

Abstract
Background and Purpose
Epigallocatechin-3-gallate (EGCG) is a component of green tea known to have chemo-preventative effects on several cancers. However, EGCG has limited clinical application, which necessitates the development of a more effective EGCG prodrug as an anticancer agent.
Experimental Approach
Derivatives of EGCG were evaluated for their stability and anti-tumour activity in human chronic myeloid leukaemia (CML) K562 and KBM5 cells.
Key Results
EGCG-mono-palmitate (EGCG-MP) showed most prolonged stability compared with other EGCG derivatives. EGCG-MP exerted greater cytotoxicity and apoptosis in K562 and KBM5 cells than the other EGCG derivatives. EGCG-MP induced Src-homology 2 domain-containing tyrosine phosphatase 1 (SHP-1) leading decreased oncogenic protein BCR-ABL and STAT3 phosphorylation in CML cells, compared with treatment with EGCG. Furthermore, EGCG-MP reduced phosphorylation of STAT3 and survival genes in K562 cells, compared with EGCG. Conversely, depletion of SHP-1 or application of the tyrosine phosphatase inhibitor pervanadate blocked the ability of EGCG-MP to suppress phosphorylation of BCR-ABL and STAT3, and the expression of survival genes downstream of STAT3. In addition, EGCG-MP treatment more effectively suppressed tumour growth in BALB/c athymic nude mice compared with untreated controls or EGCG treatment. Immunohistochemistry revealed increased caspase 3 and SHP-1 activity and decreased phosphorylation of BCR-ABL in the EGCG-MP-treated group relative to that in the EGCG-treated group.
Conclusions and Implications
EGCG-MP induced SHP-1-mediated inhibition of BCR-ABL and STAT3 signalling in vitro and in vivo more effectively than EGCG. This derivative may be a potent chemotherapeutic agent for CML treatment.
Tables of Links
| TARGETS | |
|---|---|
| Enzymes | |
| BCR-ABL kinase | |
| Caspase 3 | |
| Caspase 8 | |
| Caspase 9 | |
| Cdk4, cyclin-dependent kinase 4 | |
| PARP, poly(ADP-ribose)polymerase |
| LIGANDS | |
|---|---|
| Curcumin | |
| Epigallocatechin-3-gallate | |
| Imatinib | |
| Resveratrol |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (Alexander et al., 2013).
Introduction
Chronic myeloid leukaemia (CML) is a myeloproliferative disorder defined as the uncontrolled amplification of granulocytic cells that do not have the ability to differentiate. The BCR-ABL translocation, known as the Philadelphia chromosome, is a characteristic of CML that results from a specific chromosomal abnormality (Rangatia and Bonnet, 2006). The BCR-ABL protein has tyrosine kinase activity and activates survival genes, such as Mcl-1 and survivin, and cell cycle regulator genes such as cyclin E, cdk2, cdk4 and p27 via BCR-ABL/STAT3 activation in CML cells (Aichberger et al., 2005; Coppo et al., 2006).
The protein tyrosine phosphatases (PTPs), including the non-receptor PTP, SHP-1 or PTP1C, are key regulators of cytokine/protein tyrosine kinase-induced signalling (Jiao et al., 1996; Paulson et al., 1996; Tonks and Neel, 1996; Liedtke et al., 1998). There is growing evidence that SHP-1 negatively regulates BCR-ABL activity, which suppresses CML in vitro and in vivo (Liedtke et al., 1998; Amin et al., 2007). The STAT3 pathway is another well-documented SHP-1 regulatory signal known to be oncogenic in a variety of human cancers (Gouilleux-Gruart et al., 1996; Chai et al., 1997; Garcia et al., 1997; Bromberg et al., 1999). Thus, STAT3 activation is an important factor mediating the pathogenesis of acute myeloid leukaemia (Chai et al., 1997) and SHP-1 physically interacts with JAK3 to inhibit JAK3/STAT3 signalling in anaplastic large-cell lymphoma (Han et al., 2006a,b,). Thus, targeting STAT3 via SHP-1 is regarded as a promising strategy for cancer therapy (Lim et al., 2000).
Epigallocatechin-3-gallate (EGCG), a major component of green tea polyphenols, has antioxidant (Hu et al., 2009), anti-inflammatory (Peairs et al., 2010), and anti-tumour (Davenport et al., 2010; Wang et al., 2011; Lee et al., 2012; Tang et al., 2012) activity in various cancer cells through the regulation of major cell signalling proteins such as the EGF receptor (Masuda et al., 2001; Ma et al., 2014) and STAT3. EGCG interacts directly with the STAT3 SH2 domain (Wang et al., 2013) to stimulate caspase activation (Gupta et al., 2004). In spite of these clearly beneficial effects in vitro, EGCG has not been successful in clinical trials because of its poor membrane permeability, low chemical stability and rapid metabolism (Mereles and Hunstein, 2011). In addition, the molecular mechanisms mediating EGCG regulation of BCR-ABL and STAT3 signalling remain to be elucidated.
In the current study, the anti-tumour mechanisms of EGCG and some of its derivatives, including; EGCG-mono-octanoate (EGCG-MO), EGCG-mono-laurate (EGCG-ML) and EGCG-mono-palmitate (EGCG-MP), were compared in human CML K562 and KBM5 cells as EGCG prodrugs, with improved efficacy and stability.
Methods
Chemicals
EGCG was purchased from Sigma (St. Louis, MO, USA). The EGCG derivatives, EGCG-MO, EGCG-ML and EGCG-MP were prepared by lipase-catalysed trans-esterification according to a method reported previously (Matsumura et al., 2008). EGCG and its derivatives were dissolved in DMSO, stored at −70°C, and diluted in culture medium for experiments.
Stability measurement of EGCG and EGCG derivatives using HPLC
EGCG and EGCG fatty acid derivatives were analysed by reverse-phase HPLC using a JASCO PU-2080 plus pump system (JASCO, Tokyo, Japan), a JASCO UV-2075 detector, a AS-2055 plus intelligent autosampler, a MX-2080-32 Mixer, a DG-2080-53 degasser, a GL Science Inertsil WP 300 C-18 column (150 × 4.6 mm; GL Sciences, Rolling Hills Estates, CA, USA), 5 μm C-18 column. Each compound was dissolved in DMSO and adjusted to a concentration of 64 mM. These solutions were serially diluted to 128 nM with 0.2% DMSO containing DMEM (Sigma-Aldrich Co., St. Louis, MO, USA). Each sample was incubated at 37°C and the aliquots were removed over time. HPLC analysis of each compound was performed using the mixture of solution A [0.01% trifluoroacetic acid (TFA) in water] and solution B (0.01% TFA in acetonitrile). Gradient for the analysis: 0 → 0% B in 3 min, 0 → 40% B in 18 min, 40 → 100% B in 22 min, 100% B for 4 min, 100 → 0% B in 28 min was used at a flow rate of 1.5 mL·min−1. Chromatographic peaks were detected with an UV detector at a wavelength of 265 nm.
Cell culture
K562 and KBM-5 human CML cells were purchased from the American Type Culture Collection, and maintained in RPMI1640 medium supplemented with 10% FBS, 2 μM L-glutamine and penicillin/streptomycin.
Cytotoxicity assay
Cytotoxicity of EGCG (Sigma), EGCG-MO, EGCG-ML and EGCG-MP were evaluated using a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. Cells were seeded onto 96-well microplates at a density of 2 × 104 per well and treated with various concentrations of EGCG, EGCG-MO, EGCG-ML and EGCG-MP (0, 5, 10, 20 or 40 μM) for 24 h. The cells were incubated with MTT solution (1 mg·mL−1) for 2 h and MTT lysis buffer (20% SDS and 50% dimethylformamide) overnight. OD was measured using a microplate reader (Molecular Devices Co., Sunnyvale, CA, USA) at 570 nm. Cell viability was calculated as a percentage of viable cells in EGCG and its derivatives-treated group versus untreated control using the following equation: cell viability(%) = [OD(drug) − OD(blank)]/[OD(control) − OD(blank)] × 100.
TUNEL assay
TUNEL assay was performed using the Dead End™ fluorometric TUNEL assay kit (Promega, Madison, WI, USA) according to the manufacturer's instructions. K562 cells were treated with 30 μM of EGCG and EGCG-MP for 24 h, plated onto a poly-l-lysine-coated slide and fixed in 4% methanol-free formaldehyde solution for 25 min at 4°C and washed twice with PBS for 5 min. The cells were then permeabilized in 0.2% Triton X-100 for 5 min and equilibrated in equilibration buffer. After removing the equilibration buffer, K562 cells were treated with a terminal deoxyribonucleotidyltransferase enzyme buffer containing fluorescein-12-dUTP for 1 h at 37°C in a dark humidified chamber then terminated by washing with 2× SSC for 15 min at room temperature. The slides were mounted with mounting medium containing DAPI (VECTOR, Burlingame, CA, USA) and visualized under an Axio vision 4.0 fluorescence microscope (Carl Zeiss, Inc., Weimar, Germany).
Western blotting
K5632 cells treated with 30 μM EGCG or 7.5, 15.0 and 30.0 μM EGCG-MP for 24 h were lysed in RIPA buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1% NP-40, 0.25% deoxycholic acid-Na, 1 M EDTA, 1 mM Na3VO4, 1 mM NaF and protease inhibitor cocktail). Protein samples were quantified using the Bio-Rad DC protein assay kit II (Bio-Rad, Hercules, CA, USA), separated by electrophoresis on 8–15% SDS-PAGE gels and electrotransferred onto a Hybond enhanced chemiluminescence (ECL) transfer membrane (Amersham Pharmacia, Piscataway, NJ, USA). After blocking with 1–5% non-fat skim milk, the membrane was probed with antibodies for phospho-IκBα, IκBα, cleaved caspase-3, cleaved caspase-9, PARP, Bcl-xL, Mcl-1L (Cell Signaling Technology, Danvers, MA, USA), cyclin D1, cyclin E, CDK2, CDK4, survivin, p21, lamin B (Santa Cruz Biotechnologies, Santa Cruz, CA, USA), and β-actin (Sigma-Aldrich Co.) and exposed to HRP-conjugated secondary anti-mouse or rabbit antibodies (AbD Serotec, Kidlington, UK). Protein expression was examined using an ECL system (Amersham Pharmacia).
Cell cycle analysis
K562 cells were exposed to 7.5, 15.0 and 30.0 μM EGCG-MP for 24 h and cell cycle analysis was performed by propidium iodide (PI) staining. Cells were fixed in 70% ethanol, incubated with 0.1% RNase A in PBS at 37°C for 30 min and resuspended in PBS containing 25 μg·mL−1 of PI for 30 min at room temperature. The stained cells were analysed for DNA content using a FACSCalibur instrument (Becton Dickinson, Franklin Lakes, NJ, USA) with the Cell Quest software (Becton Dickinson).
siRNA transfection assay
K562 cells were transiently transfected with a validated scrambled control siRNA or specific siRNA for SHP-1 (Santa Cruz Biotechnology, Santa Cruz, CA, USA) using Interferin™ transfection reagent (Polyplus-transfection Inc., New York, NY). Briefly, the mixture of siRNA and Interferin transfection reagent was incubated for 10 min, added to each well at an initial concentration of 40 nM and incubated at 37°C for 24 h before drug treatment.
K562 xenograft mouse model
All animal care and experimental procedures complied with the policy of the Animal Care and Use Committee of Kyung Hee University and was approved by them [Ref IRB; KHUASP(SE)-13-032]. All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010). A total of 15 animals were used in the experiments described here.
Four-week-old male BALB/c athymic nude mice (Daehan BioLink-DBL, Chungcheongbuk-do, Korea) were housed in an animal facility at 23 ± 2°C and 60 ± 10% humidity in a light-controlled (12 h, 07:00–19:00 h) environment. All materials were sterilized by UV light before treatment. K563 cells (1 × 107) were mixed with Matrigel (50%, in 200 μL; Becton Dickinson, Bedford, MA, USA) and s.c. injected into the right flank of 6-week-old mice. When the tumours reached a volume of 50 mm3, the mice were randomized into three groups (Control, EGCG and EGCG-MP; n = 4 per group) and injected i.p. with 15 mg·kg−1 of EGCG or EGCG-MP dissolved in 4% Tween-80 five times per week. The tumour volumes (length × width2 × 0.5236) and tumour weights were measured and body weights monitored twice per week for 3 weeks.
Immunohistochemistry (IHC)
IHC was performed in tumour sections using the indirect avidin/biotin-enhanced HRP method. Antigen retrieval was performed after deparaffinization and dehydration of the tissue sections by microwave for 10 min in 10 mM citrate buffer. Tumour sections were cooled on the bench for 30 min, treated with 3% hydrogen peroxide in methanol for 10 min, and blocked with 6% horse serum for 30 min at room temperature. Sections were then incubated with the primary antibodies of cleaved caspase3, SHP-1 and p-BCR-ABL at 4°C overnight in a humidified chamber. Sections were washed in PBS and incubated with secondary antibody biotinylated goat anti-rabbit (1:150, Vector Laboratories, Burlingame, CA, USA) or biotinylated rabbit anti-rat IgG (1:150, Abcam, Boston, MA, USA) for 40 min in a humidified chamber. After washing the antibodies were detected with the Vector ABC complex/HRP kit (Vector Laboratories), and developed with 3,3′-diaminobenzidine tetrahydrochloride. For semiquantitation, 10 photomicrographs (200×) were obtained with a CCD camera, avoiding gross necrotic areas.
Data analysis
All data are presented as means ± SD. Statistical analysis of the data was performed using the SigmaPlot version 12 software (Systat Software, Inc., San Jose, CA, USA). One-way anova was used for comparisons of multiple groups. Student's t-test was used for comparison between two groups. A significant difference was indicated at P < 0.05 between the control and EGCG or EGCG derivatives-treated groups. All experiments were repeated at least three times.
Results
Stability of EGCG, EGCG-MO, EGCG-ML and EGCG-MP and their cytotoxic effects in K562 and KBM5 CML cells
To evaluate the stability of the EGCG derivatives EGCG-MO, EGCG-ML and EGCG-MP (Figure 1A) in cultured cells, we performed HPLC analysis in DMEM culture medium. We found that 50% of EGCG-MP remained after ∼23 min in culture medium, while 50% of EGCG-ML, EGCG-MO and EGCG remained after ∼8 and 3 min respectively. Likewise, 20% of EGCG-MP remained after 116 min, while 20% of EGCG-ML, EGCG-MO and EGCG remained after ∼73.5, 16.5 and 4.5 min respectively. These results indicated that EGCG-MP was more stable than the other EGCG derivatives, including EGCG (Figure 1B). Also, we checked the intracellular levels of EGCG and EGCG-MP in K562 cells by HPLC analysis. As shown in Figure 1C and 1D, approximately 1.5% of EGCG-MP was detected in K562 cells after exposure to 40 μM EGCG-MP, while there was no peak of EGCG in K562 cells, incubated under the same conditions. In contrast, we observed approximately 0.39% of EGCG only when increasing its concentration up to 200 μM. Next, the cytotoxic effect of EGCG and its derivatives was evaluated by MTT assay in K562 and KBM5 cells exposed to 0, 10, 20, 40 or 80 μM for 24 h. As shown in Figure 1C, EGCG-ML and EGCG-MP exhibited greater dose-dependent cytotoxicity in K562 and KBM5 cells, compared with EGCG or EGCG-MO (Figure 1E and F), and EGCG-MP was the most effective EGCG derivative in K562 cells.
Figure 1.
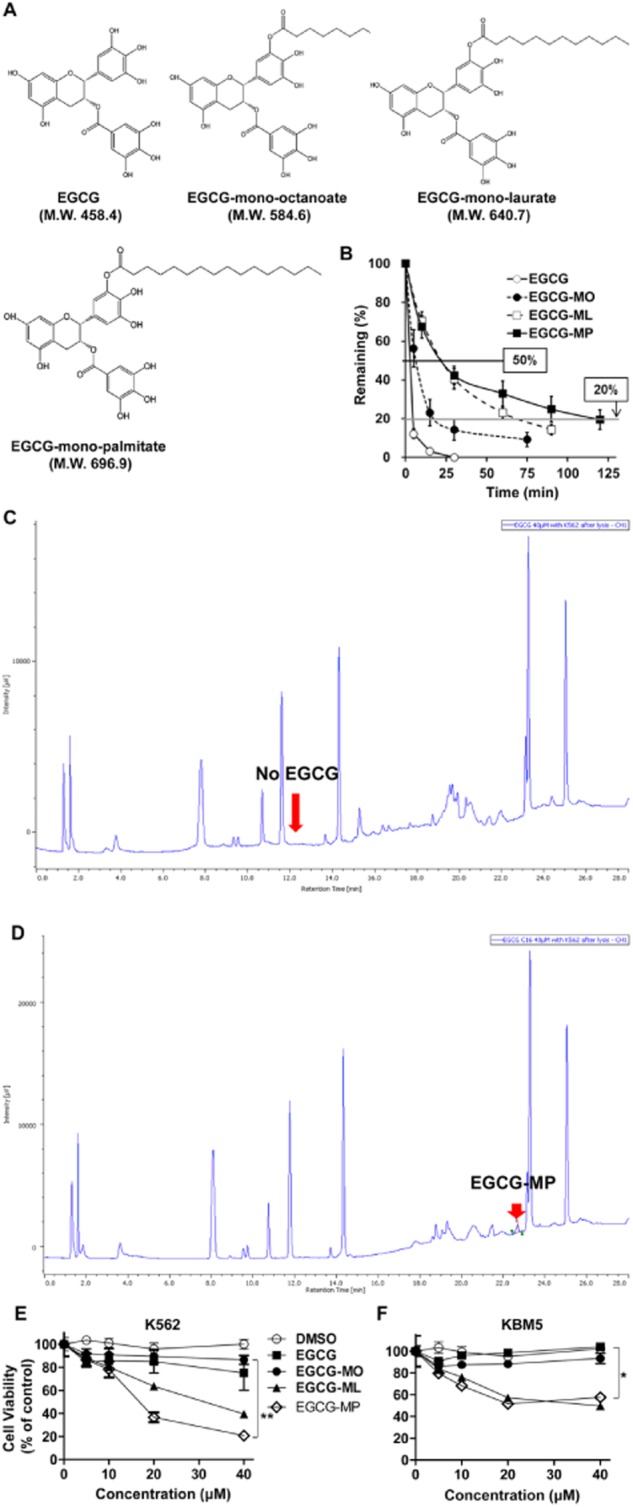
Cytotoxic effects of EGCG and its derivatives in K562 and KBM5 CML cells. (A) Chemical structures of the EGCG derivatives, EGCG (MW = 458.4), EGCG-MO (MW = 584.6), EGCG-ML (MW = 640.7) and EGCG-MP (MW = 696.9). (B) Stability of EGCG and its derivatives in culture medium was determined by HPLC. (C and D) A suspension of K562 cells at 2.0 × 105 cells·mL−1 in RPMI1640 medium was centrifuged. Then 1 mL of EGCG (40 or 200 μM) or EGCG-MP (40 μM) solution were added to the cells and incubated at 37°C with 5% CO2 for 30 min and individually dispensed to two filter-tip syringes [using 0.45 μm HLS (HTTP Live Streaming)-DISC 13 filter] and finally lysed with 1% lysis buffer for HPLC analysis. (E) K562 cells and (F) KBM5 cells were treated with 0, 5, 10, 20 and 40 μM of EGCG or EGCG derivatives for 24 h. Cell viability was measured by MTT assay. Data are presented as means ± SD. *P < 0.05; **P < 0.01, significantly different from untreated control.
EGCG-MP induced apoptosis more efficiently than EGCG or the other EGCG derivatives
To confirm whether the cytotoxicity of EGCG derivatives EGCG-MO, EGCG-ML and EGCG-MP in K562 cells was induced by apoptosis, we performed cell cycle analysis. As shown in Figure 2A, EGCG-MP increased the sub-G1 apoptotic portion to 16.6% of K562 cells, which was greater than the proportions induced by EGCG (5.5%), EGCG-MO (3.7%) or EGCG-ML (7%). Generally, apoptosis is induced via two pathways, a cell death extrinsic pathway and a mitochondria-dependent intrinsic pathway. Western blotting revealed that EGCG-MP cleaved PARP and activated caspase-9 and caspase-3 more than the other derivatives at the same concentrations in K562 cells. The effect was comparable to that of a high concentration (100 μM) of EGCG (Figure 2B). To verify apoptosis induced by EGCG-MP and EGCG in K562 cells, a TUNEL assay was performed. EGCG-MP increased the proportion of TUNEL-positive cells to 22.9%, compared with 7.5% in EGCG-treated cells and 2.6% in control K562 cells (Figure 3B). Consistent with this finding, cell cycle analysis showed that EGCG-MP at concentrations of 7.5, 15 and 30 μM caused 1.8, 3.7 and 14.9% increases in the sub-G1 apoptotic population, respectively, compared with the untreated control (0.47%) (Figure 3C). Moreover, 15 μM EGCG-MP induced the cleavage of apoptosis-related factors, such as caspase-9 and PARP, at levels comparable to those induced by 30 μM EGCG (Figure 3D).
Figure 2.

Effect of EGCG and its derivatives on the sub-G1 population and apoptotic protein expression in K562 cells. (A) Effect of EGCG and its derivatives on the apoptotic sub-G1 population of K562 cells. Cells were treated with EGCG and its derivatives at 30 μM for 24 h, stained with PI and the cell cycle analysed by flow cytometry. Data are presented as means ± SD. **P < 0.01, significantly different from untreated controls. (B) Effects of EGCG and its derivatives on caspase 9/3 and PARP in K562 cells. K562 cells were treated with EGCG (30 and 100 μM), EGCG-MO, EGCG-ML and EGCG-MP (30 μM) for 24 h. Cell lysates were prepared and analysed by Western blotting for caspase 9, cleaved caspase 3 and PARP.
Figure 3.
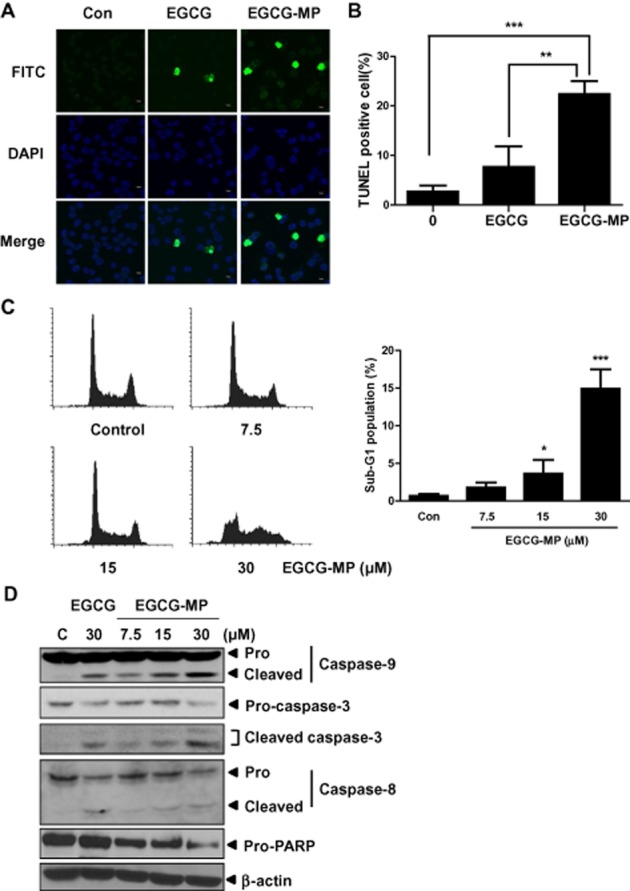
Effect of EGCG and EGCG-MP on TUNEL-positive cells and the cleavage of caspase 9/3 and PARP in K562 cells. (A) The apoptotic effects of EGCG and EGCG-MP in K562 cells were analysed by TUNEL assay. Green spots represent apoptotic positive cells. (B) A histogram representing TUNEL assay results. Data are presented as means ± SD. **P < 0.01, ***P < 0.001, significantly different as indicated. (C) Effect of EGCG-MP on the apoptotic sub-G1 population of K562 cells. Cells were treated with EGCG-MP (0, 7.5, 15 or 30 μM) for 24 h, stained with PI and the cell cycle analysed by flow cytometry. Data are presented as means ± SD. *P < 0.05; ***P < 0.001, significantly different from untreated controls. (D) Effects of EGCG and EGCG-MP on caspase 9/3 and PARP in K562 cells. K562 cells were treated with EGCG or EGCG-MP (0, 7.5, 15 or 30 μM) for 24 h. Cell lysates were prepared and analysed by Western blotting for caspase 9, caspase 3, cleaved caspase 3, caspase 8 and PARP in K562 cells.
EGCG-MP suppressed the phosphorylation of BCR-ABL via induction of SHP-1 in K562 cells
To verify the molecular target of EGCG-MP, Western blotting of K562 cells was performed. EGCG-MP markedly up-regulated SHP-1, dose- and time-dependently, in K562 cells, compared with EGCG (Figure 4A and B). We confirmed that EGCG-MP decreased phosphorylation of BCR-ABL in a dose- and time-dependent fashion, more effectively than EGCG (Figure 4C and D), which is supported by earlier reports that EGCG inhibited BCR-ABL activity by suppressing SHP-1 phosphorylation (Davenport et al., 2010). Similarly, EGCG-MP suppressed the expression of BCR-ABL and also attenuated the phosphorylation of SHP-1 in KBM5 cells (Figure 4E).
Figure 4.
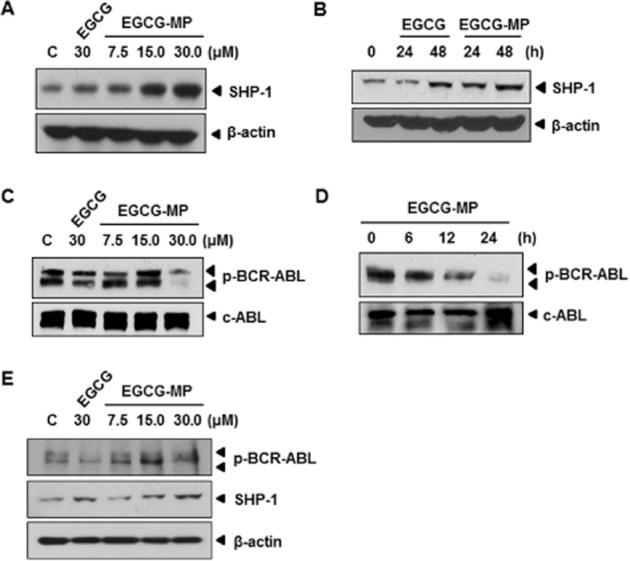
Effect of EGCG-MP on SHP-1 and p-BCR-ABL signalling in K562 cells. (A) Concentration dependency of the effect of EGCG and EGCG-MP on SHP-1 expression in K562 cells. Cells were treated with 30 μM of EGCG or the indicated concentration (7.5, 15 or 30 μM) of EGCG-MP and Western blotting was performed. (B) Time-dependency of the effect of 30 μM EGCG and EGCG-MP on SHP-1 expression in K562 cells treated for 24 or 48 h. (C) Concentration dependency of the effect of EGCG and EGCG-MP on p-BCR-ABL expression in K562 cells. (D) Time-dependency of the effect of 30 μM EGCG-MP on p-BCR-ABL in K562 cells. (E) Effects of 30 μM EGCG and various concentrations (7.5, 15 and 30 μM) of EGCG-MP on p-BCR-ABL and SHP-1 in KBM5 cells.
Inhibition of SHP-1 by a specific siRNA and pervanadate reversed the EGCG-MP-mediated decrease in BCR-ABL phosphorylation in K562 cells
To comfirm that decreased phosphorylation of BCR-ABL and STAT3 by EGCG-MP was mediated specifically by SHP-1, a siRNA against SHP-1 or the tyrosine phosphatase inhibitor pervanadate were added to EGCG-MP-treated K562 cells. As shown in Figure 5A and 5C, silencing SHP-1 or treatment with pervanadate blocked the suppression of phosphorylation of BCR-ABL and STAT3 by EGCG-MP in K562 cells. Similarly, the suppression of STAT3 downstream molecules such as Cdk4 and survivin by EGCG-MP was rescued by pervanadate treatment in K562 cells (Figure 5D). In contrast, EGCG at 30 μM weakly affected the expression of Cdk4, p-STAT3 and p-BCR-ABL in K562 cells (data not shown). Similarly, silencing of SHP-1 using siRNA transfection method suppressed the cytotoxicity of EGCG-MP in K562 cells, as measured by MTT assay (Figure 5B). Also, we confirmed that pervanadate or EGCG treatment did not affect the phosphorylation of BCR-ABL and STAT3 in K562 cells (Figure 5E).
Figure 5.
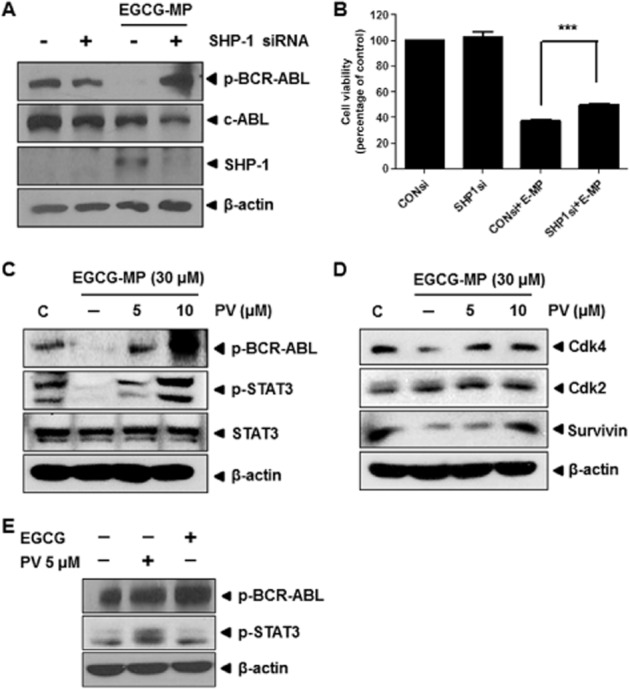
Silencing of SHP-1 or pervanadate treatment blocks the decreased phosphorylation of BCR-ABL and STAT3 in EGCG-MP treated K562 cells. (A) Effect of siRNA-mediated SHP-1 knockdown on p-BCR-ABL in EGCG-MP-treated K562 cells. Cells were transfected with siRNA for 48 h prior to EGCG-MP treatment. C, untreated control; (−), siRNA control. (B) Effect of siRNA-mediated SHP-1 knockdown on cell viability in EGCG-MP-treated K562 cells. Cells were transfected with siRNA for 48 h prior to EGCG-MP treatment. Data are presented as means ± SD. ***P < 0.001, significantly different as indicated. (C) Effect of pervanadate on p-BCR-ABL and p-STAT3 in EGCG-MP-treated K562 cells. Cells were pre-treated with pervanadate for 30 min prior to a 24-h treatment with EGCG-MP. (D) Effect of EGCG-MP on survival genes downstream of STAT3 in K562 cells. (E) Effect of pervanadate or EGCG on p-BCR-ABL and p-STAT3 in K562 cells. Cells were treated with pervanadate or EGCG for 24 h and Western blotting was performed.
EGCG-MP suppressed the growth of tumour in athymic nude mice
To confirm the in vitro anti-tumour activity of EGCG and EGCG-MP, an animal study was performed in BALB/c athymic nude mice. EGCG-MP effectively suppressed the volumes (Figure 6A), size (Figure 6B) and weights (Figure 6C) of K562 tumours implanted in BALB/c athymic nude mice, compared with the untreated control and EGCG, without affecting body weight loss (Figure 6D).
Figure 6.
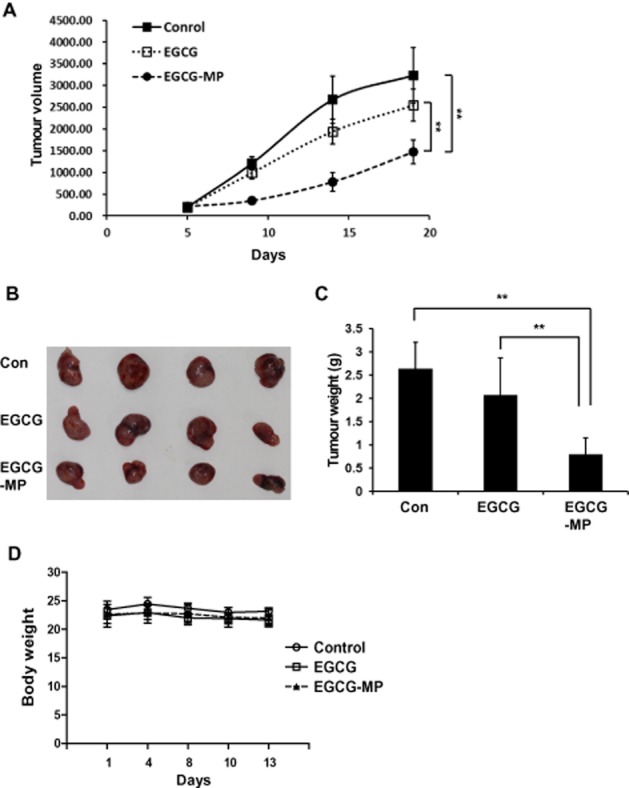
Effect of EGCG-MP on tumour growth in BALB/c athymic nude mice. Mice were inoculated with K562 for 2 weeks prior to s.c. abdominal injection of 15 mg·kg−1 EGCG-MP dissolved in 4% Tween-80 five times per week for 13 days. (A) Tumour volume, (B) tumour size, (C) tumour weights and (D) body weights of mice were recorded. Data are expressed as means ± SE, n = 4 mice per group. **P < 0.01, significantly different as indicated.
EGCG-MP increased the expression of caspase 3 and SHP-1 and decreased the phosphorylation of BCR-ABL in K562 tumour tissues
To confirm that EGCG-MP induced apoptosis and SHP-1, as well as suppressing phosphorylation of BCR-ABL (Figures 3D and 4), we performed IHC in EGCG- or EGCG-MP-treated tumour tissues. Induction of SHP-1 in the EGCG-MP treated group was detected at levels which were three- and eightfold higher than in the EGCG and control group respectively (Figure 7A and B). In addition, EGCG-MP induced five- and 10-fold decreases in BCR-ABL phosphorylation compared with the EGCG and control groups respectively (Figure 7A and B). Furthermore, expression of the apoptotic biomarker caspase 3 was increased in the EGCG-MP-treated group, compared with the untreated group. These data indicate that EGCG-MP induced SHP-1, leading to a decrease in p-BCR-ABL expression and activation of apoptosis in CML tumours in vivo.
Figure 7.
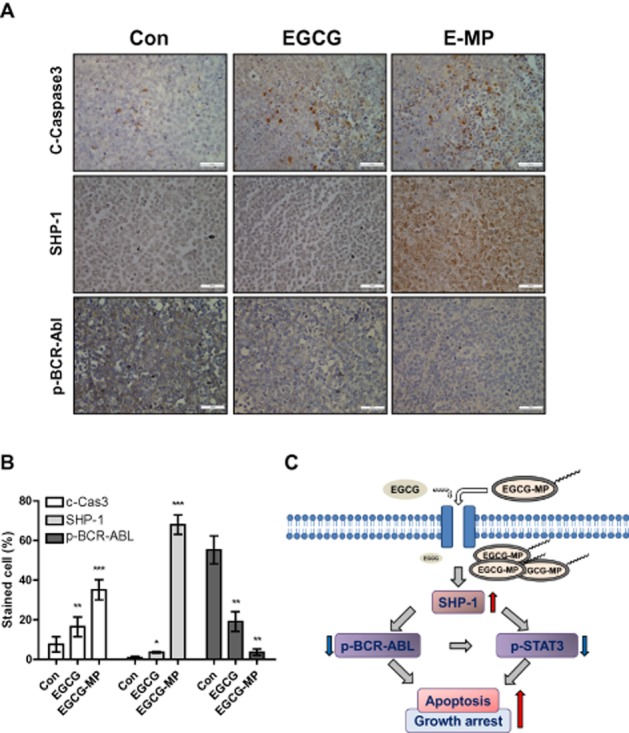
Induction of apoptosis and SHP-1 and suppression of p-BCR-ABL expression in K562-inoculated tumour tissues. (A) Immunohistochemical analysis of cleaved caspase 3, SHP-1 and p-BCR-ABL in CML tumor tissues from mice. (B) The expression levels of cleaved caspase 3, SHP-1 and p-BCR-ABL were quantified by the ratio of stained cell numbers versus total counted numbers. *P < 0.05; **P < 0.01; ***P < 0.001, significantly different from untreated control. (C) Scheme for antitumor mechanism of EGCG-MP via induction of SHP-1 and inhibition of p-BCR-ABL and p-STAT3 in CML.
Discussion
CML is a progressive and often fatal myeloproliferative disorder, and treatment with imatinib, a tyrosine kinase inhibitor specific for BCR-ABL, has been seen as a major breakthrough in CML therapy (Druker, 2002; Jabbour et al., 2010). Although most patients respond to first-line imatinib therapy, resistance (Volpe et al., 2009; Nair et al., 2012), toxicity (Faber et al., 2006) or intolerance has caused some patients to fail to respond to the standard dose. Thus, to optimize therapeutic benefit, physicians choose customized therapy based on each patient's historical response, adverse-event tolerance level, risk factors and the administration of anti-cancer herbal agents with low toxicity, which are attractive either alone or in combination with imatinib (Radujkovic et al., 2006; Walz and Sattler, 2006; Burthem et al., 2007; Murray-Rust and Rzepa, 2011; Agarwal et al., 2012).
Shishodia et al. reported that curcumin (isolated from the rhizome of the plant Curcuma longa) inhibited proliferation of primary CML cells and down-regulated STAT5 mRNA levels and activity (Shishodia et al., 2007). Lin et al. suggested that 16-hydroxycleroda-3,13-dien-15,16-olide induced apoptosis via a reduction in polycomb repressive complex-mediated gene silencing and the reactivation of downstream tumour suppressor gene expression (Lin et al., 2011). Likewise, Banerjee Mustafi et al. claimed that resveratrol inhibited BCR-ABL and Akt activity, but stimulated ERK 1/2 activity, which inhibited transcriptional activity of heat shock transcription factor 1 and Hsp70 production in K562 cells (Banerjee Mustafi et al., 2010).
Similarly, EGCG from green tea has been shown to exert potent anti-tumour activity in several types of leukaemia (Lung et al., 2002; Lee et al., 2004), prostate cancer (Siddiqui et al., 2011; Lee et al., 2012), cervical cancer (Ahn et al., 2003), colon cancer (Shimizu et al., 2005), lung cancer (Milligan et al., 2009; Wang et al., 2011; Liu et al., 2012), breast cancer (Sen and Chatterjee, 2011) and pancreatic cancer (Tang et al., 2012). However, clinical application of EGCG has been hampered by its limited solubility and efficacy in patients with cancer, underlining the need for the development of potent EGCG derivatives with improved metabolic stability and greater anti-cancer efficacy.
In our survey of EGCG derivatives, EGCG-MP was more stable than the other EGCG derivatives, including EGCG itself, which was supported by earlier evidence that the maximum plasma concentrations of EGCG were 34.7 ± 22.9 ng·mL−1 in 1.3–1.6 h after oral administration of EGCG at a dose of 2 mg·kg−1 and its elimination half-life was only 3.4 ± 0.3 h in human subjects (Lee et al., 2002). Also, EGCG-MP was the most potent cytotoxic agent in K562 and KBM5 cells, implying that EGCG-MP has potent anti-tumour activity for CML. Cell cycle analysis in K562 cells showed that EGCG-MP increased the sub-G1 population, fragmented apoptotic bodies, activated caspase 9/3 and cleaved PARP, indicating that the cytotoxicity of EGCG-MP was due to induction of apoptosis in these cells. The anti-cancer effects of EGCG-MP were mediated via induction of SHP-1 and may reflect the increased stability (Figure 1B) and permeability (Tanaka et al., 1998). Even low-dose treatment with 15 μM EGCG-MP significantly induced SHP-1 compared with 30 μM EGCG, which had weak effects on K562 cells (Figure 4A), suggesting EGCG-MP to be a more potent therapeutic agent for CML.
There is accumulating evidence that STAT3 is one of the major downstream targets of SHP-1 (Han et al., 2006a) and is involved in a variety of cellular events, such as apoptosis and cell survival. In the current study, EGCG-MP suppressed the phosphorylation of STAT3 in a concentration-dependent manner and induced the expression of SHP-1 in a concentration- and time-dependent manner in K562 cells, implying that the cytotoxic and apoptotic effects of EGCG-MP were mediated by inhibition of STAT3. In agreement with this, EGCG-MP attenuated the expression of STAT3-related survival genes such as Bcl-2, Bcl-xL, Mcl-1 and survivin in K562 cells. A variety of cellular responses are elicited by cell cycle-related genes (Viallard et al., 2001; Nixon et al., 2005). Here, EGCG-MP attenuated the expression of cell cycle-associated genes such as cyclin D1 and cyclin E, Cdk4 and activated the cyclin D1 inhibitor p21 (Chassot et al., 2008) in K562 cells, indicating that the regulation of cell cycle genes is critically involved in EGCG-MP induced cell death in K562 cells.
It is well documented that BCR-ABL, the first abnormal oncogene characterized in the Philadelphia chromosome (Hai et al., 2014), activates STAT3 signalling for CML (Coppo et al., 2006; Nair et al., 2012; Sayed et al., 2014). Here EGCG-MP significantly suppressed the phosphorylation of BCR-ABL in a dose- and time-dependent manner more effectively than EGCG in K562 cells. Silencing of SHP-1 or treatment with the tyrosine phosphatase inhibitor pervanadate blocked the ability of EGCG-MP to suppress phosphorylation of BCR-ABL and STAT3 and the expression of STAT3 downstream survival genes such as Cdk4 and survivin in K562 cells. These results demonstrate that EGCG-MP suppressed phosphorylation of BCR-ABL via SHP-1 or a tyrosine kinase-mediated pathway, which supports the induction of SHP-1 following exposure of K562 cells to differentiating agents (Tauchi et al., 1997; Bruecher-Encke et al., 2001; Hai et al., 2014). However, the role of SHP-2 in anti-tumour activity of ECCG-MP during CML treatment needs to be further studied as SHP-2 has been closely associated with BCR-ABL (Zhu et al., 2005; Chen et al., 2007; Tossidou et al., 2008).
EGCG-MP suppressed the increase of K562 cell tumour weight and volume in BALB/c athymic nude mice more effectively than in the untreated control or EGCG-treated group. Moreover, the IHC of tumour tissues showed that EGCG-MP was more effective than EGCG alone in stimulating the expression of cleaved caspase 3 and SHP-1 and inhibiting the expression of BCR-ABL. EGCG-MP also exhibited more potent anti-tumour effects than EGCG in terms of cytotoxicity, sub-G1 accumulation, and expression of apoptosis-related proteins. These results support the in vivo efficacy of EGCG-MP for CML treatment.
In conclusion, our findings suggested that EGCG-MP showed greater stability and induced SHP-1-mediated inhibition of BCR-ABL and STAT3 signalling in vitro and in vivo more effectively than EGCG (Figure 7C). This derivative could thus represent a potent chemotherapeutic agent for CML treatment.
Acknowledgments
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korean Government (MEST) (no. 2012-0005755 and 2014R1A2A10052872).
Glossary
- BCR-ABL
breakpoint cluster region-Abelson murine leukaemia viral oncogene homologue 1
- CML
chronic myeloid leukaemia
- ECL
enhanced chemiluminescence
- EGCG
epigallocatechin-3-gallate
- EGCG-ML
epigallocatechin-3-gallate mono-laurate
- EGCG-MO
epigallocatechin-3-gallate mono-octanoate
- EGCG-MP
epigallocatechin-3-gallate-mono-palmitate
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- PI
propidium iodide
- PTP
protein tyrosine phosphatase
- SH2 domain
Src-homology 2 domain
- SHP-1
SH2-containing tyrosine phosphatase 1
- TFA
trifluoroacetic acid
Author contributions
J. H. J., E.-J. C., S.-H. K., M.-S. J., D.-B. J., E.-O. K., H. L. and N. K. performed the research; J. H. J. and M. Y. designed the research study; K. K. and S.-H. K. contributed essential reagents and tools; J. H. J., M. Y., B. K. and S. K. S. analysed the data; M. Y., J. H. J., E.-J. C. and S.-H. K. wrote the paper.
Conflict of interest
Authors declare that they have no conflict of interest.
References
- Agarwal A, Fleischman AG, Petersen CL, Mackenzie R, Luty S, Loriaux M, et al. Effects of plerixafor in combination with BCR-ABL kinase inhibition in a murine model of CML. Blood. 2012;120:2658–2668. doi: 10.1182/blood-2011-05-355396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn WS, Huh SW, Bae SM, Lee IP, Lee JM, Namkoong SE, et al. A major constituent of green tea, EGCG, inhibits the growth of a human cervical cancer cell line, CaSki cells, through apoptosis, G(1) arrest, and regulation of gene expression. DNA Cell Biol. 2003;22:217–224. doi: 10.1089/104454903321655846. [DOI] [PubMed] [Google Scholar]
- Aichberger KJ, Mayerhofer M, Krauth MT, Skvara H, Florian S, Sonneck K, et al. Identification of mcl-1 as a BCR/ABL-dependent target in chronic myeloid leukemia (CML): evidence for cooperative antileukemic effects of imatinib and mcl-1 antisense oligonucleotides. Blood. 2005;105:3303–3311. doi: 10.1182/blood-2004-02-0749. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Enzymes. Br J Pharmacol. 2013;170:1797–1867. doi: 10.1111/bph.12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amin HM, Hoshino K, Yang H, Lin Q, Lai R, Garcia-Manero G. Decreased expression level of SH2 domain-containing protein tyrosine phosphatase-1 (Shp1) is associated with progression of chronic myeloid leukaemia. J Pathol. 2007;212:402–410. doi: 10.1002/path.2178. [DOI] [PubMed] [Google Scholar]
- Banerjee Mustafi S, Chakraborty PK, Raha S. Modulation of Akt and ERK1/2 pathways by resveratrol in chronic myelogenous leukemia (CML) cells results in the downregulation of Hsp70. PLoS ONE. 2010;5:e8719. doi: 10.1371/journal.pone.0008719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bromberg JF, Wrzeszczynska MH, Devgan G, Zhao Y, Pestell RG, Albanese C, et al. Stat3 as an oncogene. Cell. 1999;98:295–303. doi: 10.1016/s0092-8674(00)81959-5. [DOI] [PubMed] [Google Scholar]
- Bruecher-Encke B, Griffin JD, Neel BG, Lorenz U. Role of the tyrosine phosphatase SHP-1 in K562 cell differentiation. Leukemia. 2001;15:1424–1432. doi: 10.1038/sj.leu.2402214. [DOI] [PubMed] [Google Scholar]
- Burthem J, Rees-Unwin K, Mottram R, Adams J, Lucas GS, Spooncer E, et al. The rho-kinase inhibitors Y-27632 and fasudil act synergistically with imatinib to inhibit the expansion of ex vivo CD34(+) CML progenitor cells. Leukemia. 2007;21:1708–1714. doi: 10.1038/sj.leu.2404762. [DOI] [PubMed] [Google Scholar]
- Chai SK, Nichols GL, Rothman P. Constitutive activation of JAKs and STATs in BCR-Abl-expressing cell lines and peripheral blood cells derived from leukemic patients. J Immunol. 1997;159:4720–4728. [PubMed] [Google Scholar]
- Chassot AA, Lossaint G, Turchi L, Meneguzzi G, Fisher D, Ponzio G, et al. Confluence-induced cell cycle exit involves pre-mitotic CDK inhibition by p27(Kip1) and cyclin D1 downregulation. Cell Cycle. 2008;7:2038–2046. doi: 10.4161/cc.7.13.6233. [DOI] [PubMed] [Google Scholar]
- Chen J, Yu WM, Daino H, Broxmeyer HE, Druker BJ, Qu CK. SHP-2 phosphatase is required for hematopoietic cell transformation by BCR-ABL. Blood. 2007;109:778–785. doi: 10.1182/blood-2006-04-019141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppo P, Flamant S, De Mas V, Jarrier P, Guillier M, Bonnet ML, et al. BCR-ABL activates STAT3 via JAK and MEK pathways in human cells. Br J Haematol. 2006;134:171–179. doi: 10.1111/j.1365-2141.2006.06161.x. [DOI] [PubMed] [Google Scholar]
- Davenport A, Frezza M, Shen M, Ge Y, Huo C, Chan TH, et al. Celastrol and an EGCG pro-drug exhibit potent chemosensitizing activity in human leukemia cells. Int J Mol Med. 2010;25:465–470. doi: 10.3892/ijmm_00000366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Druker BJ. Inhibition of the BCR-ABL tyrosine kinase as a therapeutic strategy for CML. Oncogene. 2002;21:8541–8546. doi: 10.1038/sj.onc.1206081. [DOI] [PubMed] [Google Scholar]
- Faber E, Nausova J, Jarosova M, Egorin MJ, Holzerova M, Rozmanova S, et al. Intermittent dosage of imatinib mesylate in CML patients with a history of significant hematologic toxicity after standard dosing. Leuk Lymphoma. 2006;47:1082–1090. doi: 10.1080/10428190600565057. [DOI] [PubMed] [Google Scholar]
- Garcia R, Yu CL, Hudnall A, Catlett R, Nelson KL, Smithgall T, et al. Constitutive activation of Stat3 in fibroblasts transformed by diverse oncoproteins and in breast carcinoma cells. Cell Growth Differ. 1997;8:1267–1276. [PubMed] [Google Scholar]
- Gouilleux-Gruart V, Gouilleux F, Desaint C, Claisse JF, Capiod JC, Delobel J, et al. STAT-related transcription factors are constitutively activated in peripheral blood cells from acute leukemia patients. Blood. 1996;87:1692–1697. [PubMed] [Google Scholar]
- Gupta S, Hastak K, Afaq F, Ahmad N, Mukhtar H. Essential role of caspases in epigallocatechin-3-gallate-mediated inhibition of nuclear factor kappa B and induction of apoptosis. Oncogene. 2004;23:2507–2522. doi: 10.1038/sj.onc.1207353. [DOI] [PubMed] [Google Scholar]
- Hai A, Kizilbash NA, Zaidi SH, Alruwaili J, Shahzad K. Differences in structural elements of BCR-Abl oncoprotein isoforms in chronic myelogenous leukemia. Bioinformation. 2014;10:108–114. doi: 10.6026/97320630010108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Y, Amin HM, Franko B, Frantz C, Shi X, Lai R. Loss of SHP1 enhances JAK3/STAT3 signaling and decreases proteosome degradation of JAK3 and NPM-ALK in ALK+ anaplastic large-cell lymphoma. Blood. 2006a;108:2796–2803. doi: 10.1182/blood-2006-04-017434. [DOI] [PubMed] [Google Scholar]
- Han Y, Amin HM, Frantz C, Franko B, Lee J, Lin Q, et al. Restoration of shp1 expression by 5-AZA-2′-deoxycytidine is associated with downregulation of JAK3/STAT3 signaling in ALK-positive anaplastic large cell lymphoma. Leukemia. 2006b;20:1602–1609. doi: 10.1038/sj.leu.2404323. [DOI] [PubMed] [Google Scholar]
- Hu J, Zhou D, Chen Y. Preparation and antioxidant activity of green tea extract enriched in epigallocatechin (EGC) and epigallocatechin gallate (EGCG) J Agric Food Chem. 2009;57:1349–1353. doi: 10.1021/jf803143n. [DOI] [PubMed] [Google Scholar]
- Jabbour E, Kantarjian H, Cortes J. Chronic myeloid leukemia and second-generation tyrosine kinase inhibitors: when, how, and which one? Semin Hematol. 2010;47:344–353. doi: 10.1053/j.seminhematol.2010.06.002. [DOI] [PubMed] [Google Scholar]
- Jiao H, Berrada K, Yang W, Tabrizi M, Platanias LC, Yi T. Direct association with and dephosphorylation of Jak2 kinase by the SH2-domain-containing protein tyrosine phosphatase SHP-1. Mol Cell Biol. 1996;16:6985–6992. doi: 10.1128/mcb.16.12.6985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MJ, Maliakal P, Chen L, Meng X, Bondoc FY, Prabhu S, et al. Pharmacokinetics of tea catechins after ingestion of green tea and (−)-epigallocatechin-3-gallate by humans: formation of different metabolites and individual variability. Cancer Epidemiol Biomarkers Prev. 2002;11:1025–1032. [PubMed] [Google Scholar]
- Lee YH, Kwak J, Choi HK, Choi KC, Kim S, Lee J, et al. EGCG suppresses prostate cancer cell growth modulating acetylation of androgen receptor by anti-histone acetyltransferase activity. Int J Mol Med. 2012;30:69–74. doi: 10.3892/ijmm.2012.966. [DOI] [PubMed] [Google Scholar]
- Lee YK, Bone ND, Strege AK, Shanafelt TD, Jelinek DF, Kay NE. VEGF receptor phosphorylation status and apoptosis is modulated by a green tea component, epigallocatechin-3-gallate (EGCG), in B-cell chronic lymphocytic leukemia. Blood. 2004;104:788–794. doi: 10.1182/blood-2003-08-2763. [DOI] [PubMed] [Google Scholar]
- Liedtke M, Pandey P, Kumar S, Kharbanda S, Kufe D. Regulation of BCR-Abl-induced SAP kinase activity and transformation by the SHPTP1 protein tyrosine phosphatase. Oncogene. 1998;17:1889–1892. doi: 10.1038/sj.onc.1202117. [DOI] [PubMed] [Google Scholar]
- Lim YM, Wong S, Lau G, Witte ON, Colicelli J. BCR/ABL inhibition by an escort/phosphatase fusion protein. Proc Natl Acad Sci U S A. 2000;97:12233–12238. doi: 10.1073/pnas.210253497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin YH, Lee CC, Chang FR, Chang WH, Wu YC, Chang JG. 16-hydroxycleroda-3,13-dien-15,16-olide regulates the expression of histone-modifying enzymes PRC2 complex and induces apoptosis in CML K562 cells. Life Sci. 2011;89:886–895. doi: 10.1016/j.lfs.2011.09.011. [DOI] [PubMed] [Google Scholar]
- Liu LC, Tsao TC, Hsu SR, Wang HC, Tsai TC, Kao JY, et al. EGCG inhibits transforming growth factor-beta-mediated epithelial-to-mesenchymal transition via the inhibition of Smad2 and Erk1/2 signaling pathways in nonsmall cell lung cancer cells. J Agric Food Chem. 2012;60:9863–9873. doi: 10.1021/jf303690x. [DOI] [PubMed] [Google Scholar]
- Lung HL, Ip WK, Wong CK, Mak NK, Chen ZY, Leung KN. Anti-proliferative and differentiation-inducing activities of the green tea catechin epigallocatechin-3-gallate (EGCG) on the human eosinophilic leukemia EoL-1 cell line. Life Sci. 2002;72:257–268. doi: 10.1016/s0024-3205(02)02236-1. [DOI] [PubMed] [Google Scholar]
- Ma YC, Li C, Gao F, Xu Y, Jiang ZB, Liu JX, et al. Epigallocatechin gallate inhibits the growth of human lung cancer by directly targeting the EGFR signaling pathway. Oncol Rep. 2014;31:1343–1349. doi: 10.3892/or.2013.2933. [DOI] [PubMed] [Google Scholar]
- Masuda M, Suzui M, Weinstein IB. Effects of epigallocatechin-3-gallate on growth, epidermal growth factor receptor signaling pathways, gene expression, and chemosensitivity in human head and neck squamous cell carcinoma cell lines. Clin Cancer Res. 2001;7:4220–4229. [PubMed] [Google Scholar]
- Matsumura K, Kaihatsu K, Mori S, Cho HH, Kato N, Hyon SH. Enhanced antitumor activities of (−)-epigallocatechin-3-O-gallate fatty acid monoester derivatives in vitro and in vivo. Biochem Biophys Res Commun. 2008;377:1118–1122. doi: 10.1016/j.bbrc.2008.10.128. [DOI] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mereles D, Hunstein W. Epigallocatechin-3-gallate (EGCG) for clinical trials: more pitfalls than promises? Int J Mol Sci. 2011;12:5592–5603. doi: 10.3390/ijms12095592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milligan SA, Burke P, Coleman DT, Bigelow RL, Steffan JJ, Carroll JL, et al. The green tea polyphenol EGCG potentiates the antiproliferative activity of c-Met and epidermal growth factor receptor inhibitors in non-small cell lung cancer cells. Clin Cancer Res. 2009;15:4885–4894. doi: 10.1158/1078-0432.CCR-09-0109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray-Rust P, Rzepa HS. CML: evolution and design. J Cheminform. 2011;3:44. doi: 10.1186/1758-2946-3-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair RR, Tolentino JH, Hazlehurst LA. Role of STAT3 in transformation and drug resistance in CML. Front Oncol. 2012;2:30. doi: 10.3389/fonc.2012.00030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nixon C, Chambers G, Ellsmore V, Campo MS, Burr P, Argyle DJ, et al. Expression of cell cycle associated proteins cyclin A, CDK-2, p27kip1 and p53 in equine sarcoids. Cancer Lett. 2005;221:237–245. doi: 10.1016/j.canlet.2004.08.039. [DOI] [PubMed] [Google Scholar]
- Paulson RF, Vesely S, Siminovitch KA, Bernstein A. Signalling by the W/Kit receptor tyrosine kinase is negatively regulated in vivo by the protein tyrosine phosphatase Shp1. Nat Genet. 1996;13:309–315. doi: 10.1038/ng0796-309. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, et al. NC-IUPHAR. The IUPHAR/BPS Guide to PHARMACOLOGY: an expert-driven knowledge base of drug targets and their ligands. Nucl Acids Res. 2014;42(Database Issue):D1098–D1106. doi: 10.1093/nar/gkt1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peairs A, Dai R, Gan L, Shimp S, Rylander MN, Li L, et al. Epigallocatechin-3-gallate (EGCG) attenuates inflammation in MRL/lpr mouse mesangial cells. Cell Mol Immunol. 2010;7:123–132. doi: 10.1038/cmi.2010.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radujkovic A, Topaly J, Fruehauf S, Zeller WJ. Combination treatment of imatinib-sensitive and -resistant BCR-ABL-positive CML cells with imatinib and farnesyltransferase inhibitors. Anticancer Res. 2006;26:2169–2177. [PubMed] [Google Scholar]
- Rangatia J, Bonnet D. Transient or long-term silencing of BCR-ABL alone induces cell cycle and proliferation arrest, apoptosis and differentiation. Leukemia. 2006;20:68–76. doi: 10.1038/sj.leu.2403999. [DOI] [PubMed] [Google Scholar]
- Sayed D, Badrawy H, Gaber N, Khalaf MR. p-Stat3 and bcr/abl gene expression in chronic myeloid leukemia and their relation to imatinib therapy. Leuk Res. 2014;38:243–250. doi: 10.1016/j.leukres.2013.11.012. [DOI] [PubMed] [Google Scholar]
- Sen T, Chatterjee A. Epigallocatechin-3-gallate (EGCG) downregulates EGF-induced MMP-9 in breast cancer cells: involvement of integrin receptor alpha5beta1 in the process. Eur J Nutr. 2011;50:465–478. doi: 10.1007/s00394-010-0158-z. [DOI] [PubMed] [Google Scholar]
- Shimizu M, Deguchi A, Hara Y, Moriwaki H, Weinstein IB. EGCG inhibits activation of the insulin-like growth factor-1 receptor in human colon cancer cells. Biochem Biophys Res Commun. 2005;334:947–953. doi: 10.1016/j.bbrc.2005.06.182. [DOI] [PubMed] [Google Scholar]
- Shishodia S, Chaturvedi MM, Aggarwal BB. Role of curcumin in cancer therapy. Curr Probl Cancer. 2007;31:243–305. doi: 10.1016/j.currproblcancer.2007.04.001. [DOI] [PubMed] [Google Scholar]
- Siddiqui IA, Asim M, Hafeez BB, Adhami VM, Tarapore RS, Mukhtar H. Green tea polyphenol EGCG blunts androgen receptor function in prostate cancer. FASEB J. 2011;25:1198–1207. doi: 10.1096/fj.10-167924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka T, Kusano R, Kouno I. Synthesis and antioxidant activity of novel amphipathic derivatives of tea polyphenol. Bioorg Med Chem Lett. 1998;8:1801–1806. doi: 10.1016/s0960-894x(98)00311-4. [DOI] [PubMed] [Google Scholar]
- Tang SN, Fu J, Shankar S, Srivastava RK. EGCG enhances the therapeutic potential of gemcitabine and CP690550 by inhibiting STAT3 signaling pathway in human pancreatic cancer. PLoS ONE. 2012;7:e31067. doi: 10.1371/journal.pone.0031067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tauchi T, Ohyashiki K, Yamashita Y, Sugimoto S, Toyama K. SH2-containing phosphotyrosine phosphatase SHP-1 is involved in BCR-ABL signal transduction pathways. Int J Oncol. 1997;11:471–475. doi: 10.3892/ijo.11.3.471. [DOI] [PubMed] [Google Scholar]
- Tonks NK, Neel BG. From form to function: signaling by protein tyrosine phosphatases. Cell. 1996;87:365–368. doi: 10.1016/s0092-8674(00)81357-4. [DOI] [PubMed] [Google Scholar]
- Tossidou I, Dangers M, Koch A, Brandt DT, Schiffer M, Kardinal C. Tyrosine phosphatase SHP-2 is a regulator of p27(Kip1) tyrosine phosphorylation. Cell Cycle. 2008;7:3858–3868. doi: 10.4161/cc.7.24.7260. [DOI] [PubMed] [Google Scholar]
- Viallard JF, Lacombe F, Belloc F, Pellegrin JL, Reiffers J. [Molecular mechanisms controlling the cell cycle: fundamental aspects and implications for oncology] Cancer Radiother. 2001;5:109–129. doi: 10.1016/s1278-3218(01)00087-7. [DOI] [PubMed] [Google Scholar]
- Volpe G, Panuzzo C, Ulisciani S, Cilloni D. Imatinib resistance in CML. Cancer Lett. 2009;274:1–9. doi: 10.1016/j.canlet.2008.06.003. [DOI] [PubMed] [Google Scholar]
- Walz C, Sattler M. Novel targeted therapies to overcome imatinib mesylate resistance in chronic myeloid leukemia (CML) Crit Rev Oncol Hematol. 2006;57:145–164. doi: 10.1016/j.critrevonc.2005.06.007. [DOI] [PubMed] [Google Scholar]
- Wang H, Bian S, Yang CS. Green tea polyphenol EGCG suppresses lung cancer cell growth through upregulating miR-210 expression caused by stabilizing HIF-1alpha. Carcinogenesis. 2011;32:1881–1889. doi: 10.1093/carcin/bgr218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Ren X, Deng C, Yang L, Yan E, Guo T, et al. Mechanism of the inhibition of the STAT3 signaling pathway by EGCG. Oncol Rep. 2013;30:2691–2696. doi: 10.3892/or.2013.2743. [DOI] [PubMed] [Google Scholar]
- Zhu XZ, Yu YZ, Fang YM, Liang Y, Lu QH, Xu RZ. [Overexpression of Shp-2 is associated with the unlimited growth and apoptosis resistance of p210 BCR-ABL-mediated chronic myeloid leukemia] Zhonghua Yi Xue Za Zhi. 2005;85:1903–1906. [PubMed] [Google Scholar]