelet activity and consequent suppression of blood monocytosis, as a similar modulation of these parameters was exerted by clopidogrel (Figure 7A–F). Because we observed that the two anti-platelet drugs differed substantially in their ability to regulate systemic and vascular endothelial expression of netrin-1 (Figure 6), we postulated that aspirin-mediated suppression of monocyte arterial accumulation could be mediated through its effects on netrin-1. In order to test this, we examined adoptively transferred monocyte trafficking in the brachiocephalic artery of ApoE−/− mice fed with a HFD and co-treated with aspirin for 8 weeks in vivo, either in the presence or absence of netrin-1 blockade using Unc5b-Fc chimera antibody or isotype control respectively. Atherosclerotic vessels of aspirin-treated ApoE−/− mice, harvested 36 h after injection of exogenous radiolabelled monocytes and assessed by autoradiography, exhibited an increase in monocyte accumulation in those receiving the netrin-1 blocking antibody compared with those receiving the isotype control (Figure 7G).
Discussion and conclusions
Aspirin and clopidogrel are widely used for cardiovascular prophylaxis. Our demonstration that both drugs suppressed the blood monocytosis that accompanies atheroma development in ApoE−/− mice provides good evidence for a systemic anti-inflammatory effect of both anti-platelet drugs. However, the fact that only aspirin was able to reduce plaque size and inflammation suggests that this drug exerts beneficial effects on atherogenesis over and above its anti-platelet action. Our data both in vitro and in vivo suggest an important role in this regard for netrin-1 modulation, whose up-regulation by aspirin at a systemic level leads to reduced arterial accumulation of monocytes.
Aspirin-dependent down-regulation of several endothelial molecules involved in monocyte trafficking within the atheroma has been demonstrated previously (Weber et al., 1995; Pierce et al., 1996; Dunoyer-Geindre et al., 2004; Yang et al., 2004). However, our data showing a modulatory activity of aspirin on endothelial-derived netrin-1 and the key role of secreted netrin-1 in preventing monocyte vascular recruitment are entirely novel. In vitro, we found that the sole blockade of netrin-1 was able to entirely abolish the monocyte chemorepellent action of activated endothelial cells induced by aspirin, irrespective of any potential effect of the drug on other endothelial-derived adhesive/chemotactic molecules. Similarly, our in vivo experiments of adoptively transferred monocytes in ApoE−/− mice demonstrated that inhibition of netrin-1 enhances monocyte trafficking into atherosclerotic lesions in aspirin-treated animals. Moreover, we found that up-regulation of netrin-1 during atherosclerosis development, in response to aspirin treatment, was not detrimental for plaque progression, but rather beneficial and was associated with a reduction in the inflammatory component of lesions (comprising both monocyte and macrophage content) that was not observable with clopidogrel treatment. In accordance with our findings, it has previously been reported that plaque development can be delayed in a murine model of atherosclerosis by netrin-1 gene therapy (Khan et al., 2011). On the other hand, van Gils et al. (2012) proposed selective down-regulation of haematopoietic netrin-1 as a strategy to favour monocyte/macrophage egress from plaques, with a consequent putative beneficial effect on atheroma development. Such a strategy may certainly be relevant in the later stages of disease, where large numbers of myeloid cells have already accumulated in the plaque. However, the evidence from the present study suggests that, at least in the earlier stages of atherosclerosis, enhancement of vascular netrin-1 expression (reportedly down-regulated under pro-atherogenic conditions; van Gils et al., 2013) may prevent monocyte infiltration and macrophage accumulation in nascent plaques and hence reduce their size and lipid content. As Ly6Chigh monocytes appear to be the main precursors of plaque macrophages (Swirski et al., 2007), it is likely that the ability of aspirin to decrease atherosclerosis progression is primarily due to its negative modulatory action on arterial accumulation of this particular subset of cells. Indeed, clopidogrel, which also reduced the Ly6Clow but not the Ly6Chigh cell component in plaques, albeit with less efficacy than aspirin, did not exert any beneficial effect on plaque size, macrophage or lipid content.
As discussed above, aspirin is known to modulate the endothelial expression of adhesion molecules. We have not investigated this here and so cannot exclude the possibility that arterial infiltration of monocytes may have been affected by a concomitant pharmacological action of aspirin on endothelial-derived molecules other than netrin-1. Having said that, clopidogrel also exerts anti-inflammatory properties on the vasculature (Li et al., 2007; Hoving et al., 2011), but in our study clopidogrel was less effective than aspirin in reducing the monocyte/macrophage component of arterial lesions.
Our data also provide entirely novel insights on the mechanism(s) by which aspirin regulates endothelial netrin-1. We have demonstrated that two different isoforms of netrin-1, full length and truncated, were expressed by the vascular endothelium, and that these exhibited important differences in their gene regulation under either physiological or pro-inflammatory conditions. Specifically, we demonstrated that TNF-α modified the balance between the two isoforms of netrin-1, in terms of both gene expression and protein production, leading to NF-κB-dependent preferential transcription of truncated netrin-1 over full-length netrin-1, with consequent over-expression of the protein within nuclei and concomitant reduction in the synthesis of secreted netrin-1. The NF-κB inhibitor Bay 11-6075 reduced the TNF-α-dependent over-expression of nuclear netrin-1 and restored the balance between the two isoforms, both in terms of transcript level and protein production. Aspirin, salicylic acid, indomethacin and TSA were all able to prevent the nuclear netrin-1 increase induced by TNF-α, attributable to their ability to suppress NF-κB activation and consequent gene transcription of the truncated protein. However, these compounds also directly modified the level of secreted netrin-1, although to different extents. Unlike the NF-κB-dependent transcription of the nuclear isoform, we found that gene induction of full-length secreted netrin-1 was NF-κB independent and that that chromatin remodelling, as caused by increased histone acetylation in aspirin/salicylic acid or TSA-treated cells, enhanced its synthesis at the transcriptional level. The COX inhibition exerted by aspirin is likely to be responsible as indomethacin exerted a similar action to aspirin in terms of NF-κB inhibition, histone acetylation and netrin-1 release into HUVEC supernatant. The action of TSA, an HDAC inhibitor, on netrin-1 was entirely COX independent. Although we found that TSA reduced PGE2 synthesis in response to TNF-α treatment, such an effect was most likely because of the NF-κB inhibitory action of TSA in TNF-α-treated cells, with consequent suppression of the NF-κB-dependent gene induction of COX-2. Of note, unlike aspirin, TSA increased the production of full-length netrin-1 in endothelial cells either in the presence or absence of TNF-α, most likely because of the extent of H3 acetylation that was considerably higher, compared with that observed in aspirin or salicylic acid-treated HUVECs. This resulted in a level of gene transcription of the secreted isoform of netrin-1 that was 40 times higher than untreated cells, more than 10 times the increase observed with aspirin or indomethacin. The relevance of histone acetylation in the context of atherosclerosis has been previously reported, such that TSA retards atherosclerosis development (Zampetaki et al., 2010; Zhou et al., 2011) by acting on endothelial function (Inoue et al., 2006; Wang et al., 2007). It is known that histone acetylation enables access to genes by transcription factors. Our data suggest that aspirin alters the balance between histone acetylation and deacetylation in favour of the former, thereby promoting transcription of full-length netrin-1 in TNF-α-treated cells.
The fact that aspirin alone was not able to induce any significant change in the level of netrin-1 and of acetylated H3, compared with untreated cells is highly suggestive of a concerted action of TNF-α and aspirin co-treatment on both these events. In keeping with this, our study of HAT and HDAC activities revealed that aspirin augmented the increase in HAT activity induced by TNF-α alone, most likely because of a direct effect on HAT activity as demonstrated in nuclear extracts. The simultaneous suppression of HDAC, albeit through an indirect action of aspirin on the enzyme, resulted in an imbalance between histone acetylation and deacetylation. In our in vivo model, study of HAT and HDAC activities showed that systemic inflammation, and particularly platelet activity, was crucial in determining the status of histone acetylation within the arterial wall. Although acting through different pharmacological mechanisms, the anti-platelet effect exerted by clopidogrel or aspirin was associated with roughly equal attenuation of arterial HDAC activity, thus demonstrating the crucial role played by platelet activation on this parameter. However, the concomitant increase in HAT activity exerted by aspirin was not seen in clopidogrel-treated animals, thereby giving rise to a significant modification of the HDAC/HAT ratio only in the group of mice treated with aspirin. In support of our in vitro data showing the relevance of histone acetylation to secreted netrin-1 synthesis, we observed that blood and endothelial netrin-1 levels were increased in aspirin- but not clopidogrel-treated animals in vivo.
In conclusion, we have provided evidence for selective and differential regulation of the secreted and nuclear isoforms of netrin-1 in endothelial cells. The suppression of endothelial-derived netrin-1 under pro-inflammatory conditions only relates to the secreted isoform of the protein, with consequent enhancement of arterial infiltration by monocytes. Aspirin increases netrin-1 synthesis by the vascular endothelium. This effect, which was COX dependent, was mediated by an epigenetic regulation of netrin-1 that occurred only in the presence of a pro-inflammatory endothelial state. As a result, aspirin preserved the synthesis of secreted netrin-1 under pro-inflammatory conditions, thus counteracting monocyte/macrophage plaque accumulation. Future clinical investigations are needed to verify the applicability of these findings to human atherosclerosis, as well as to non-cardiovascular diseases including neurological disorders (Rajasekharan and Kennedy, 2009; Izzi and Charron, 2011) and cancer (Delloye-Bourgeois et al., 2012; Kefeli et al., 2012; Ko et al., 2012) where aspirin has also been reported to exert protective actions (Algra and Rothwell, 2012; Rothwell et al., 2014,b2012a).
Author contributions
G. P. and A. F. developed the hypothesis and experimental design, analysed the data and wrote the manuscript. G. P. performed the experimental work. C. W. contributed to the in vitro work. A. P., M. C., M. E. A. and R. M. B. contributed to the in vivo work. B. L. and A. A. contributed to the histological analyses. All authors contributed to the writing of the manuscript.
Conflict of interest
The authors declare no competing financial interests.
Glossary
- H3
histone 3
- HAoEC
human aortic endothelial cell
- HAT
histone acetyltransferase
- HDAC
histone deacetylase
- HFD
high-fat diet
- TSA
trichostatin A
Supporting Information
Figure S1 Characterization of netrin-1 isoforms synthesized by HUVECs.
{kind=link}
Figure S2 Expression of netrin-1 and NF-κB activation in HAoECs.
{kind=link}
Figure S3 Monocyte characterization in ApoE−/− and wild-type (ApoE+/+) mice.
{kind=link}
A full description of materials and methods used in the study is available in the Supplementary File.
References
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Enzymes. Br J Pharmacol. 2013;170:1797–1867. doi: 10.1111/bph.12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Algra AM, Rothwell PM. Effects of regular aspirin on long-term cancer incidence and metastasis: a systematic comparison of evidence from observational studies versus randomised trials. Lancet Oncol. 2012;13:518–527. doi: 10.1016/S1470-2045(12)70112-2. [DOI] [PubMed] [Google Scholar]
- Barnes PJ, Adcock IM, Ito K. Histone acetylation and deacetylation: importance in inflammatory lung diseases. Eur Respir J. 2009;25:552–563. doi: 10.1183/09031936.05.00117504. [DOI] [PubMed] [Google Scholar]
- Delloye-Bourgeois C, Goldschneider D, Paradisi A, Therizols G, Belin S, Hacot S, et al. Nucleolar localization of a netrin-1 isoform enhances tumor cell proliferation. Sci Signal. 2012;5:ra57. doi: 10.1126/scisignal.2002456. [DOI] [PubMed] [Google Scholar]
- Dunoyer-Geindre S, Kruithof EK, Boehlen F, Satta-Poschung N, Reber G, de Moerloose P. Aspirin inhibits endothelial cell activation induced by antiphospholipid antibodies. J Thromb Haemost. 2004;2:1176–1181. doi: 10.1111/j.1538-7836.2004.00801.x. [DOI] [PubMed] [Google Scholar]
- Gerszten RE, Tager AM. The monocyte in atherosclerosis–should I stay or should I go now? Engl J Med. 2012;366:1734–1736. doi: 10.1056/NEJMcibr1200164. [DOI] [PubMed] [Google Scholar]
- van Gils JM, Derby MC, Fernandes LR, Ramkhelawon B, Ray TD, Rayner KJ, et al. The neuroimmune guidance cue netrin-1 promotes atherosclerosis by inhibiting the emigration of macrophages from plaques. Nat Immunol. 2012;13:136–143. doi: 10.1038/ni.2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Gils JM, Ramkhelawon B, Fernandes L, Stewart MC, Guo L, Seibert T, et al. Endothelial expression of guidance cues in vessel wall homeostasis dysregulation under proatherosclerotic conditions. Arterioscler Thromb Vasc Biol. 2013;33:911–919. doi: 10.1161/ATVBAHA.112.301155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harter PN, Zinke J, Scholz A, Tichy J, Zachskorn C, Kvasnicka HM, et al. Netrin-1 expression is an independent prognostic factor for poor patient survival in brain metastases. PLoS ONE. 2014;9:e92311. doi: 10.1371/journal.pone.0092311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoving S, Heeneman S, Gijbels MJ, te Poele JA, Pol JF, Gabriels K, et al. Anti-inflammatory and anti-thrombotic intervention strategies using atorvastatin, clopidogrel and knock-down of CD40L do not modify radiation-induced atherosclerosis in ApoE null mice. Radiother Oncol. 2011;101:100–108. doi: 10.1016/j.radonc.2011.09.019. [DOI] [PubMed] [Google Scholar]
- Inoue K, Kobayashi M, Yano K, Miura M, Izumi A, Mataki C, et al. Histone deacetylase inhibitor reduces monocyte adhesion to endothelium through the suppression of vascular cell adhesion molecule-1 expression. Arterioscler Thromb Vasc Biol. 2006;26:2652–2659. doi: 10.1161/01.ATV.0000247247.89787.e7. [DOI] [PubMed] [Google Scholar]
- Izzi L, Charron F. Midline axon guidance and human genetic disorders. Clin Genet. 2011;80:226–234. doi: 10.1111/j.1399-0004.2011.01735.x. [DOI] [PubMed] [Google Scholar]
- Kefeli U, Yildirim ME, Aydin D, Madenci OC, Yasar N, Sener N, et al. Netrin-1 concentrations in patients with advanced gastric cancer and its relation with treatment. Biomarkers. 2012;17:663–667. doi: 10.3109/1354750X.2012.709882. [DOI] [PubMed] [Google Scholar]
- Khan JA, Cao M, Kang BY, Liu Y, Mehta JL, Hermonat PL. Systemic human Netrin-1 gene delivery by adeno-associated virus type 8 alters leukocyte accumulation and atherogenesis in vivo. Gene Ther. 2011;18:437–444. doi: 10.1038/gt.2010.155. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. Animal research: Reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kircher MF, Grimm J, Swirski FK, Libby P, Gerszten RE, Allport JR, et al. Noninvasive in vivo imaging of monocyte trafficking to atherosclerotic lesions. Circulation. 2008;117:388–395. doi: 10.1161/CIRCULATIONAHA.107.719765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko SY, Dass CR, Nurgali K. Netrin-1 in the developing enteric nervous system and colorectal cancer. Trends Mol Med. 2012;18:544–554. doi: 10.1016/j.molmed.2012.07.001. [DOI] [PubMed] [Google Scholar]
- Li M, Zhang Y, Ren H, Zhang Y, Zhu X. Effect of clopidogrel on the inflammatory progression of early atherosclerosis in rabbits model. Atherosclerosis. 2007;194:348–356. doi: 10.1016/j.atherosclerosis.2006.11.006. [DOI] [PubMed] [Google Scholar]
- Ly NP, Komatsuzaki K, Fraser IP, Tseng AA, Prodhan P, Moore KJ, et al. Netrin-1 inhibits leukocyte migration in vitro and in vivo. Proc Natl Acad Sci U S A. 2005;102:14729–14734. doi: 10.1073/pnas.0506233102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinsey TA. Therapeutic potential for HDAC inhibitors in the heart. Annu Rev Pharmacol Toxicol. 2012;52:303–319. doi: 10.1146/annurev-pharmtox-010611-134712. [DOI] [PubMed] [Google Scholar]
- Mirakaj V, Thix CA, Laucher S, Mielke C, Morote-Garcia JC, Schmit MA, et al. Netrin-1 dampens pulmonary inflammation during acute lung injury. Am J Respir Crit Care Med. 2010;181:815–824. doi: 10.1164/rccm.200905-0717OC. [DOI] [PubMed] [Google Scholar]
- Paradisi A, Maisse C, Bernet A, Coissieux MM, Maccarrone M, Scoazec JY, et al. NF-kappaB regulates netrin-1 expression and affects the conditional tumor suppressive activity of the netrin-1 receptors. Gastroenterology. 2008;135:1248–1257. doi: 10.1053/j.gastro.2008.06.080. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, et al. NC-IUPHAR. The IUPHAR/BPS Guide to PHARMACOLOGY: an expert-driven knowledge base of drug targets and their ligands. Nucl. Acids Res. 2014;42(Database Issue):D1098–D1106. doi: 10.1093/nar/gkt1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce JW, Read MA, Ding H, Luscinskas FW, Collins T. Salicylates inhibit I kappa B-alpha phosphorylation, endothelial-leukocyte adhesion molecule expression, and neutrophil transmigration. J Immunol. 1996;156:3961–3969. [PubMed] [Google Scholar]
- Pons D, de Vries FR, van den Elsen PJ, Heijmans BT, Quax PH, Jukema JW. Epigenetic histone acetylation modifiers in vascular remodelling: new targets for therapy in cardiovascular disease. Eur Heart J. 2009;30:266–277. doi: 10.1093/eurheartj/ehn603. [DOI] [PubMed] [Google Scholar]
- Potteaux S, Gautier EL, Hutchison SB, van Rooijen N, Rader DJ, Thomas MJ, et al. Suppressed monocyte recruitment drives macrophage removal from atherosclerotic plaques of Apoe-/- mice during disease regression. J Clin Invest. 2011;121:2025–2036. doi: 10.1172/JCI43802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajasekharan S, Kennedy TE. The netrin protein family. Genome Biol. 2009;10:239–243. doi: 10.1186/gb-2009-10-9-239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramkhelawon B, Hennessy EJ, Ménager M, Ray TD, Sheedy FJ, Hutchison S, et al. Netrin-1 promotes adipose tissue macrophage retention and insulin resistance in obesity. Nat Med. 2014;20:377–384. doi: 10.1038/nm.3467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothwell PM, Wilson M, Price JF, Belch JF, Meade TW, Mehta Z. Effect of daily aspirin on risk of cancer metastasis: a study of incident cancers during randomised controlled trials. Lancet. 2012b;379:1591–1601. doi: 10.1016/S0140-6736(12)60209-8. [DOI] [PubMed] [Google Scholar]
- Rothwell PM, Price JF, Fowkes FG, Zanchetti A, Roncaglioni MC, Tognoni G, et al. Short-term effects of daily aspirin on cancer incidence, mortality, and non-vascular death: analysis of the time course of risks and benefits in 51 randomised controlled trials. Lancet. 2012a;379:1602–1612. doi: 10.1016/S0140-6736(11)61720-0. [DOI] [PubMed] [Google Scholar]
- Swirski FK, Libby P, Aikawa E, Alcaide P, Luscinskas FW, Weissleder R, et al. Ly-6Chi monocytes dominate hypercholesterolemia-associated monocytosis and give rise to macrophages in atheromata. J Clin Invest. 2007;117:195–205. doi: 10.1172/JCI29950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tadagavadi RK, Wang W, Ramesh G. Netrin-1 regulates Th1/Th2/Th17 cytokine production and inflammation through UNC5B Receptor and protects kidney against ischemia–reperfusion injury. J Immunol. 2010;185:3750–3758. doi: 10.4049/jimmunol.1000435. [DOI] [PubMed] [Google Scholar]
- Wang J, Mahmud SA, Bitterman PB, Huo Y, Slungaard A. Histone deacetylase inhibitors suppress TF-kappaB-dependent agonist-driven tissue factor expression in endothelial cells and monocytes. J Biol Chem. 2007;282:28408–28418. doi: 10.1074/jbc.M703586200. [DOI] [PubMed] [Google Scholar]
- Weber C, Erl W, Pietsch A, Weber PC. Aspirin inhibits nuclear factor kappa B mobilization and monocyte adhesion in stimulated human endothelial cells. Circulation. 1995;91:1914–1917. doi: 10.1161/01.cir.91.7.1914. [DOI] [PubMed] [Google Scholar]
- Yang YY, Hu CJ, Chang SM, Tai TY, Leu SJ. Aspirin inhibits monocyte chemoattractant protein-1 and interleukin-8 expression in TNF-alpha stimulated human umbilical vein endothelial cells. Atherosclerosis. 2004;174:207–213. doi: 10.1016/j.atherosclerosis.2004.01.024. [DOI] [PubMed] [Google Scholar]
- Zampetaki A, Zeng L, Margariti A, Xiao Q, Li H, Zhang Z, et al. Histone deacetylase 3 is critical in endothelial survival and atherosclerosis development in response to disturbed flow. Circulation. 2010;121:132–142. doi: 10.1161/CIRCULATIONAHA.109.890491. [DOI] [PubMed] [Google Scholar]
- Zhou B, Margariti A, Zeng L, Xu Q. Role of histone deacetylases in vascular cell homeostasis and arteriosclerosis. Cardiovasc Res. 2011;90:413–420. doi: 10.1093/cvr/cvr003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Characterization of netrin-1 isoforms synthesized by HUVECs.
Figure S2 Expression of netrin-1 and NF-κB activation in HAoECs.
Figure S3 Monocyte characterization in ApoE−/− and wild-type (ApoE+/+) mice.
A full description of materials and methods used in the study is available in the Supplementary File.