

Abstract
Background and Purpose
The aim of this study was to devise a nanoemulsified carrier system (CopNEC) to improve the oral delivery of amphotericin B (AmB) by increasing its oral bioavailability and synergistically enhance its antileishmanial activity with copaiba oil (Cop).
Experimental Approach
The AmB encapsulated NEC (CopNEC-AmB) comprised of Cop, d-α-tocopheryl polyethylene glycol 1000 succinate and phosphatidylcholine was prepared by high-pressure homogenization method. Stability study of CopNEC-AmB was carried out in simulated gastric fluid and simulated intestinal fluid. The CopNEC-AmB and plain AmB were compared as regards their in vitro antileishmanial activity, pharmacokinetics, organ distribution and toxicity.
Key Results
The optimal CopNEC-AmB had a small globule size, low polydispersity index, high ζ potential and encapsulation efficiency. The high resolution transmission electron microscopy illustrated spherical particle geometry with homogeny in their sizes. The optimal CopNEC-AmB was found to be stable in gastrointestinal fluids showing insignificant changes in globule size and encapsulation efficiency. The AUC0–48 value of CopNEC-AmB in rats was significantly improved showing 7.2-fold higher oral bioavailability than free drug. The in vitro antileishmanial activity of CopNEC-AmB was significantly higher than that of the free drug as Cop synergistically enhanced the antileishmanial effect of AmB by causing drastic changes in the morphology of Leishmania parasite and rupturing its plasma membrane. The CopNEC-AmB showed significantly less haemolytic toxicity and cytotoxicity and did not change the histopathology of kidney tissues as compared with AmB alone.
Conclusions and Implications
This prototype CopNEC formulation showed improved bioavailability and had a non-toxic synergistic effect on the antileishmanial activity of AmB.
Introduction
Parasites of the genus Leishmania cause a wide spectrum of human infections ranging from the disfiguring mucosal and cutaneous forms of the disease to the life-threatening visceral form (Boelaert et al., 2000; Murray et al., 2005). The parasites have developed a variety of adaptive mechanisms to evade the vertebrate host immune responses, including survival within the host macrophage (Cunningham, 2002; Kima, 2007) and leishmaniasis mainly affects poor countries where research and the development of new drugs have been seriously neglected (Trouiller et al., 2002). Hence, there is a need for new drugs and/or a delivery system for treating leishmaniasis. For many years, pentavalent antimonials have been the therapy of choice for leishmaniasis, but now the parasites have developed resistance to these drugs. In addition, amphotericin B (AmB) is a standard drug for treating several cases of visceral and cutaneous leishmaniasis caused by flagellated protozoa of the genus Leishmania (Bern et al., 2006; Asthana et al., 2013a). The major drawbacks of AmB treatment are its acute (fever, chilling, rigour) and subacute (mainly nephrotoxicity) toxicity and the need to administer the drug i.v. This need for parenteral delivery results in a high cost of treatment and limits the use of AmB in developing countries. Thus, oral administration of AmB is an appealing idea, as it could overcome the acute and subacute toxicity associated with i.v. delivery. At the same time, it would improve the quality of life for patients and reduce the associated cost of treatment. However, AmB belongs to the class IV category of drugs according to the biopharmaceutics classification system (Amidon et al., 1995), that is it has a low solubility and low membrane permeability due to its amphoteric nature. These two properties, aggravated by its poor stability in the gastric environment, are the primary factors responsible for its poor bioavailability when administered orally. Although its poor stability in the acidic gastric environment could be relatively easily addressed by, for example, by use of an enteric coating, the low solubility and poor membrane permeability of AmB present a significant challenge for researchers. The plasma concentrations of AmB following oral administration are very low in humans (Hofstra et al., 1982), rats (Robbie et al., 1999) and mice (Halde et al., 1957).
Therefore, the aqueous solubility, membrane permeability and oral bioavailability of AmB need to be enhanced through appropriate delivery systems. In recent years, improvements in the oral delivery of AmB have been sought by using different formulations, such as lipid-based cochleates (Delmas et al., 2002), nanosuspension (Kayser et al., 2003), lipid nanospheres and a self-emulsifying drug delivery system (Pham et al., 2013). The nanoemulsified carrier has shown promise as a carrier system for the oral delivery of poor water soluble and poor membrane permeable drugs. It is prepared by mixing a surfactant, co-surfactant, oil and drug that form an oil-in-water emulsion in aqueous medium after homogenization and form nanocarrier below 100 nm which is anticipated to enhance oral bioavailability by different mechanisms (Wu et al., 2006; Pham et al., 2013).
Consequently, there is an urgent need to discover new drug delivery formulations/excipients that are effective against leishmaniasis and eradicate Leishmania parasite completely without toxicity. Copaiba oil (Cop) has been used in folk medicine since the 19th century (Brandao et al., 2008) and has been used to treat leishmaniasis, as cited in several ethnopharmacological studies (Kvist et al., 2006; Dos Santos et al., 2012). The Cop contains diterpenes, sesquiterpenes and β-caryophyllene that cause antileishmanial activity against Leishmania parasites (Santos et al., 2008; 2013; Dos Santos et al., 2012; Soares et al., 2013). In the present study, we used Cop to prepare a nanoemulsified carrier that will enhance the oral bioavailability of AmB and at the same time provide an additionalt antileishmanial effect leading to a synchronized delivery system, which is highly effective against leishmaniasis and is economical to use and devoid of any toxicity.
Thus, the aim of this study was to prepare and evaluate AmB encapsulated in a nanoemulsified carrier (CopNEC-AmB) for oral administration. The system has been developed using Cop as an integral component to impart synergistic antileishmanial activity and improved oral bioavailability. To the best of our knowledge, we are the first to explore the antileishmanial effects of Cop in an oral drug delivery system.
Methods
Materials, parasite, cell line and animals
Cop from Copaifera, South American spp., tween 80, labrasol, d-α-tocopheryl polyethylene glycol 1000 succinate (TPGS), pluronic F-68, pluronic F-127, propylene glycol, span 80, 3-[4, 5-dimethylthiazol-2-yl]-2, 5-diphenyltetrazolium bromide (MTT), pepsin, pancreatin and dialysis membrane (MW 12 KD) were purchased from Sigma-Aldrich (St. Louis, MO, USA). Dimethylacetamide (DMAc), HPLC grade methanol and acetonitrile were from SD Fine-Chem Ltd (Mumbai, India). L-α-phosphatidyl choline, soya lecithin (PC) was from HiMedia Laboratory Pvt. Ltd. (Mumbai, India). AmB was kindly provided as a gift sample from Emcure Pharmaceutical Ltd. (Pune, India). All other reagents were of analytical grade.
The World Health Organization reference strain of Leishmania donovani (MHOM/IN/80/Dd8) was used for both in vitro experiments. Leishmania parasites and the macrophage cell line J774A were maintained in RPMI-1640 medium (Sigma-Aldrich) and Caco-2 cells were maintained in DMEM (Sigma-Aldrich), supplemented with 10% heat-inactivated FBS, 100 U·mL−1 penicillin and 100 μg·mL−1 streptomycin at 37°C in a humidified atmosphere of 5% (v v−1) CO2/air mixture.
Wistar male rats (200 ± 5 g, for pharmacokinetic analysis) and male mice (25 ± 3 g, for kidney toxicity) were used to study the effect of AmB formulations with prior approval of the Animal Ethics Committee of Council of Scientific and Industrial Research-Central Drug Research Institute and according to the regulations of the Council for the Purpose of Control and Supervision of Experiments on Animals, Ministry of Social Justice and Empowerment, Government of India. The Indian Animal Ethics Committee approval number is CDRI/2012/38.
Preparation of CopNEC-AmB and Cop containing nanoemulsified carrier (CopNEC)
The CopNEC-AmB was prepared through a high-pressure homogenization method using Microfluidizer Microfluidics International Corporation, Westwood, MA, USA). For this method, Cop, AmB dissolved in DMAc and co-surfactant dissolved in ethanol were mixed together for 30 min under stirring to prepare the oil phase, followed by complete solvent evaporation under vacuum for several hours in a rotary evaporator (Buchi, Rotavapor R-210, Flawil, Switzerland). The surfactant was dissolved in double distilled water into another beaker to form an aqueous phase. The oil phase was added dropwise into the aqueous phase under constant stirring. Subsequently the dispersed phase was homogenized using a microfluidizer. The prepared formulation was stored at room temperature (RT). To get the optimal formulation, various test formulations (F 1 to F 15) were prepared using different pharmaceutical and process parameters as shown in Table 1. The CopNEC was prepared using the method described earlier but was devoid of AmB.
Table 1.
Effect of various process parameters on size, PDI, ζ potential and encapsulation efficiency of CopNEC-AmB formulation
| Formulation code | Type of surfactant | Type of co surfactant | Ratio of surfactant : co-surfactant | Homogenizer pressure (MPa) | Number of cycles | oil | Ratio of drug : oil | Size (nm) | PDI | ζ potential | Encapsulation efficiency (% w/w) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| F 1 | Tween 80 | PC | 1:1 | 50 | 15 | Cop | 1:20 | 151 ± 15 | 0.13 ± 0.03 | (−)34.4 ± 1.8 | 90.8 ± 2.8 |
| F 2 | TPGS | PC | 1:1 | 50 | 15 | Cop | 1:20 | 127 ± 21 | 0.11 ± 0.02 | (−)38.5 ± 2.7 | 91.9 ± 1.4 |
| F 3 | Labrasol | PC | 1:1 | 50 | 15 | Cop | 1:20 | 143 ± 26 | 0.15 ± 0.07 | (−)36.6 ± 1.4 | 89.3 ± 1.6 |
| F 4 | PF68 | PC | 1:1 | 50 | 15 | Cop | 1:20 | 162 ± 34 | 0.15 ± 0.08 | (−)37 ± 1.9 | 89.6 ± 1.8 |
| F 5 | PF127 | PC | 1:1 | 50 | 15 | Cop | 1:20 | 158 ± 28 | 0.12 ± 0.04 | (−)35.6 ± 1.7 | 88.7 ± 2.6 |
| F 6 | TPGS | Span 80 | 1:1 | 50 | 15 | Cop | 1:20 | 144 ± 24 | 0.12 ± 0.03 | (−)25.4 ± 2.7 | 84.7 ± 3.7 |
| F 7 | TPGS | PG | 1:1 | 50 | 15 | Cop | 1:20 | 165 ± 32 | 0.16 ± 0.02 | (−)16.2 ± 1.3 | 82.8 ± 2.6 |
| F 8 | TPGS | PC | 1:0.25 | 50 | 15 | Cop | 1:20 | 172 ± 47 | 0.11 ± 0.08 | (−)30.8 ± 2.8 | 85.8 ± 3.4 |
| F 9 | TPGS | PC | 1:0.5 | 50 | 15 | Cop | 1:20 | 169 ± 46 | 0.12 ± 0.07 | (−)34.5 ± 2.9 | 86.4 ± 2.8 |
| F 10 | TPGS | PC | 1:2 | 50 | 15 | Cop | 1:20 | 144 ± 38 | 0.11 ± 0.08 | (−)45.6 ± 2.2 | 89.5 ± 2.6 |
| F 11 | TPGS | PC | 1:1 | 50 | 15 | Cop | 1:10 | 157 ± 47 | 0.11 ± 0.06 | (−)32.7 ± 1.8 | 89.7 ± 1.4 |
| F 12 | TPGS | PC | 1:1 | 50 | 15 | Cop | 1:40 | 132 ± 26 | 0.2 ± 0.06 | (−)37.8 ± 1.6 | 90.9 ± 2.8 |
| F 13 | TPGS | PC | 1:1 | 50 | 5 | Cop | 1:20 | 155 ± 29 | 0.11 ± 0.05 | (−)38.8 ± 2.5 | 88.4 ± 1.5 |
| F 14 | TPGS | PC | 1:1 | 50 | 10 | Cop | 1:20 | 151 ± 27 | 0.12 ± 0.07 | (−)37.9 ± 2.9 | 89.3 ± 1.6 |
| F 15 | TPGS | PC | 1:1 | 50 | 20 | Cop | 1:20 | 163 ± 30 | 0.11 ± 0.09 | (−)38.6 ± 3.7 | 88.9 ± 2.4 |
Each result represents the mean ± SD (n = 3). AmB, amphotericin B; Cop, copaiba oil; NEC, nanoemulsified carrier; PC, phosphatidyl choline; PDI, polydispersity index; PF127, pluronic F127; PF68, pluronic F68; PG, propylene glycol; TPGS, d-α-tocopheryl polyethylene glycol 1000 succinate.
Characterization of CopNEC-AmB
The CopNEC-AmB was characterized on the basis of size, polydispersity index (PDI), ζ potential and encapsulation efficiency (EE).
Measurement of size, PDI and ζ potential
The cumulative mean size of CopNEC-AmB was determined by dynamic light scattering using Zetasizer (Nano ZS, Malvern Instruments, Malvern, Worcestershire, UK) after suitable dilution in double distilled water. Each measurement was carried out at 25°C after equilibration for 2 min and the mean of 20 measurements of 120 s divided into three subruns was taken. The diameter of the prepared CopNEC-AmB was calculated from the autocorrelation function of the intensity of light scattered from the particles assuming a spherical form of particles. A fresh disposable cuvette (10 mm × 10 mm × 48 mm) was used for each measurement and a multimodal type of analysis was adopted for particle size distribution. The PDI was obtained by the in-built software provided in the instrument.
To measure the physical properties of the formulations in terms of ζ potential (ξ), the electrophoretic mobilities of CopNEC-AmB were analysed using Zetasizer (Nano ZS, Malvern Instruments). The mobility, u, was converted into ζ potential (ξ) values using the Smoluchowski relation ξ = uη/∈; where η and ∈ are the viscosity and permittivity of the solution respectively. Each measurement was recorded at 25°C with prior equilibration of 2 min and an average of 20 measurements was taken. All ξ-potential measurements were performed in diluted samples without added electrolyte.
High resolution transmission electron microscopy (HRTEM)
The HRTEM (Tecnai™ G2 F20, Eindhoven, The Netherlands) of optimal CopNEC-AmB was carried out after preparing the sample (1 mg 20 mL−1) in a thin aqueous film supported on a 300-mesh copper grid and negative staining was performed using a droplet of 2% (w v−1) phosphotungstic acid. Direct imaging of dried samples was executed at 200 kV acceleration voltage using HRTEM.
Measurement of AmB
The AmB in the samples was measured by reverse-phase HPLC. The HPLC system was equipped with 10 ATVP binary gradient pumps (Shimadzu, Tokyo, Japan), a rheodyne model 7125 injector (St. Louis, MO, USA) with a 20 mL loop and SPD-M10 AVP UV detector (Shimadzu). The separation was carried out on a Lichrosphere Lichrocart C18 column (250 × 4 mm, 5 μm; Merck, Darmstadt, Germany) using acetonitrile and potassium dihydrogen phosphate buffer at pH 4 (60:40, v v−1) as mobile phase at 1.0 mL·min−1 flow rate and column effluent was detected with a UV detector at 407 nm. Data were obtained and processed using class VP software (Asthana et al., 2013b).
Determination of AmB EE
The EE was determined by extracting the drug from CopNEC-AmB using DMAc, diluted with methanol and filtered through a 0.45 μm membrane filter and analysed using HPLC method as described earlier (Asthana et al., 2013b). The removal of free drug from CopNEC-AmB was carried out by ultra-centrifugation at 40 000× g for 30 min (Thermo Scientific, Sorvall, Wx ultra 100, Langenselbold, Germany) and the pellets obtained were dried under vacuum. The percentage of EE was calculated using the following formula:
Stability studies of optimal CopNEC-AmB
The optimal CopNEC-AmB formulation was subjected to stability studies at different time points (1, 7, 15, 30 and 90 days) at 4 and 25°C. At each time point, an aliquot of CopNEC-AmB was withdrawn for analysis for any change in physicochemical parameters such as size, PDI, ζ potential, EE, viscosity and pH (Choudhury et al., 2014).
Viscosity
The change in viscosity of the formulation, subjected to stability, without dilution was measured and analysed using Bohlin visco 88 viscometer equipped with Bohlin software (Malvern Instruments). Cone and plate geometry (cp5.4°/30) was employed with 0.05 mm gap and the sample (∼1 mL) was spread to completely fill this gap (Wanis et al., 1993). For each sample, continuous variation of shear rate (80–400 s−1) was applied at 25°C.
pH measurements
The change in pH of the formulation kept for the stability study was measured using a pH meter (Thermo Scientific, ORION™ 2-STAR, pH Benchtop meter) at 25°C.
Stability of optimal CopNEC-AmB in simulated gastrointestinal fluids
Stability of optimal CopNEC-AmB in simulated gastric fluid (SGF)
The SGF was prepared by dissolving 2.0 g of sodium chloride and 3.2 g of purified pepsin in 7.0 mL of hydrochloric acid and double distilled water up to 1000 mL. The assessment of the stability of CopNEC-AmB (1 mg·mL−1) was performed in triplicate; the samples were incubated in SGF at a 1:10 (v v−1) dilution at 37°C. The size and EE of CopNEC-AmB were measured in SGF after incubation for different time intervals (0, 15, 30, 60 and 120 min) (Wasan et al., 2009).
Stability of optimal CopNEC-AmB in simulated intestinal fluid (SIF)
The SIF was prepared according to US Pharmacopeia 33-28NF (2010) and was composed of 6.8 g of monobasic potassium phosphate in 250 mL of water, 77 mL of 0.2 N sodium hydroxide and 500 mL of water. Ten grams of pancreatin were added and the resulting solution was adjusted with 0.2 N sodium hydroxide or 0.2 N hydrochloric acid to a pH of 6.8 ± 0.1 and finally diluted to 1000 mL. The assessment of the stability of CopNEC-AmB (1 mg·mL−1) was performed in triplicate; the samples were incubated in SIF at a 1:50 (v v−1) dilution at 37°C. The size and EE of CopNEC-AmB were measured in SIF after incubation for different time intervals (0, 2, 4, 6 and 8 h) (Wasan et al., 2009).
Tagging of AmB with FITC
The AmB was tagged with FITC according to our previously reported method (Gupta et al., 2014a) to perform an in vitro uptake study. In brief, 10 mg of AmB and 5 mg of FITC were dissolved in 2 mL DMAc followed by addition of 200 μL triethylamine as base catalyst in 5 mL round bottom flask. The reaction mixture was stirred for 2 h at RT and thereafter 10 mL ethyl acetate was added to precipitate the final product. The final product was separated by centrifugation at 18 000× g for 10 min and dried over a desiccant under vacuum. Thin layer chromatography (TLC) of the product, AmB and FITC was performed using a mobile phase composed of ethyl acetate : methanol in a ratio of 2:3 to identify the formation of FITC tagged AmB (FAmB).
In vitro uptake study of CopNEC-FAmB
FAmB was encapsulated in the CopNEC by a method described above using FAmB instead of AmB to prepare FAmB encapsulated CopNEC (CopNEC-FAmB). The Caco-2 stable cell cultures were incubated with a FITC tagged formulation (CopNEC-FAmB) and FAmB at 10 μg·mL−1 equivalent concentration for 6 h at 37°C and 5% CO2 in DMEM. After incubation, the samples were transferred into vials and relative fluorescence was measured by flow cytometry (Becton Dickinson, Oxford, UK) at λEX (495 nm) and λEM (525 nm).
Pharmacokinetic and biodistribution study
Experimental design
The Wistar rats were deprived of food (fasted) for 12 h after the administration of AmB with free access to drinking water. Nine animals weighing around 200 g were allocated into the following three treatment groups: oral administration of 10 mg·kg−1 AmB (n = 3), oral administration of 10 mg·kg−1 CopNEC-AmB (n = 3) and oral administration of 5 mg·kg−1 CopNEC-AmB (n = 3). Blood (0.2 mL) was obtained from the retro-orbital plexus of the rat at 10 min pre-dose and 1, 2, 4, 6, 8, 12, 24 and 48 h post-dosing from all groups and serum was separated by centrifugation (2000× g, 10 min and 15°C). The withdrawn blood was replaced by an equal volume of normal saline to prevent hypovolaemia. The animals were killed at 6, 24 and 48 h following administration of AmB formulations and liver, spleen, lung and right kidney were harvested for biodistribution analysis of drug. Plasma and tissue samples were stored at −80°C until analysis. The total number of animals used in our experiments are 21. All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010).
Analytical procedures
Tissues were washed with double distilled water, dried by blotting on tissue papers and homogenized in acetonitrile (1 g of tissue 2 mL−1 of acetonitrile) using a homogenizer (IKA® T25 digital ULTRA-TURRAX®, Kengeri, Bangalore, Karnataka, India) for 3–4 min over an ice bath and left to stand for 30 min then centrifuged at 10 000× g for 10 min. The 200 μL of acetonitrile was added to each 200 μL of serum and tissue homogenates and was allowed to stand for 30 min for complete extraction of AmB followed by centrifugation at 10 000× g for 10 min (Wasan et al., 2010). The aliquots of 20 μL of serum and tissue samples were analysed based on a previously published validated isocratic analytical method (Asthana et al., 2013b). Calibration curves of AmB were linear in the range of 0–10 μg·mL−1 for serum samples and 0–10 μg·g−1 for tissue samples. The limit of quantification of AmB was 10 ng·mL−1 for serum samples and 20 ng·g−1 for tissue samples.
In vitro anti-amastigote assay
The activity of CopNEC-AmB and AmB against intracellular amastigotes was evaluated using a protocol described previously (Mookerjee Basu et al., 2008). The infection of J774A macrophages (1 × 105 cells per well) in 24-well plates was carried out by adding metacyclic-stage promastigotes, expressing fluorescent protein, in the ratio of 10 parasites per macrophage and incubated for 12 h at 37°C and 5% CO2. After the incubation, culture plates were washed thrice with PBS (pH 7.4) to remoe non-phagocytosed promastigotes and resupplemented with RPMI-1640 complete medium. The infected macrophages were treated with CopNEC-AmB and AmB at different equivalent drug concentrations. To assess the effect of CopNEC and Cop, the same amount as present in the formulation was added in triplicate for 48 h. Then, J774A cells were washed twice with PBS (pH 7.4), removed and quantified by flow cytometry equipped with a 20 mW argon laser with excitation at 488 nm and emission at 515 nm followed by multiparametric data analysis by Kaluza analysis software (Beckman Coulter, Atlanta GA, USA). The inhibition of parasite growth evoked by the different formulations was measured by comparing the fluorescence levels of drug-treated parasites with that of untreated control parasites (Mookerjee Basu et al., 2008) and the concentrations of the formulations that inhibited the growth of the parasites by 50% and 90% (IC50 and IC90) were calculated using GraphPad Prism6 (GraphPad Software Inc., La Jolla, CA, USA).
In vitro toxicity studies
Study of haemolytic activity
The haemolytic activity of the different formulations (CopNEC-AmB and AmB) containing AmB equivalent to 5, 10 and 20 μg·mL−1 and CopNEC and Cop were determined on erythrocytes (Gupta et al., 2014b). The erythrocytes (RBCs) were collected from the blood of Wistar rats by centrifugation for 10 min at 2000× g, washing thrice with isotonic PBS (pH 7.4) and were dispersed in PBS to obtain 3% (w v−1) haematocrit. Subsequently, an aliquot of 200 μL of the RBC suspension was mixed in 1 mL of formulation containing 5, 10 and 20 μg·mL−1 equivalent of AmB (CopNEC-AmB and AmB). Similarly, RBCs were incubated with CopNEC and Cop at 37°C for 4 h in a shaking water bath at 100 r.p.m. After incubation, the RBC samples were removed by centrifugation for 10 min at 2000× g and the supernatant was analysed for the extent of haemolysis at 540 nm using a multiwell microplate reader (BIO-TEK, Model-Power wave XS, Crailsheim, Germany). The percentage of haemolysis was calculated from the haemoglobin released into the supernatants by 100 × (Abss − Abs0)/(Abs)100 where Abss is the absorbance of the sample, Abs0 is the average absorbance of the saline and Abs100 is the average absorbance of the double distilled water.
Cytotoxicity against non-infected J774A macrophages
Cytotoxicity was determined by the MTT proliferation assay for different formulations of CopNEC-AmB, CopNEC, Cop and AmB at 5, 10 and 20 μg·mL−1 equivalent AmB concentrations (Gupta et al., 2014b). For measurement of cytotoxicity, J774A macrophages (5 × 104 cells per well) in RPMI-1640 medium were placed in to 96-well plates and allowed to adhere overnight. The medium was replaced with fresh medium and incubated in triplicate with all the different formulations at 37 ± 1°C for 24 h. After incubation, the medium of the J774A cells was replaced with RPMI-1640 containing MTT (500 μg·mL−1) followed by incubation for an additional 4 h, to allow for the MTT to be reduced and to form purple formazan crystals by viable cells. Twenty microlitres of DMSO was added to each well to ensure complete solubilization of formazan crystals. The OD was measured by a multiwell microplate reader at 570 nm and relative viability (%) of the cells subjected to the different formulations was calculated relative to the control wells containing cell culture medium without formulation (OD of the control well as 100% viability).
Mitochondrial membrane potential determination
The Leishmania promastigotes were subjected to treatment with CopNEC-AmB, AmB, CopNEC and Cop for different time points (0, 6, 24 and 48 h), incubated for 7 min with 10 mM JC-1 (Sigma-Aldrich) at 37°C, washed and resuspended in medium. The relative mitochondrion potential (Δψm) value was calculated from the ratio of fluorescence at 590 to 530 nm (Dey and Moraes, 2000).
In vivo toxicity
This was measured in four groups (n = 3): they received either CopNEC-AmB (10 mg·kg−1), AmB (10 mg·kg−1), Cop (0.2 mL·kg−1) or normal saline (control) p.o. in a constant volume of 0.2 mL for each group, daily for 15 days. Twelve hours after the last treatment, mice were killed and kidney tissues were collected and preserved in 10% formalin, embedded in paraffin, sectioned and stained with haematoxylin and eosin for histopathological examination.
Statistical analysis
All results are presented as mean ± SD of three independent measurements. Statistical significance of different formulations were analysed by one-way anova followed by the Tukey–Kramer multiple comparison test, using InStat software (GraphPad Software Inc.) and P < 0.05 was considered significant in all cases.
Results
Preparation, optimization and characterization of CopNEC-AmB
To obtain optimal nanoemulsified carrier system various surfactants, co-surfactant and their ratios were carefully evaluated in terms of size, PDI, ζ potential and EE, as listed in Table 1. Initially, the types of surfactant were evaluated for size, PDI, ζ potential and EE keeping other parameters such as surfactant to co-surfactant ratio and oil homogenization parameters (50 MPa, 15 cycles) constant. The mixture of TPGS, PC and Cop (F 2) shown in Table 1 provided the lowest size and PDI with highest ζ potential and comparable EE. Secondly, the highest encapsulation 91.9 ± 1.4% was observed with PC besides span 80 and propylene glycol as co-surfactant had only 84.7 ± 3.7% and 82.8 ± 2.6%, respectively, during screening of type of co-surfactant. The ζ potential was also highest (−)38.5 ± 2.7 mV in PC formulation (F 2) compared with F 6 and F 7 while size and PDI were comparable in all three formulations. In F 2, PC was used, which is phospholipidic in nature and provides increased EE because of its hydrophobic interaction and association with bilayer membranes (Were et al., 2003). The TPGS and PC ratio was also optimal in F 2, F 8, F 9 and F 10 formulations but it was observed that the isotropic mixture (1:1) of surfactant and co-surfactant provided better particle characteristics. The drug to Cop ratio was also evaluated (F 2, F 11 and F 12 formulations) and showed insignificant change in EE but particle characteristics were much better in the F 2 formulation. Lastly, at a microfluidizer pressure of 50 MPa, an increase in the number of cycles led to an initial decrease in the size, but after 15 cycles the size was found to be increased while PDI, ζ potential and EE remained comparable as showed in Table 1. It was observed that increasing the homogenization pressure beyond 50 MPa, size and PDI changed because of increased mechanical energy, which leads to aggregation.
The optimal parameters, for preparation of CopNEC-AmB, selected were isotropic mixture (1:1) of TPGS and PC, Cop, drug to Cop ratio 1:20 and homogenization parameters 15 cycles at 50 MPa of microfluidizer. The parameters of CopNEC-AmB mentioned earlier have optimal particle characteristics that is size 127 ± 21 nm, PDI 0.11 ± 0.02 and ζ potential (−)38.5 ± 2.7 with EE 91.9 ± 1.4% w w−1. The HRTEM also supports particle characteristics of formulation that illustrates spherical particle geometry with homogeny in their sizes for optimal CopNEC-AmB as shown in Figure 1. A good correlation was visualized between the particle size obtained by HRTEM and that measured with a ζ sizer.
Figure 1.
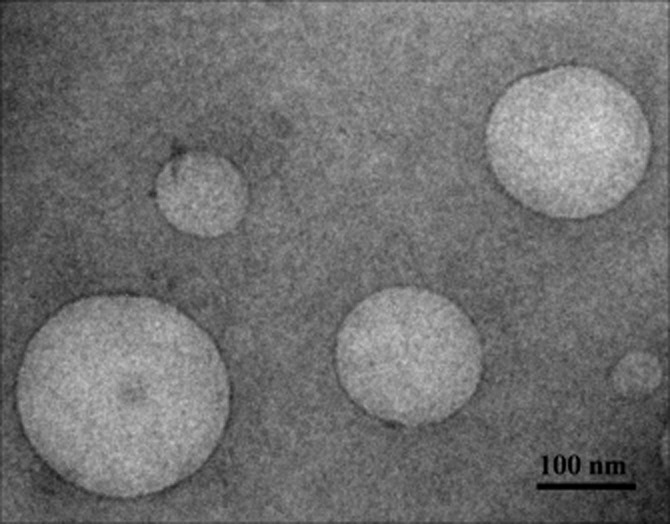
HRTEM microphotographs of optimal CopNEC-AmB.
In vitro stability study
Time and temperature-dependent stability of CopNEC-AmB were monitored on the basis of variation in different parameters as shown in Table 2. The CopNEC-AmB was found to be stable under all conditions and there were no significant changes in size, PDI, ζ potential, EE, viscosity and pH at 25 and 4°C during the 3 month storage period. It should be noted that no phase separation of the formulation was observed during 90 days of incubation at 25 and 4°C. The vitamin E TPGS in the formulation provided tremendous stability to CopNEC-AmB and the rate of drug loss was also significantly decreased from the formulation (Wasan et al., 2010). The CopNEC-AmB is intended to be administered orally, so stability of AmB in gastrointestinal fluid plays a critical role for development of an oral drug delivery system. It was observed (Figure 2) that size and EE were not changed over a period of time and CopNEC-AmB was found to be stable in SIF and SGF due to the presence of TPGS and PC which contributed to a higher EE and restricted particle coalescence as compared with other surfactants in the formulation (data not shown) (Zhang et al., 2012).
Table 2.
Stability study of optimal formulation F 2 at different temperatures
| Temperature | Time (days) | Size (nm) | PDI | ζ potential (mV) | Encapsulation efficiency (% w w−1) | pH | Viscosity (η) |
|---|---|---|---|---|---|---|---|
| 25°C | 1 | 126 ± 27 | 0.22 ± 0.08 | −40.7 ± 2.3 | 91.9 ± 1.2 | 7.2 ± 0.0 | 0.181 ± 0.003 |
| 7 | 127 ± 29 | 0.23 ± 0.09 | −40.8 ± 2.9 | 91.6 ± 1.7 | 7.2 ± 0.0 | 0.181 ± 0.007 | |
| 15 | 128 ± 37 | 0.22 ± 0.04 | −40.5 ± 3.6 | 91.3 ± 1.9 | 7.2 ± 0.0 | 0.181 ± 0.006 | |
| 30 | 129 ± 40 | 0.21 ± 0.07 | −40.2 ± 3.1 | 90.9 ± 2.1 | 7.2 ± 0.0 | 0.180 ± 0.006 | |
| 90 | 131 ± 46 | 0.21 ± 0.05 | −41.7 ± 4.5 | 90.3 ± 1.8 | 7.2 ± 0.0 | 0.180 ± 0.005 | |
| 4°C | 1 | 126 ± 25 | 0.23 ± 0.07 | −39.5 ± 2.0 | 91.9 ± 1.6 | 7.2 ± 0.0 | 0.181 ± 0.006 |
| 7 | 123 ± 29 | 0.22 ± 0.06 | −40.6 ± 3.4 | 91.8 ± 1.3 | 7.2 ± 0.0 | 0.181 ± 0.009 | |
| 15 | 121 ± 32 | 0.21 ± 0.08 | −40.5 ± 3.8 | 91.6 ± 1.4 | 7.2 ± 0.0 | 0.181 ± 0.004 | |
| 30 | 123 ± 37 | 0.20 ± 0.07 | −38.1 ± 4.3 | 91.2 ± 1.5 | 7.2 ± 0.0 | 0.181 ± 0.009 | |
| 90 | 121 ± 39 | 0.21 ± 0.09 | −39.7 ± 5.7 | 90.9 ± 1.8 | 7.2 ± 0.0 | 0.181 ± 0.007 |
Each result represents mean ± SD (n = 3). PDI, polydispersity index.
Figure 2.
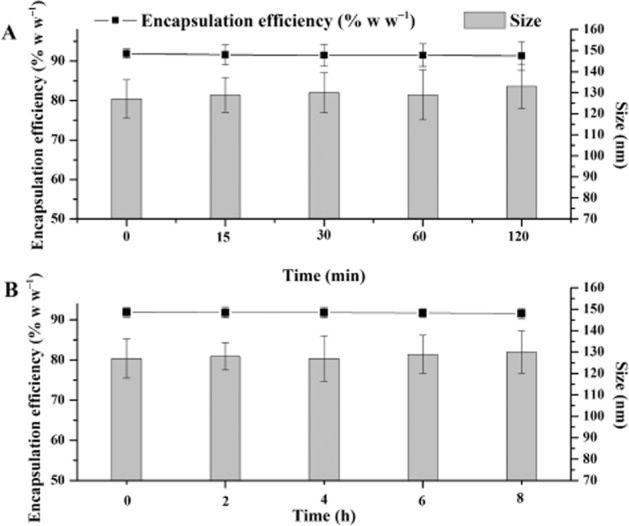
The stability of optimal CopNEC-AmB in (A) SGF and (B) SIF at 37°C showed changes in size and encapsulation efficiency over predetermined time points. Data represent the mean ± SD of three independent experiments, each of which was performed in triplicate.
In vitro uptake of CopNEC-FAmB
AmB is not inherently fluorescent in visible light, thus, AmB was tagged with FITC and entrapped in CopNEC. FAmB was characterized using TLC where the retention factor value of FAmB was slightly higher than AmB and far less than FITC (Jain and Kumar, 2010). The in vitro uptake of CopNEC-FAmB (mean 501 FL1-H) was significantly (P < 0.05) higher as compared with FAmB (mean 248 FL1-H) at a similar concentration as shown in Figure 3. The CopNEC is a particulate drug delivery system which is easily taken up by Caco-2 cell monolayer (Aderem and Underhill, 1999; Alexis et al., 2008). The tagged CopNEC-FAmB showed significantly higher uptake (∼2.1) (P < 0.05) in contrast to tagged FAmB in Caco-2 cells as indicated by the significantly higher absorption into the systemic circulation via membranous epithelial cells (M-cells) of the Peyer's patches. It has also been reported that lipid-raft-dependent and clathrin-mediated endocytosis by Caco-2 cell monolayer is evidence for efficient uptake of particles (Gupta et al., 2009).
Figure 3.
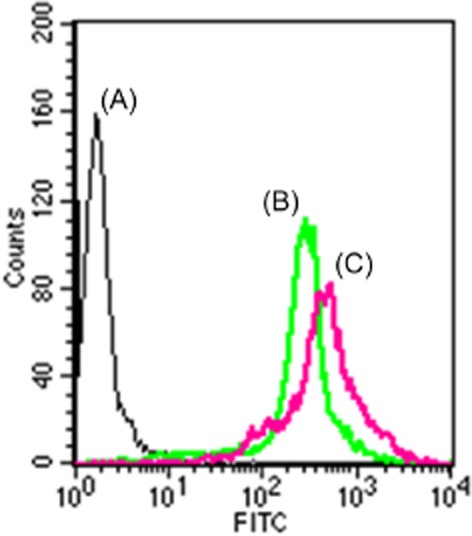
In vitro uptake study of (A) control, (B) FAmB and (C) CopNEC-FAmB on Caco-2 cell lines after 6 h of incubation.
Pharmacokinetics and biodistribution of CopNEC-AmB
AmB plasma concentrations following single oral administration of CopNEC-AmB at doses of 10 and 5 mg·kg−1 and AmB in water at 10 mg·kg−1 dose are shown in Figure 4. The pharmacokinetics of AmB in the formulations were significant different following oral administration (Table 1). The oral administration of CopNEC-AmB resulted in a higher AmB concentration in plasma as compared with oral administration of plain AmB over a period of 48 h. However, after 6 h of oral administration, plasma concentrations of AmB from CopNEC-AmB declined as it was preferentially distributed in the liver and spleen due to physical uptake of the particulate formulations (at about 127 nm).
Figure 4.
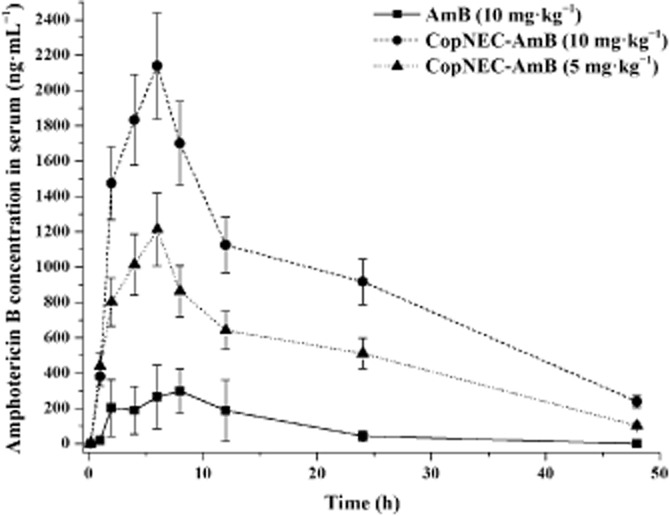
The serum concentration time profile of the various formulations after oral administration in rat. Data represent the mean ± SD of three independent experiments, each of which was performed in triplicate.
The results of the tissue distribution of AmB formulations at the different time points, 6, 24 and 48 h, following oral administration are presented in Table 4. At 6 h, the AmB concentration following oral administration of 10 mg·kg−1 CopNEC-AmB, 5 mg·kg−1 CopNEC-AmB and plain AmB in liver was 16 350 ± 4738, 15 968 ± 4152 and 5661 ± 1273 ng·g−1 respectively. While after 6 h, CopNEC-AmB value in liver was lesser 3687 ± 542 and 2305 ± 376 ng·g−1 for 10 and 5 mg·kg−1, respectively, as it was redistributed towards the spleen as shown in Table 4. The AmB concentration at 24 h, following oral administration of 10 mg·kg−1 CopNEC-AmB, 5 mg·kg−1 CopNEC-AmB and plain AmB in spleen was 6657 ± 973, 6634 ± 1208 and 2108 ± 804 ng·g−1 and in kidney was 1384 ± 137, 295 ± 42 and 1608 ± 148 ng·g−1 respectively. In addition, the level of CopNEC-AmB in lung tissues was slightly higher as compared with free AmB, as shown in Table 4.
Table 4.
Biodistribution of AmB formulations after oral administration
| AmB equivalent concentration in formulation | Concentration of AmB in liver (ng·g−1) | Concentration of AmB in spleen (ng·g−1) | Concentration of AmB in lung (ng·g−1) | Concentration of AmB in kidney (ng·g−1) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 6 h | 24 h | 48 h | 6 h | 24 h | 48 h | 6 h | 24 h | 48 h | 6 h | 24 h | 48 h | |
| AmB (10 mg·kg−1) | 5 661 ± 1 273 | 1348 ± 217 | 175 ± 28 | 819 ± 116 | 2108 ± 804 | 129 ± 27 | 47 ± 8 | 125 ± 26 | 17 ± 2 | 15 717 ± 843 | 1608 ± 148 | 202 ± 37 |
| CopNEC-AmB (10 mg·kg−1) | 16 350 ± 4 738 | 3687 ± 542 | 2525 ± 189 | 2872 ± 343 | 6657 ± 973 | 581 ± 83 | 76 ± 9 | 345 ± 49 | 95 ± 16 | 3 812 ± 184 | 1384 ± 137 | 179 ± 26 |
| CopNEC-AmB (5 mg·kg−1) | 15 968 ± 4 152 | 2305 ± 376 | 1923 ± 273 | 1124 ± 215 | 6634 ± 1208 | 411 ± 72 | 49 ± 6 | 147 ± 23 | 51 ± 11 | 1 403 ± 57 | 295 ± 42 | 151 ± 18 |
Each value represents the mean ± SD (n = 3). AmB, amphotericin B; Cop, copaiba oil; NEC, nanoemulsified carrier.
In vitro antileishmanial activity
These results are presented in the form of IC50 as well as IC90 values (Figure 5). The IC50 value of CopNEC-AmB, plain AmB, blank CopNEC and Cop was 0.018 ± 0.004 (P < 0.05), 0.214 ± 0.06 and 0.33 ± 0.08, 0.38 ± 0.11 μg·mL−1 respectively. The difference in IC50 between CopNEC and Cop was negligible, which suggests that antileishmanial activity is only due to Cop and the other excipients in the CopNEC formulation; PC and TPGS have no role in the in vitro activity against leishmania parasite.
Figure 5.
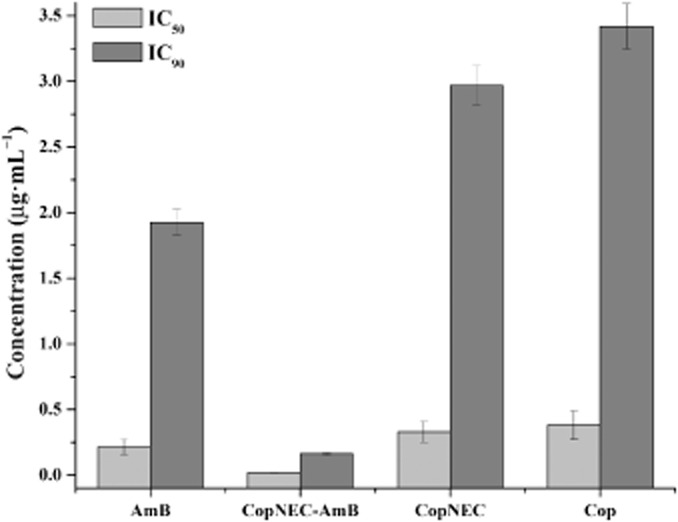
In vitro antileishmanial activity (IC50 and IC90) of CopNEC-AmB, AmB, CopNEC and Cop in Leishmania donovani amastigote-infected macrophages observed after 48 h of incubation (n = 3).
Mitochondrial membrane potential
Many studies suggest that the mitochondrion is a possible target for programmed cell death by apoptosis in Leishmania, occurring via the loss of Δψm (Sudhandiran and Shaha, 2003; dos Santos et al., 2011). The rapid fall of relative Δψm [measured from ratio of J aggregate (for intact mitochondria) to J monomer (for damaged mitochondria)] was observed from 0 to 48 h post-treatment of all formulations as shown in Figure 6. However, the maximum drop in Δψm occurred with the Cop-based formulations (CopNEC-AmB, CopNEC and Cop) compared with free AmB at all time points (Figure 6).
Figure 6.
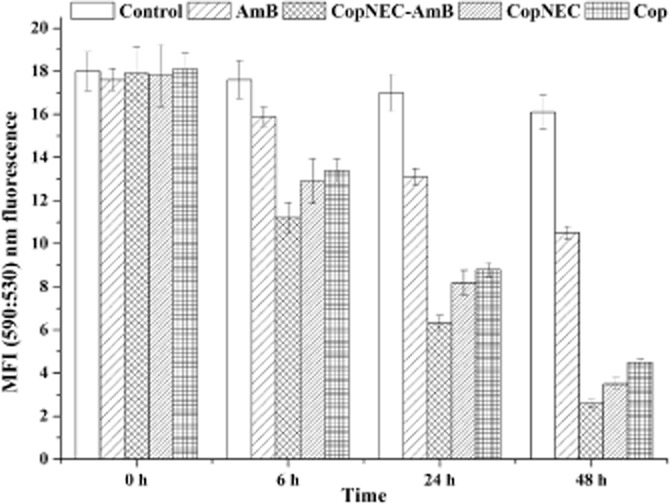
Decrease in mitochondrial potential (Δψm) in promastigotes following treatment with AmB, CopNEC-AmB, CopNEC and Cop for the different times (0, 6, 24 and 48 h) and staining with the potentiometric probe JC-1 (10 mM). Δψm values are expressed as the ratio of mean fluorescence intensity (MFI) at 590:530 nm fluorescence.
In vitro toxicity
Haemolysis study
Leishmaniasis treatment with conventional AmB formulation leads to the development of anaemic conditions in patients mainly because of active haemolysis and that occurs by pore formation and change of electrolyte balance in erythrocytes. AmB is well known for its haemolysis and this is a major drawback. We have also observed similar effects in our present study. The CopNEC-AmB, CopNEC and Cop exhibited haemotoxicity up to 18.20 ± 1.27%, 9.8 ± 0.69% and 14.6 ± 1.02%, respectively, whereas AmB showed haemotoxicity up to 69.8 ± 4.89% as depicted in Figure 7A.
Figure 7.

The effect of CopNEC-AmB and AmB equivalent to AmB concentrations of 5, 10 and 20 μg·mL−1 on (A) RBCs collected from Wistar rat's blood and (B) J774A macrophage cell line (n = 3). Histopathology of kidney from treated mice after an oral dose of normal saline in control group (C), 10 mg·kg−1 AmB in AmB group (D), 10 mg·kg−1 AmB in CopNEC-AmB group and 0.2 mL·kg−1 AmB in Cop group (F).
Cytotoxicity in J774A macrophage cells
The MTT proliferation assay for cell viability has been described as a suitable method for detection of biomaterial toxicity (Mosmann, 1983). The cytotoxicity induced by various concentrations of the test formulations is shown in Figure 7B. The order of cytotoxicity of the formulations against the J774A cell line was AmB > CopNEC-AmB > Cop > CopNEC.
Animal toxicity after repeated administration
The histopathological analysis of kidney tissues revealed a normal pattern in control, CopNEC-AmB and Cop groups as shown in Figure 7C, E, F. However, in the AmB group, the kidney tissue showed necrotic tubules and their subsequent fragmentation was observed as shown in Figure 7D, which varied in level from critical to wide ranging.
Discussion
In the present study, we have developed a lipid-based nanoemulsified carrier system for increasing the stability of AmB in gastrointestinal fluids and improving its pharmacokinetics to make it suitable for oral administration. The CopNEC-AmB was also investigated for its synergistic increase in antileishmanial activity as compared with plain AmB and Cop.
The poor solubility, oral bioavailability and toxicity of AmB are intrinsic limitations for it as a first-line treatment for leishmaniasis. The poor solubility and oral bioavailability of AmB were addressed by formulating AmB in a lipid-based nanoemulsified carrier system. In this lipid-based nanoemulsified carrier system, the AmB was solubilized in the oily core of Cop and/or at the interface of the nanocarrier structure (Narang et al., 2007). Various pharmaceutical parameters are responsible for particle characteristics such as type of surfactant and co-surfactant, surfactant to co-surfactant ratio, drug to oil ratio and concentration of oil in the formulation. In this study, the effect of size, PDI, ζ potential and EE of CopNEC-AmB was investigated by varying one parameter at a time and keeping the others constant. The TPGS has a hydrophilic domain comprising of polyethylene glycol (PEG) and a hydrophobic domain of tocopherol succinate (Mondal et al., 2013) giving it a large surface area making it suitable as a nanoemulsified carrier for an amphiphilic drug such as AmB, as compared with the other surfactants used in this study. Therefore, TPGS provides a smaller PDI in the F 2 formulation. The F 2 formulation contains PC, which as a result of its lipidic nature increases the ζ potential and EE compared with span 80 and propylene glycol (Were et al., 2003). Moreover, TPGS is a potent efflux pump (e.g. P-gp, MRP1 and BCRP) inhibitor that acts through inhibition of substrate-induced efflux pump without inducing significant ATPase activity on its own (Collnot et al., 2007) and has been utilized against multidrug resistance strains of Leishmania parasite (Collnot et al., 2006). Furthermore, the TPGS component of the matrix material has been routinely used as a stabilizer and improves a drug's oral bioavailability.
The evaluation of stability in gastrointestinal transit is somewhat difficult because the composition, volume, dynamics, motility and the gastrointestinal transit vary among patients; apart from this, food, disease or drug intake might modify the transit conditions (McConnell et al., 2008). Due to this, it was difficult to select in vitro media to predict fate of CopNEC-AmB in vivo. Due to the variable characteristics of gastrointestinal transit, several simulated fluids are used to test the solubility of the drug but none have been developed to measure the stability of the nanocarriers. Accordingly, the stability of CopNEC-AmB was analysed in two different media. Firstly, it was measured in the SGF having a pH of 1.2, which contains pepsin, a digestive protease that simulates fasting conditions in the stomach. As shown in Figure 2A, CopNEC-AmB was stable in SGF for 2 h, the size of the nanocarrier was not altered and 1–2% of the initial amount of encapsulated AmB was released. As CopNEC-AmB is composed of TPGS (mixture of PEG and tocopherol succinate) on its surface, it was anticipated that this would protect AmB and PC from acidic degradation by lowering the free energy at the interface between the two phases and resisting coalescence and flocculation (Tobio et al., 2000; Vila et al., 2002). The CopNEC-AmB was found to be stable in both stomach and intestinal fluids. The size and EE of the CopNEC-AmB nanocarriers were preserved over a period of 8 h after contact with SIF, as shown in Figure 2B. The same results were observed in previous studies of nanoparticles coated with PEG, where they were protected from the rapid degradation induced by the enzymatic activity of pancreatin (Tobio et al., 2000). Consequently, the TPGS being at the surface of the CopNEC-AmB protected the particles against pancreatin enzyme activity. CopNEC-AmB was also found to be stable when kept at different temperatures for a period of 3 months. It did not show any sign of phase separation and drug precipitation because of the presence of TPGS which acts as stabilizer.
The stability of AmB in the CopNEC formulation during gastrointestinal transit was reflected in in vivo pharmacokinetic studies, which revealed 4.08- and 7.2-fold increase in Cmax and a 5.81- and 10.54-fold increase in the AUC0–48 in the case of lower (5 mg·kg−1) and higher (10 mg·kg−1) doses of CopNEC-AmB, respectively, as compared with the drug alone (Figure 4). The lower AUC of AmB was due to its instability or metabolism in the gut and its poor permeability (Robbie et al., 1999). Usually nanoparticulate systems have been reported to be absorbed intact into the systemic circulation via the Peyer's patches (M-cells), but the net absorption of the material via this transport was reported to be extremely low (Kagan et al., 2007). In our case, a significantly higher concentration was achieved with both 10 and 5 mg·kg−1 after oral administration because CopNEC-AmB was protected from the acidic environment. Also this carrier promoted the absorption of AmB in the stomach (Fan et al., 2013), by affecting the fluidity of the enterocyte membrane, thereby improving the permeability and cellular uptake of this formulation, as shown in Figure 3. The plasma concentration of AmB following oral administration of the higher dose (10 mg·kg−1) was slightly higher (not twice) than the smaller dose (5 mg·kg−1) as shown in Figure 4. This may be because the co-administration of PC and TPGS delayed gastric empting and reduced peristaltic movement (Gershkovich et al., 2009). The mean residence time and terminal half-life of 10 mg·kg−1 CopNEC-AmB was significantly longer as compared with 5 mg·kg−1 CopNEC-AmB, as shown in Table 3. This is an example of ‘flip-flop’ kinetics due to a prolonged absorption phase caused by a higher volume of PC and TPGS in 10 mg·kg−1 CopNEC-AmB in our rat model.
Table 3.
Pharmacokinetic parameters of AmB formulations
| AmB equivalent concentration in formulation | Cmax (ng·mL−1) | AUC0–48 (h × ng·mL−1) | CL (mL·h−1·kg−1) | MRT (h) | t1/2 (h) |
|---|---|---|---|---|---|
| AmB (10 mg·kg−1) | 297.2 ± 47.4 | 4 102.7 ± 889.3 | 2525.7 ± 647.5 | 11.0 ± 2.4 | 5.9 ± 0.9 |
| CopNEC-AmB (5 mg·kg−1) | 1213.1 ± 109.8 | 23 868.1 ± 2 183.6 | 388.4 ± 34.4 | 19.7 ± 3.1 | 14.6 ± 2.4 |
| CopNEC-AmB (10 mg·kg−1) | 2139.9 ± 127.2 | 43 222.7 ± 6 537.8 | 207.4 ± 30.6 | 22.0 ± 3.7 | 15.9 ± 1.7 |
Each result represents the mean ± SD (n = 3). AmB, amphotericin B; CL, clearance; Cop, copaiba oil; MRT, mean residence time; NEC, nanoemulsified carrier.
The tissue distribution of CopNEC-AmB at 10 mg·kg−1 dose was slightly higher in liver and spleen than with 5 mg·kg−1. This may be due to the fact that TPGS and PC shifted the preferential distribution of CopNEC-AmB 10 mg·kg−1 towards kidney tissues (Gershkovich et al., 2009; 2010) compared to that seen with plain AmB, as shown in Table 4. Moreover, CopNEC-AmB showed a significantly higher distribution in the liver and spleen than plain AmB, as shown in Table 4. This was probably caused by the nanocarrier CopNEC-AmB being associated with increased phagocytosis (Asthana et al., 2013a), as shown in Figure 3, and TPGS in the formulation enhances its uptake by macrophages (Zou and Gu, 2013). On the other hand, CopNEC-AmB showed slightly higher distribution in lung tissues as compared with free AmB at equivalent doses. The AmB concentration in the liver and spleen was significantly higher 48 h following oral administration of CopNEC, while the concurrent concentration in plasma was very low. This might be due to the fact that AmB, once it reaches the liver and spleen, remains there for a prolonged period of time, supported by multicompartmental analysis of the data (Smith et al., 2007), slowly redistributing back to the bloodstream and subsequently being slowly eliminated from the body, as shown in Table 3. The CopNEC-AmB showed a different biodistribution pattern from AmB due to the higher affinity of AmB to bind to plasma proteins and lipoproteins (Hong et al., 2007). It was reported that different excipients in the formulations of AmB, such as Peceol/DSPE-PEG2000 (Gershkovich et al., 2009) and poloxamer 188 (Echevarria et al., 2000), affect the distribution characteristics of the drug in the body.
The in vitro antileishmanial activity of CopNEC-AmB was significantly improved as compared with AmB (P < 0.05) as shown in Figure 5. Our data also illustrate that CopNEC could be a potential carrier for antileishmanial drugs into macrophages, and interestingly, CopNEC also had a synergistic action on antileishmanial activity by itself because of the antileishmanial property of Cop. Cop has been reported to have potent antileishmanial activity due to β-caryophyllene (Santos et al., 2008) that is metabolized by the macrophages to an active molecule with leishmanicidal activity (Izumi et al., 2012). The β-caryophyllene alone did not show antileishmanial activity against amastigote-like forms but in Cop, both sesquiterpene and diterpene are present, which might have affected the active component by a synergistic effect (Dos Santos et al., 2012). Additionally, fractionation of Cop results in fractions that are less active than the crude oil itself (Lima et al., 2003). The Cop causes drastic changes in the morphology of Leishmania parasite with rupture of the plasma membrane. Additionally, it is also responsible for the protein denaturation of the cell surface of Leishmania parasite (Dos Santos et al., 2012). Many studies suggest that cell surface (carbohydrates associated with lipids to form glycolipids) of parasitic protozoa plays an important role in various processes including cell recognition, cell adhesion, regulation of cell growth, expression of surface antigens and receptors (De Souza, 1995; Naderer et al., 2004). Consequently, surface changes caused by Cop treatment may affect the parasite–host interaction and thus reduce the infection. The blank CopNEC showed significant (P < 0.01) antileishmanial activity compared with plain AmB (Figure 5). CopNEC with AmB was significantly active as compared with plain AmB. The activity of the former was enhanced several fold because of considerable changes in the morphology and ultrastructural damage of mitochondria, observed as a change in mitochondrial potential (shown in Figure 6), and denaturation of the plasma membrane of Leishmania parasites due to Cop (Dos Santos et al., 2012). In accord with this, the maintenance of mitochondrion properties such as the mitochondrial membrane potential (ΔΨm), synthesis of ATP and oxidative phosphorylation, is essential for the survival of Leishmania because these parasites have a single mitochondrion (Sen et al., 2004; Roy et al., 2008). The CopNEC is also responsible for the loss of mitochondrial membrane potential, the swelling of mitochondria and cytoplasmic vacuolization that lead to the death of the parasites (Dos Santos et al., 2012). Hence, the use of Cop in the CopNEC-AmB formulation synergistically enhanced the antileishmanial activity of AmB (Park et al., 2011; Eid et al., 2012). This activity of CopNEC-AmB leads to a decrease in the amount of AmB required to treat infected animals which resulted in a reduction of drug-related toxicities.
Figure 7A illustrates the haemolytic effect of AmB, which was high even at the minimum concentration used in the experiment; this was expected because AmB lyses RBCs by an active process involving pore formation. The CopNEC-AmB showed a smaller haemolytic effect possibly due to encapsulation of AmB inside the emulsified core of the formulation and, moreover, the presence of a low concentration of dimeric and multimeric aggregated forms of AmB reduced the degree of haemolysis (Gupta et al., 2014b). The CopNEC caused less haemolysis than plain Cop because Cop forms the core of the CopNEC formulation. The results showed that CopNEC-AmB was haemocompatible and is likely to exert minimum haemolysis in vivo. Our findings also suggest that CopNEC-AmB was less cytotoxic (higher % cell viability) than AmB, as shown in Figure 7B. This is in agreement with the previous results showing that the aggregated form of AmB is more toxic against human erythrocytes and other cultured mammalian cells than the monomeric form of the drug (Legrand et al., 1992). This was further supported by histopathological analysis of kidney tissue, which revealed a normal pattern in control, CopNEC-AmB and Cop groups, as depicted in Figure 7C, E, F, whereas in the AmB group it showed patchy tubular epithelial necrosis and glomerular debris that varied in extent from focal to extensive, as shown in Figure 7D. The reduced toxicity of CopNEC-AmB was further explained by the pharmacokinetic and biodistribution profile of the CopNEC-AmB, as much less of this compound was distributed in the kidney tissue compared to AmB alone. Furthermore, oral administration of CopNEC-AmB decreased its nephrotoxicity as the plasma creatinine level was reduced (data not shown) compared with that after plain AmB. It was also reported in literature that acute single doses of Cop up to 2 g·kg−1 do not cause genotoxic and mutagenic effects in mice (Almeida et al., 2012).
Hence, the antileishmanial activity of Cop and its ability to enhance drug uptake/absorption effectively means it has a synergistic effect on the killing of L. donovani parasites when present in the CopNEC-AmB formulation, which would achieve a favourable therapeutic effect. This also means that a smaller amount of AmB is required for therapeutic treatment and subsequently a decrease in dose-related toxicity. Furthermore, the oral administration of a drug is the preferred choice for all groups of patients as this also reduces its toxicity.
Conclusion
A novel approach for enhancing the oral bioavailability of AmB was employed based on the use of a nanoemulsified carrier system. The formulation was stable throughout the gastrointestinal tract; therefore, it could be used as an alternative strategy to enteric coating, which is at present used to protect AmB from acidic degradation in the stomach. According to the pharmacokinetics and tissue distribution data, the oral CopNEC formulation of AmB could have potential as an improved therapeutic treatment of visceral leishmaniasis. Moreover, our formulation contained Cop, an antileishmanial component which synergistically enhanced parasite clearance in equivalent doses of CopNEC-AmB compared with plain AmB in a biologically safe therapeutic regimen. In a nutshell, CopNEC-AmB could be a highly effective and low-cost alternative to the drugs currently available for treating leishmaniasis and fungal infections.
Acknowledgments
P. K. G. thankfully acknowledges the Council of Scientific and Industrial Research (CSIR), New Delhi, India for awarding a fellowship. The authors thank the Electron Microscope Facility at Department of Anatomy, All India Institute of Medical Sciences (New Delhi, India) for providing the electron microscope analysis. The authors thank the CSIR network project BIOCERAM (ESC 0103) for their financial support. This is CDRI manuscript number 8959.
Glossary
- AmB
amphotericin B
- Cop
copaiba oil
- CopNEC
copaiba oil containing nanoemulsified carrier
- CopNEC-AmB
AmB encapsulated nano-emulsified carrier
- CopNEC-FAmB
FAmB encapsulated CopNEC
- CPCSEA
Council for the Purpose of Control and Supervision of Experiments on Animals
- DMAc
dimethylacetamide
- EE
entrapment efficiency
- FAmB
FITC-tagged AmB
- HRTEM
high-resolution transmission electron microscopy
- IAEC
Indian Animal Ethics Committee
- MTT
(3-[4,5-dimethylthiazol-2-yl]-2, 5-diphenyltetrazolium bromide)
- PC
L-α-phosphatidyl choline
- PDI
poly dispersity index
- PEG
poly ethylene glycol
- PF68
pluronic F-68
- PF127
pluronic F-127
- SGF
simulated gastric fluid
- SIF
simulated intestinal fluid
- TPGS
d-alpha tocopheryl polyethylene glycol 1000 succinate
Author contributions
P. R. M. and A. D. conceived, designed and facilitated the study and the ethical approval; P.K.G. conducted the formulation and characterization of CopNECs; P.K.G., A.J., S.A., V.T.B., N.S., P.S. and M.S. managed the in vivo and in vitro studies involving Leishmania donovani; P. R. M., A. D., S.K.R. and P. K.G. analysed the data and prepared the first draft of the paper; P. R. M., A. D., S.A. and P.K.G. all reviewed and critically appraised the paper; P. R. M. had the primary responsibility for the final content of the paper; and all authors agreed on the final version.
Conflict of interest
None to declare.
References
- Aderem A, Underhill DM. Mechanisms of phagocytosis in macrophages. Annu Rev Immunol. 1999;17:593–623. doi: 10.1146/annurev.immunol.17.1.593. [DOI] [PubMed] [Google Scholar]
- Alexis F, Pridgen E, Molnar LK, Farokhzad OC. Factors affecting the clearance and biodistribution of polymeric nanoparticles. Mol Pharm. 2008;5:505–515. doi: 10.1021/mp800051m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almeida MR, Darin JD, Hernandes LC, de Souza Ramos MF, Antunes LM, de Freitas O. Genotoxicity assessment of copaiba oil and its fractions in Swiss mice. Genet Mol Biol. 2012;35:664–672. doi: 10.1590/S1415-47572012005000052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amidon GL, Lennernas H, Shah VP, Crison JR. A theoretical basis for a biopharmaceutic drug classification: the correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm Res. 1995;12:413–420. doi: 10.1023/a:1016212804288. [DOI] [PubMed] [Google Scholar]
- Asthana S, Gupta PK, Chaurasia M, Dube A, Chourasia MK. Polymeric colloidal particulate systems: intelligent tools for intracellular targeting of antileishmanial cargos. Expert Opin Drug Deliv. 2013a;10:1633–1651. doi: 10.1517/17425247.2013.838216. [DOI] [PubMed] [Google Scholar]
- Asthana S, Jaiswal AK, Gupta PK, Pawar VK, Dube A, Chourasia MK. Immunoadjuvant chemotherapy of visceral leishmaniasis in hamsters using amphotericin B-encapsulated nanoemulsion template-based chitosan nanocapsules. Antimicrob Agents Chemother. 2013b;57:1714–1722. doi: 10.1128/AAC.01984-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bern C, Adler-Moore J, Berenguer J, Boelaert M, den Boer M, Davidson RN, et al. Liposomal amphotericin B for the treatment of visceral leishmaniasis. Clin Infect Dis. 2006;43:917–924. doi: 10.1086/507530. [DOI] [PubMed] [Google Scholar]
- Boelaert M, Criel B, Leeuwenburg J, Van Damme W, Le Ray D, Van der Stuyft P. Visceral leishmaniasis control: a public health perspective. Trans R Soc Trop Med Hyg. 2000;94:465–471. doi: 10.1016/s0035-9203(00)90055-5. [DOI] [PubMed] [Google Scholar]
- Brandao MG, Zanetti NN, Oliveira P, Grael CF, Santos AC, Monte-Mor RL. Brazilian medicinal plants described by 19th century European naturalists and in the Official Pharmacopoeia. J Ethnopharmacol. 2008;120:141–148. doi: 10.1016/j.jep.2008.08.004. [DOI] [PubMed] [Google Scholar]
- Choudhury H, Gorain B, Karmakar S, Biswas E, Dey G, Barik R, et al. Improvement of cellular uptake, in vitro antitumor activity and sustained release profile with increased bioavailability from a nanoemulsion platform. Int J Pharm. 2014;460:131–143. doi: 10.1016/j.ijpharm.2013.10.055. [DOI] [PubMed] [Google Scholar]
- Collnot EM, Baldes C, Wempe MF, Hyatt J, Navarro L, Edgar KJ, et al. Influence of vitamin E TPGS poly(ethylene glycol) chain length on apical efflux transporters in Caco-2 cell monolayers. J Control Release. 2006;111:35–40. doi: 10.1016/j.jconrel.2005.11.005. [DOI] [PubMed] [Google Scholar]
- Collnot EM, Baldes C, Wempe MF, Kappl R, Huttermann J, Hyatt JA, et al. Mechanism of inhibition of P-glycoprotein mediated efflux by vitamin E TPGS: influence on ATPase activity and membrane fluidity. Mol Pharm. 2007;4:465–474. doi: 10.1021/mp060121r. [DOI] [PubMed] [Google Scholar]
- Cunningham AC. Parasitic adaptive mechanisms in infection by leishmania. Exp Mol Pathol. 2002;72:132–141. doi: 10.1006/exmp.2002.2418. [DOI] [PubMed] [Google Scholar]
- De Souza W. Structural organization of the cell surface of pathogenic protozoa. Micron. 1995;26:405–430. doi: 10.1016/0968-4328(95)00010-0. [DOI] [PubMed] [Google Scholar]
- Delmas G, Park S, Chen ZW, Tan F, Kashiwazaki R, Zarif L, et al. Efficacy of orally delivered cochleates containing amphotericin B in a murine model of aspergillosis. Antimicrob Agents Chemother. 2002;46:2704–2707. doi: 10.1128/AAC.46.8.2704-2707.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dey R, Moraes CT. Lack of oxidative phosphorylation and low mitochondrial membrane potential decrease susceptibility to apoptosis and do not modulate the protective effect of Bcl-x(L) in osteosarcoma cells. J Biol Chem. 2000;275:7087–7094. doi: 10.1074/jbc.275.10.7087. [DOI] [PubMed] [Google Scholar]
- Dos Santos AO, Ueda-Nakamura T, Dias Filho BP, da Veiga Junior VF, Nakamura CV. Copaiba oil: an alternative to development of new drugs against leishmaniasis. Evid Based Complement Alternat Med. 2012;2012:898419. doi: 10.1155/2012/898419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Echevarria I, Barturen C, Renedo MJ, Troconiz IF, Dios-Vieitez MC. Comparative pharmacokinetics, tissue distributions, and effects on renal function of novel polymeric formulations of amphotericin B and amphotericin B-deoxycholate in rats. Antimicrob Agents Chemother. 2000;44:898–904. doi: 10.1128/aac.44.4.898-904.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eid SY, El-Readi MZ, Wink M. Digitonin synergistically enhances the cytotoxicity of plant secondary metabolites in cancer cells. Phytomedicine. 2012;19:1307–1314. doi: 10.1016/j.phymed.2012.09.002. [DOI] [PubMed] [Google Scholar]
- Fan Z, Wu J, Fang X, Sha X. A new function of Vitamin E-TPGS in the intestinal lymphatic transport of lipophilic drugs: enhancing the secretion of chylomicrons. Int J Pharm. 2013;445:141–147. doi: 10.1016/j.ijpharm.2013.01.070. [DOI] [PubMed] [Google Scholar]
- Gershkovich P, Wasan EK, Lin M, Sivak O, Leon CG, Clement JG, et al. Pharmacokinetics and biodistribution of amphotericin B in rats following oral administration in a novel lipid-based formulation. J Antimicrob Chemother. 2009;64:101–108. doi: 10.1093/jac/dkp140. [DOI] [PubMed] [Google Scholar]
- Gershkovich P, Wasan EK, Sivak O, Li R, Zhu X, Werbovetz KA, et al. Visceral leishmaniasis affects liver and spleen concentrations of amphotericin B following administration to mice. J Antimicrob Chemother. 2010;65:535–537. doi: 10.1093/jac/dkp465. [DOI] [PubMed] [Google Scholar]
- Gupta GK, Kansal S, Misra P, Dube A, Mishra PR. Uptake of biodegradable gel-assisted LBL nanomatrix by Leishmania donovani-infected macrophages. AAPS PharmSciTech. 2009;10:1343–1347. doi: 10.1208/s12249-009-9334-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta PK, Asthana S, Jaiswal AK, Kumar V, Verma A, Shukla P, et al. Exploitation of lectinized lipo-polymerosome bearing amphotericin B to target macrophages for effective management of visceral leishmaniasis. Bioconjug Chem. 2014a;25:1091–1102. doi: 10.1021/bc500087h. [DOI] [PubMed] [Google Scholar]
- Gupta PK, Jaiswal AK, Kumar V, Verma A, Dwivedi P, Dube A, et al. Covalent functionalized self-assembled lipo-polymerosome bearing amphotericin B for better management of leishmaniasis and its toxicity evaluation. Mol Pharm. 2014b;11:951–963. doi: 10.1021/mp400603t. [DOI] [PubMed] [Google Scholar]
- Halde C, Newcomer VD, Wright ET, Sternberg TH. An evaluation of amphotericin B in vitro and in vivo in mice against Coccidioides immitis and Candida albicans, and preliminary observations concerning the administration of amphotericin B to man. J Invest Dermatol. 1957;28:217–231. doi: 10.1038/jid.1957.26. discussion, 231–232. [DOI] [PubMed] [Google Scholar]
- Hofstra W, de Vries-Hospers HG, van der Waaij D. Concentrations of amphotericin B in faeces and blood of healthy volunteers after the oral administration of various doses. Infection. 1982;10:223–227. doi: 10.1007/BF01666915. [DOI] [PubMed] [Google Scholar]
- Hong Y, Shaw PJ, Tattam BN, Nath CE, Earl JW, Stephen KR, et al. Plasma protein distribution and its impact on pharmacokinetics of liposomal amphotericin B in paediatric patients with malignant diseases. Eur J Clin Pharmacol. 2007;63:165–172. doi: 10.1007/s00228-006-0240-x. [DOI] [PubMed] [Google Scholar]
- Izumi E, Ueda-Nakamura T, Veiga VF, Jr, Pinto AC, Nakamura CV. Terpenes from Copaifera demonstrated in vitro antiparasitic and synergic activity. J Med Chem. 2012;55:2994–3001. doi: 10.1021/jm201451h. [DOI] [PubMed] [Google Scholar]
- Jain JP, Kumar N. Self assembly of amphiphilic (PEG)(3)-PLA copolymer as polymersomes: preparation, characterization, and their evaluation as drug carrier. Biomacromolecules. 2010;11:1027–1035. doi: 10.1021/bm1000026. [DOI] [PubMed] [Google Scholar]
- Kagan L, Gershkovich P, Mendelman A, Amsili S, Ezov N, Hoffman A. The role of the lymphatic system in subcutaneous absorption of macromolecules in the rat model. Eur J Pharm Biopharm. 2007;67:759–765. doi: 10.1016/j.ejpb.2007.04.002. [DOI] [PubMed] [Google Scholar]
- Kayser O, Olbrich C, Yardley V, Kiderlen AF, Croft SL. Formulation of amphotericin B as nanosuspension for oral administration. Int J Pharm. 2003;254:73–75. doi: 10.1016/s0378-5173(02)00686-5. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kima PE. The amastigote forms of Leishmania are experts at exploiting host cell processes to establish infection and persist. Int J Parasitol. 2007;37:1087–1096. doi: 10.1016/j.ijpara.2007.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kvist LP, Christensen SB, Rasmussen HB, Mejia K, Gonzalez A. Identification and evaluation of Peruvian plants used to treat malaria and leishmaniasis. J Ethnopharmacol. 2006;106:390–402. doi: 10.1016/j.jep.2006.01.020. [DOI] [PubMed] [Google Scholar]
- Legrand P, Romero EA, Cohen BE, Bolard J. Effects of aggregation and solvent on the toxicity of amphotericin B to human erythrocytes. Antimicrob Agents Chemother. 1992;36:2518–2522. doi: 10.1128/aac.36.11.2518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lima SR, Junior VF, Christo HB, Pinto AC, Fernandes PD. In vivo and in vitro studies on the anticancer activity of Copaifera multijuga Hayne and its fractions. Phytother Res. 2003;17:1048–1053. doi: 10.1002/ptr.1295. [DOI] [PubMed] [Google Scholar]
- McConnell EL, Fadda HM, Basit AW. Gut instincts: explorations in intestinal physiology and drug delivery. Int J Pharm. 2008;364:213–226. doi: 10.1016/j.ijpharm.2008.05.012. [DOI] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mondal S, Roy P, Das S, Halder A, Mukherjee A, Bera T. In vitro susceptibilities of wild and drug resistant Leishmania donovani amastigote stages to andrographolide nanoparticle: role of vitamin E derivative TPGS for nanoparticle efficacy. PLoS ONE. 2013;8:e81492. doi: 10.1371/journal.pone.0081492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mookerjee Basu J, Mookerjee A, Banerjee R, Saha M, Singh S, Naskar K, et al. Inhibition of ABC transporters abolishes antimony resistance in Leishmania infection. Antimicrob Agents Chemother. 2008;52:1080–1093. doi: 10.1128/AAC.01196-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods. 1983;65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- Murray HW, Berman JD, Davies CR, Saravia NG. Advances in leishmaniasis. Lancet. 2005;366:1561–1577. doi: 10.1016/S0140-6736(05)67629-5. [DOI] [PubMed] [Google Scholar]
- Naderer T, Vince JE, McConville MJ. Surface determinants of Leishmania parasites and their role in infectivity in the mammalian host. Curr Mol Med. 2004;4:649–665. doi: 10.2174/1566524043360069. [DOI] [PubMed] [Google Scholar]
- Narang AS, Delmarre D, Gao D. Stable drug encapsulation in micelles and microemulsions. Int J Pharm. 2007;345:9–25. doi: 10.1016/j.ijpharm.2007.08.057. [DOI] [PubMed] [Google Scholar]
- Park KR, Nam D, Yun HM, Lee SG, Jang HJ, Sethi G, et al. β-Caryophyllene oxide inhibits growth and induces apoptosis through the suppression of PI3K/AKT/mTOR/S6K1 pathways and ROS-mediated MAPKs activation. Cancer Lett. 2011;312:178–188. doi: 10.1016/j.canlet.2011.08.001. [DOI] [PubMed] [Google Scholar]
- Pham TT, Loiseau PM, Barratt G. Strategies for the design of orally bioavailable antileishmanial treatments. Int J Pharm. 2013;454:539–552. doi: 10.1016/j.ijpharm.2013.07.035. [DOI] [PubMed] [Google Scholar]
- Robbie G, Wu TC, Chiou WL. Poor and unusually prolonged oral absorption of amphotericin B in rats. Pharm Res. 1999;16:455–458. doi: 10.1023/a:1011961322883. [DOI] [PubMed] [Google Scholar]
- Roy A, Ganguly A, BoseDasgupta S, Das BB, Pal C, Jaisankar P, et al. Mitochondria-dependent reactive oxygen species-mediated programmed cell death induced by 3,3′-diindolylmethane through inhibition of F0F1-ATP synthase in unicellular protozoan parasite Leishmania donovani. Mol Pharmacol. 2008;74:1292–1307. doi: 10.1124/mol.108.050161. [DOI] [PubMed] [Google Scholar]
- Santos AO, Ueda-Nakamura T, Dias Filho BP, Veiga Junior VF, Pinto AC, Nakamura CV. Effect of Brazilian copaiba oils on Leishmania amazonensis. J Ethnopharmacol. 2008;120:204–208. doi: 10.1016/j.jep.2008.08.007. [DOI] [PubMed] [Google Scholar]
- dos Santos AO, Costa MA, Ueda-Nakamura T, Dias-Filho BP, da Veiga-Junior VF, de Souza Lima MM, et al. Leishmania amazonensis: effects of oral treatment with copaiba oil in mice. Exp Parasitol. 2011;129:145–151. doi: 10.1016/j.exppara.2011.06.016. [DOI] [PubMed] [Google Scholar]
- Santos AO, Izumi E, Ueda-Nakamura T, Dias-Filho BP, Veiga-Junior VF, Nakamura CV. Antileishmanial activity of diterpene acids in copaiba oil. Mem Inst Oswaldo Cruz. 2013;108:59–64. doi: 10.1590/S0074-02762013000100010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen N, Das BB, Ganguly A, Mukherjee T, Tripathi G, Bandyopadhyay S, et al. Camptothecin induced mitochondrial dysfunction leading to programmed cell death in unicellular hemoflagellate Leishmania donovani. Cell Death Differ. 2004;11:924–936. doi: 10.1038/sj.cdd.4401435. [DOI] [PubMed] [Google Scholar]
- Smith PJ, Olson JA, Constable D, Schwartz J, Proffitt RT, Adler-Moore JP. Effects of dosing regimen on accumulation, retention and prophylactic efficacy of liposomal amphotericin B. J Antimicrob Chemother. 2007;59:941–951. doi: 10.1093/jac/dkm077. [DOI] [PubMed] [Google Scholar]
- Soares DC, Portella NA, Ramos MF, Siani AC, Saraiva EM. Trans-β-Caryophyllene: an effective antileishmanial compound found in commercial copaiba oil (Copaifera spp.) Evid Based Complement Alternat Med. 2013;2013:761323. doi: 10.1155/2013/761323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudhandiran G, Shaha C. Antimonial-induced increase in intracellular Ca2+ through non-selective cation channels in the host and the parasite is responsible for apoptosis of intracellular Leishmania donovani amastigotes. J Biol Chem. 2003;278:25120–25132. doi: 10.1074/jbc.M301975200. [DOI] [PubMed] [Google Scholar]
- Tobio M, Sanchez A, Vila A, Soriano II, Evora C, Vila-Jato JL, et al. The role of PEG on the stability in digestive fluids and in vivo fate of PEG-PLA nanoparticles following oral administration. Colloids Surf B Biointerfaces. 2000;18:315–323. doi: 10.1016/s0927-7765(99)00157-5. [DOI] [PubMed] [Google Scholar]
- Trouiller P, Olliaro P, Torreele E, Orbinski J, Laing R, Ford N. Drug development for neglected diseases: a deficient market and a public-health policy failure. Lancet. 2002;359:2188–2194. doi: 10.1016/S0140-6736(02)09096-7. [DOI] [PubMed] [Google Scholar]
- Vila A, Sanchez A, Tobio M, Calvo P, Alonso MJ. Design of biodegradable particles for protein delivery. J Control Release. 2002;78:15–24. doi: 10.1016/s0168-3659(01)00486-2. [DOI] [PubMed] [Google Scholar]
- Wanis TM, Combe EC, Grant AA. Measurement of the viscosity of irreversible hydrocolloids. J Oral Rehabil. 1993;20:379–384. doi: 10.1111/j.1365-2842.1993.tb01621.x. [DOI] [PubMed] [Google Scholar]
- Wasan EK, Bartlett K, Gershkovich P, Sivak O, Banno B, Wong Z, et al. Development and characterization of oral lipid-based amphotericin B formulations with enhanced drug solubility, stability and antifungal activity in rats infected with Aspergillus fumigatus or Candida albicans. Int J Pharm. 2009;372:76–84. doi: 10.1016/j.ijpharm.2009.01.003. [DOI] [PubMed] [Google Scholar]
- Wasan EK, Gershkovich P, Zhao J, Zhu X, Werbovetz K, Tidwell RR, et al. A novel tropically stable oral amphotericin B formulation (iCo-010) exhibits efficacy against visceral Leishmaniasis in a murine model. PLoS Negl Trop Dis. 2010;4:e913. doi: 10.1371/journal.pntd.0000913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Were LM, Bruce BD, Davidson PM, Weiss J. Size, stability, and entrapment efficiency of phospholipid nanocapsules containing polypeptide antimicrobials. J Agric Food Chem. 2003;51:8073–8079. doi: 10.1021/jf0348368. [DOI] [PubMed] [Google Scholar]
- Wu W, Wang Y, Que L. Enhanced bioavailability of silymarin by self-microemulsifying drug delivery system. Eur J Pharm Biopharm. 2006;63:288–294. doi: 10.1016/j.ejpb.2005.12.005. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Tan S, Feng SS. Vitamin E TPGS as a molecular biomaterial for drug delivery. Biomaterials. 2012;33:4889–4906. doi: 10.1016/j.biomaterials.2012.03.046. [DOI] [PubMed] [Google Scholar]
- Zou T, Gu L. TPGS emulsified zein nanoparticles enhanced oral bioavailability of daidzin: in vitro characteristics and in vivo performance. Mol Pharm. 2013;10:2062–2070. doi: 10.1021/mp400086n. [DOI] [PubMed] [Google Scholar]