

Abstract
Background and Purpose
The cation channel transient receptor potential canonical (TRPC) 6 has been associated with several pathologies including focal segmental glomerulosclerosis, pulmonary hypertension and ischaemia reperfusion-induced lung oedema. We set out to discover novel inhibitors of TRPC6 channels and investigate the therapeutic potential of these agents.
Experimental Approach
A library of potential TRPC channel inhibitors was designed and synthesized. Activity of the compounds was assessed by measuring intracellular Ca2+ levels. The lead compound SAR7334 was further characterized by whole-cell patch-clamp techniques. The effects of SAR7334 on acute hypoxic pulmonary vasoconstriction (HPV) and systemic BP were investigated.
Key Results
SAR7334 inhibited TRPC6, TRPC3 and TRPC7-mediated Ca2+ influx into cells with IC50s of 9.5, 282 and 226 nM, whereas TRPC4 and TRPC5-mediated Ca2+ entry was not affected. Patch-clamp experiments confirmed that the compound blocked TRPC6 currents with an IC50 of 7.9 nM. Furthermore, SAR7334 suppressed TRPC6-dependent acute HPV in isolated perfused lungs from mice. Pharmacokinetic studies of SAR7334 demonstrated that the compound was suitable for chronic oral administration. In an initial short-term study, SAR7334 did not change mean arterial pressure in spontaneously hypertensive rats (SHR).
Conclusions and Implications
Our results confirm the role of TRPC6 channels in hypoxic pulmonary vasoregulation and indicate that these channels are unlikely to play a major role in BP regulation in SHR. SAR7334 is a novel, highly potent and bioavailable inhibitor of TRPC6 channels that opens new opportunities for the investigation of TRPC channel function in vivo.
Tables of Links
| LIGANDS | |
|---|---|
| Econazole | |
| OAG, 1-oleoyl-2-acetyl-sn-glycerol | |
| SKF96365 |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (a,bAlexander et al., 2013a, b).
Introduction
Since the discovery of transient receptor potential (TRP) cation channels in the early 1990s, significant efforts have been directed towards the development of pharmacological TRP modulators. Such tools are essential for the confirmation and extension of our current understanding of TRP channel function that mainly originates from genetic studies (Moran et al., 2011). Moreover, due to the role of certain TRP channels in human pathologies (Nilius and Owsianik, 2010; Wu et al., 2010a), these ion channels represent interesting drug targets.
A potential candidate for therapeutic intervention is the TRPC6 channel. This member of the canonical TRP subfamily is expressed in several tissues, in particular brain, kidney and vasculature. The TRPC6 channel is activated by the PLC-generated hydrolysis product diacylglycerol (Hofmann et al., 1999) and can be modulated by diverse signalling mechanisms including lipid messengers (Fleming et al., 2007; Kwon et al., 2007) and reactive oxygen species (Ding et al., 2011; Anderson et al., 2014). Thus, TRPC6 channels serve as signal integrators, coupling activation of various receptors and intracellular networks to cation – most importantly Ca2+ – entry and plasma membrane depolarization. Functional TRPC channels are most likely tetramers (Mio et al., 2005; Barrera et al., 2007) and TRPC6 channels have been shown to form homomers as well as heteromers with other subunits, especially the closely related TRPC3 and TRPC7 channels (Hofmann et al., 2002). Such multimeric assembly can create channel complexes with diverse properties which complicates the elucidation of channel function in native tissues. Nevertheless, studies in genetically modified animals and human genetic data have provided important insights into the (patho)physiological role of TRPC6 channels. TRPC6−/− mice were found to have increased vascular contractility and raised BP. This somewhat unexpected finding can be explained by the compensatory up-regulation of constitutively more active TRPC3 channels that increases basal Ca2+ influx into myocytes (Dietrich et al., 2005). Although these results highlight the interplay and importance of both TRPC3 and TRPC6 channels in the regulation of systemic vascular tone in mice, it remains unclear to date how TRPC6 channels modulate BP in a normal genetic context or in disease. In addition to systemic vasoregulation, experiments in transgenic and knock-out mice revealed several other functions of TRPC6 channels in, for example, cardiac hypertrophic signalling (Wu et al., 2010b; Seo et al., 2014), regulation of lung endothelial permeability (Weissmann et al., 2012) and platelet aggregation (Harper et al., 2013). Moreover, direct evidence for the involvement of TRPC6 channels in human disease has been found in patients with inherited focal segmental glomerulosclerosis (FSGS). In affected individuals, activating mutations of TRPC6 channels were identified as the cause of Ca2+ influx into podocytes and subsequent dysfunction of the glomerular filtration barrier (Reiser et al., 2005; Winn et al., 2005). Therefore, limiting TRPC6 channel activity may be a novel treatment option for FSGS and other conditions characterized by pathological Ca2+ entry.
It would be highly desirable to validate the therapeutic value of inhibition of TRPC6 channels using pharmacological tools. A number of compounds, including SKF96365, econazole, W7 and compound 8009-5364 (Harteneck and Gollasch, 2011; Urban et al., 2012; Bon and Beech, 2013) have been described as inhibitors of TRPC6 channel activity. However, the utility of these substances is limited due to their low potency and poor selectivity. More recently, a series of anilino-thiazole TRPC3/6 channel blockers have been described by Washburn et al. (2013). These compounds are highly potent but have a low oral bioavailability and are not suitable for chronic pharmacological studies.
We describe here a novel class of TRPC6 channel inhibitors that combine nanomolar activity with a favourable pharmacokinetic profile, which allows pharmacological investigation of channel function in vivo.
Methods
Pharmacophore modelling
The pharmacophore model was built with Catalyst (Accelrys, San Diego, CA, USA). Up to 250 conformers were generated for each training molecule. For model generation, the default parameter settings were used with the following features allowed for model generation: acceptor, basic, ring aromatic and hydrophobic features.
Chemistry
Aminoindanol derivatives with cis or trans geometries were synthesized (Figure 1B) starting from 2-bromo-1-indanones by nucleophilic substitution with amines (1.2 eq. amine in acetone, 1.6 eq. of K2CO3 as base, room temperature, 2 h), subsequent carbonyl reduction (cis: 1.5 eq. L-selectride in tetrahydrofuran (THF), 0°C – room temperature, 10–48 h; trans: 6.0 eq. NaBH4, 1.5 eq. HCl, THF/H2O, room temperature, 3–24 h) and finally O-alkylation/arylation (2.0 eq. alkyl halide, 5.0 eq. Ag2O, toluene, 85°C, 2–5 h / 3.0 eq. aryl fluoride, 5.0 eq. NaH, DMSO, 150°C, 10–24 h). Trans geometries, in particular with aryloxy substituents (R5 = aryl), were selectively accessible by epoxide opening of indene oxide with amines (1.3 eq. amine in MeCN, reflux, 10–24 h) and a Mitsunobu reaction with double inversion via an aziridinium intermediate (Freedman et al., 1991) [1.2 eq R5-OH, 2.5 eq. polymer(polysterene)-bound PPh3, 2.0 eq. diisopropyl azodicarboxylate, THF, room temperature, 10–24 h]. Stereochemically pure SAR7334 was selectively synthesized starting from (1R,2S)-indene oxide (Larrow et al., 1999).
Figure 1.
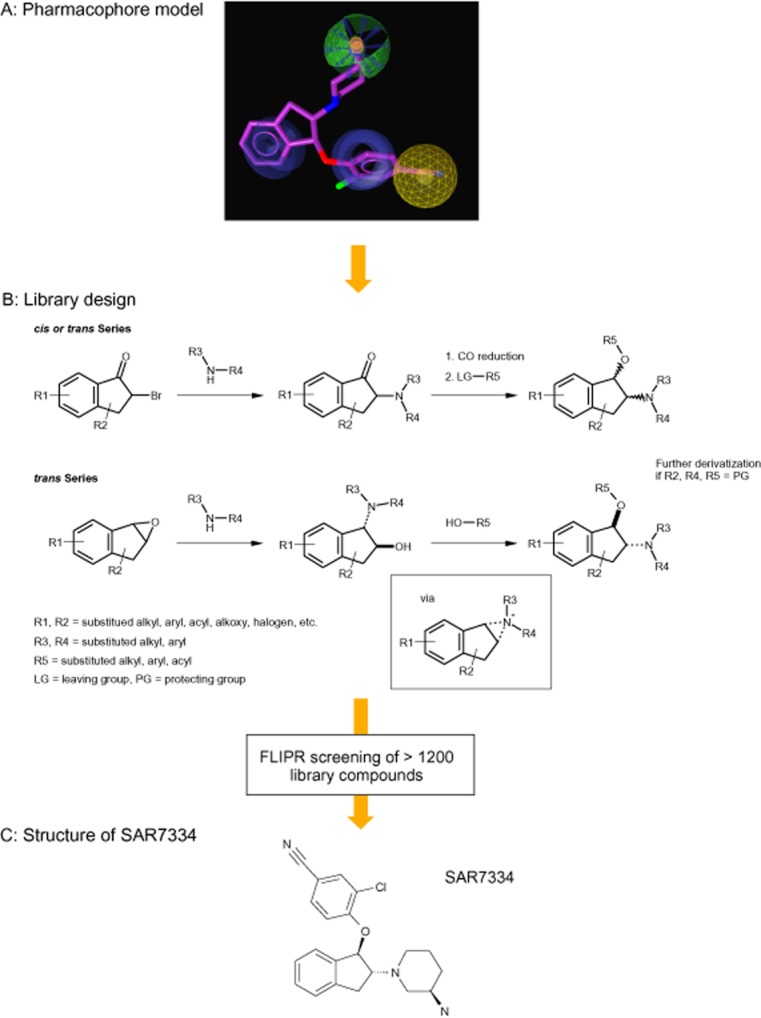
Discovery of SAR7334 using pharmacophore-guided design of focused aminoindanol libraries. Pharmacophoric information derived from a selection of known analogues of the cation channel inhibitor SKF96365 were translated into library designs and synthesized. Promising aminoindanol derivatives were further investigated with emphasis on stereochemical positioning of relevant substituents. (A) Mapping of SAR7334 to the TRPC6 pharmacophore model. The red sphere corresponds to a positive ionizable moiety, the light blue sphere indicates lipophilic features, while brown spheres mark ring aromatic features. The mapping of SAR7334 to the pharmacophore was generated with LigandScout (Inteligand Software-Entwicklungs und Consulting GmbH, Maria Enzersdorf, Austria). (B) Aminoindanol derivatives with cis or trans geometries were accessed from 2-bromo-1-indanones by nucleophilic substitution with amines, carbonyl reduction and subsequent O-alkylation/arylation. Trans geometries, in particular with aryloxy substituents (R5 = aryl) were realized by epoxide opening of indene oxide with amines and a Mitsunobu reaction with double inversion. (C) Structure of SAR7334.
Detailed information and an explicit experimental pathway for the preparation of SAR7334 is provided in Supporting Information Fig. S2.
Cell culture and cell line generation
Cells were grown at 37°C in a humidified atmosphere (5% or 7% CO2) under standard cell culture conditions. Stable HEK cell lines expressing recombinant hTRPC6 (GenBank accession number AF080394) or TRPC7 channels (GenBank accession number AJ272034) under the control of a tetracycline-inducible promoter were generated using the Flp-In T-Rex (FITR) system (Invitrogen, Karlsruhe, Germany). TRPC6 and TRPC7 HEK-FITR cells were maintained in DMEM (with GlutaMAX I, 4.5 g·L−1 glucose and 110 mg·mL−1 sodium pyruvate) supplemented with 10% (v/v) FBS (Biochrom, Berlin, Germany), 1 mM glutamine, 1 mM MEM sodium pyruvate, 40 μg·mL−1 hygromycin and 15 μg·mL−1 blasticidine HCl. Channel expression was induced by supplementing the growth medium for 18–24 h with 1 μg·mL−1 doxycycline. hTRPC3 channels (GenBank accession number NM_003305) were stably expressed in CHO cells using a proprietary high expression vector (Steinbeis – Transferzentrum für Angewandte Biologische Chemie, Mannheim, Germany) and maintained in HAM's F12 supplemented with 10% (v/v) FBS (Biochrom), 1 mM glutamine, 0.6 mg·mL−1 geneticin, 100 U·mL−1 penicillin and 100 μg·mL−1 streptomycin.
Fluo-4 measurement of intracellular calcium concentration ([Ca2+]i)
Ca2+ measurements were performed at room temperature using a fluorometric imaging plate reader (FLIPR; Molecular Devices, Sunnyvale, CA, USA). Cells grown on black poly-D-lysine-coated 96-well plates (Greiner, Frickenhausen, Germany) were washed with standard extracellular solution (140 mM NaCl, 1 mM MgCl2, 5.4 mM KCl, 2 mM CaCl2, 10 mM HEPES, 10 mM glucose, pH 7.35) and stained with dye solution (2 μM Fluo-4 AM, 0.02% pluronic F127, 0.1% BSA in standard extracellular solution) for 30 min at room temperature. The cells were rinsed and incubated with standard extracellular solution supplemented with different concentrations of the test compound or vehicle for 10 min.
Ca2+ entry into TRPC3/6/7-expressing cells was elicited by application of the diacylglycerol, 1-oleoyl-2-acetyl-sn-glycerol (OAG). For calculation of SAR7334-induced inhibition, fluorescence values were plotted over time and the AUC was considered as a measure of Ca2+ influx.
Electrophysiological techniques
Whole-cell patch-clamp measurements were performed essentially as described (Miehe et al., 2012). An intracellular pipette solution containing 120 mM CsOH, 120 mM gluconic acid, 2 mM MgCl2, 3 mM CaCl2, 5 mM Cs4-BAPTA, 10 mM HEPES (pH 7.4 adjusted with gluconic acid) and an extracellular bath solution containing 140 mM NaCl, 5.4 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 10 mM glucose, 10 mM HEPES (pH 7.4 adjusted with NaOH) were used. Cells grown on poly-L-lysine-coated coverslips were continuously superfused with extracellular solution and compounds were applied using an ALA BPS-8 perfusion system (ALA Scientific Instruments, Westbury, NY, USA). Cells were held at −70 mV and current-voltage relationships were measured every 2 or 3 s by applying voltage ramps (180 ms) from −100 to +80 mV. All experiments were performed at room temperature.
Ethics statement
All animal care and experimental procedures were performed according to Sanofi Ethical Committee guidelines and to the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health. Sanofi is an authorized institution to house and handle laboratory animals according §11 German Animal Welfare Act. Procedures were approved by the competent authorities (Regierungspräsidium Darmstadt or Regierungspräsidium Giessen). All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010). A total of 26 animals were used in the experiments described here.
Measurement of hypoxic pulmonary vasoconstriction (HPV) in isolated perfused and ventilated lungs
C57/BL6N mice were anaesthetised with xylazine and ketamine and anticoagulated with heparin as previously described (Weissmann et al., 2004; Fuchs et al., 2011). Male 6–8 weeks old mice were from Charles River Laboratories (Sulzfeld, Germany). In brief, lungs were explanted during deep anaesthesia and artificially ventilated and perfused blood free at 2 mL·min−1 at 37°C with Krebs-Henseleit buffer containing 120 mM NaCl, 4.3 mM KCl, 1.1 mM KH2PO4, 2.4 mM CaCl2, 1.3 mM MgCl2, 13.32 mM glucose, 5% (w/v) hydroxyethylamylopectin and 23.8 mM NaHCO3. The left atrial pressure was set at 2.0 mmHg. Positive pressure ventilation (250 μL tidal volume, 90 breath·min−1 and 2 cm H2O positive end-expiratory pressure) was performed with a mixture containing 21% O2, 5.3% CO2, balanced with N2 (normoxia) or 1% O2, 5.3% CO2 (hypoxia). The pressure in the pulmonary artery and in the left atrium was measured. Changes in pulmonary artery pressure directly reflect changes in pulmonary vascular tone as the lungs are perfused at constant flow. Lungs were ventilated in an alternating mode with hypoxia (10 min) or normoxia (15 min) to induce acute HPV. In such a sequence of repetitive hypoxic ventilation manoeuvres, increasing doses of SAR7334 were applied 5 min prior to the next hypoxic ventilation manoeuvre. For application the SAR7334 stock solution (2 mM in 100% DMSO) was diluted 1:100 in perfusion buffer and appropriate amounts were cumulatively added to the recirculating perfusate (15 mL). The first application was performed after the second hypoxic ventilation period. The strength of acute HPV is given as the maximum increase of pulmonary arterial pressure in each hypoxic ventilation period, referenced to the strength of the second hypoxic manoeuvre (set at 100%).
In vivo determination of SAR7334 pharmacokinetics
Plasma concentrations of SAR7334 were determined in a serial sampling study after single oral administration of the compound (250 g) in 30% glycopherol/cremophor (75/25) 70% glucose (5%) solution to male Sprague Dawley rats (Harlan Winkelmann, Borchen, Germany). From each animal, eight plasma samples (approximately 200 µL blood were taken by tail tip sampling) were collected over 24 h and stored below −15°C until analysis. After addition of the precipitant solution (acetonitrile) containing an analogous internal standard, the test item SAR7334 was detected by LC-MS/MS, using an Agilent LC (Series 1200; Agilent Technologies, Santa Clara, CA, USA) with CTC HTC PAL auto sampler (CTC Analytics AG, Zwingen, Switzerland) and a Sciex API4000 (AB Sciex, Toronto, Canada) mass spectrometer in the positive ion mode. Using a sample volume of 50 μL, the lower limit of quantitation was 2.0 ng·mL-1 and the linear range was between 2.0 and 2000 ng·mL−1.
Telemetric assessment of BP
Adult male (6 months old) spontaneously hypertensive rats (SHR; Harlan Winkelmann) were treated on two consecutive days. On day one, the animals received 1 mL·kg-1 vehicle by oral gavage. After 24 h, rats received either vehicle alone or were treated with 10 mg·kg-1 SAR7334. Telemetric measurement of BP was performed as described (Lohn et al., 2009). In brief, a telemetric device (TL11M2-C50-PXT PMP, Data Sciences International, St. Paul, MN, USA) was placed between the aorta and the vena cava and the catheter tip of the transmitter was inserted into the aorta. Systolic BP, diastolic BP and heart rate were acquired continuously at a sampling rate of 500 Hz and data were stored as 5 min averages. Mean arterial pressure was calculated from systolic and diastolic pressure and low-pass filtered using the fast Fourier transform function of the vendor software (Dataquest A.R.T. V4.0, Data Sciences International) for better visualization of time-dependent BP variations. For statistical analysis, raw data were averaged over a 6 h period starting 2 h after application of vehicle or SAR7334 (labelled ‘post’). This interval corresponded to the maximal plasma levels of SAR7334 (see Figure 6). Baseline data (labelled ‘pre’) were sampled over the same time interval on the day before treatment.
Figure 6.
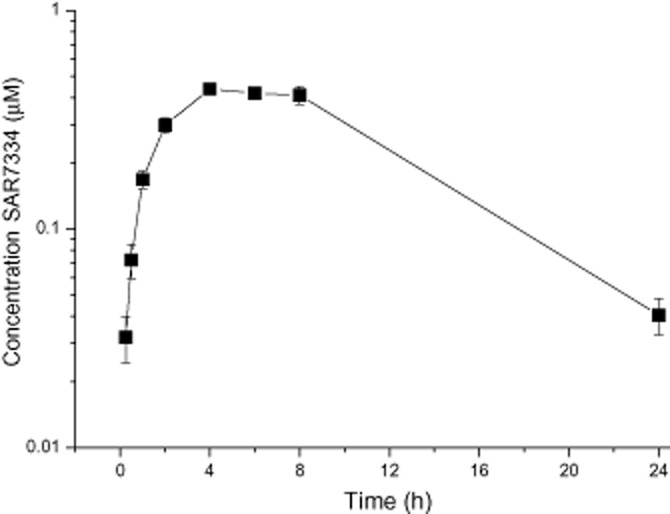
Pharmacokinetic profile of SAR7334. Plasma concentrations of SAR7334 were determined after single oral administration of 10 mg·kg−1 of the compound to male Sprague Dawley rats. Data are given as means ± SEM (n = 3).
Data analysis
Averaged data are expressed as means ± SEM. Repeated measures two-way anova was performed with GraphPad Prism (GraphPad Software Inc., La Jolla, CA, USA) to analyse treatment effects on BP. P-< 0.05 was considered as statistically significant. IC50 values were calculated with Origin 6.0 software (Microcal Software Inc., Northampton, MA, USA). Data from [Ca2+]i measurements were normalized to the area under the fluorescence curve in the absence of SAR7334 and fitted using a standard three-parameter logistic model. The relative current inhibition in patch-clamp experiments was defined as the decrease of the OAG-induced current after application of SAR7334 normalized to the maximal OAG-induced current before drug application. Dose response curves were fitted to the equation: f = 100 − (100/(1 + (C/IC50)n)); where f is the relative inhibition, C is the applied drug concentration and n is the Hill coefficient. For isolated perfused lung experiments, statistical analysis was performed by Student's t-test.
Materials
DMEM, HAM's F12, glutamine, MEM sodium pyruvate, hygromycin B, blasticidin HCl, geneticin, Fluo-4 AM, Pluronic F127 and Cs4-BAPTA were from Invitrogen; MgCl2 and MgSO4 were from Merck (Darmstadt, Germany); doxycycline was from BD Biosciences (Heidelberg, Germany), and OAG was from Avanti Polar Lipids Inc. (Alabaster, AL, USA). All other chemicals were from Sigma-Aldrich (Munich, Germany).
Results
We identified several close analogues of the cation channel inhibitor SKF96365 by similarity and substructure searches as potential inhibitors of TRPC6 channels and investigated the ability of these compounds to block TRPC6-mediated Ca2+ entry in TRPC6-HEK-FITR cells. For the substructure search, the relative position and type of aromatic rings were systematically varied. Examples of identified molecules with activities in the μM range are given in Supporting Information Fig. S1. Validated actives were subsequently used to derive a pharmacophore model with Catalyst (Kurogi and Guner, 2001; Guner et al., 2004). Next, several scaffolds and geometries were evaluated for their potential to inhibit TRPC6 activity using this pharmacophore. One of the best matching structures was the aminoindanol scaffold as illustrated with the derivative SAR7334 (Figure 1A).
Capitalizing on the variable chemistry of this structural class, we designed a library of more than 1200 diversely substituted aminoindanol derivatives with a special emphasis on the stereochemical positioning of substituents (Figure 1B). While a cis relationship at the indane was realized starting from the bromoketone, the eventually more interesting trans configuration was quantitatively accessed using indene oxide as starting material. Potential asymmetric centres in the substituents (R1-5) were introduced with the corresponding starting materials. FLIPR screening of this library identified several compounds with TRPC6 channel blocking activity. Among the most potent inhibitors was SAR7334 (Figure 1C) featuring a trans relationship of the substituents at the indane five-membered ring. Stereochemically defined access to SAR7334 is possible in four steps starting from indene (Supporting Information Fig. S2).
SAR7334 reduced TRPC6-mediated Ca2+ influx with an IC50 of 9.5 nM. TRPC3 and TRPC7 channels, the two closest TRPC6 channel homologues, were also inhibited although at significantly higher concentrations (IC50 = 282 nM and 226 nM respectively) (Figure 2).
Figure 2.

Inhibition of Ca2+ influx via diacylglycerol-sensitive TRPC channels by SAR7334. Time-dependent changes of intracellular Ca2+ concentration in Fluo-4-loaded induced TRPC6- HEK-FITR cells (A), TRPC3-CHO cells (C) and TRPC7 HEK-FITR cells (E) were measured using FLIPR. TRPC-mediated Ca2+ influx following application of 30 μM OAG (+OAG) was dose-dependently reduced in cells pre-incubated with the indicated concentrations of SAR7334. Control traces (con) were recorded during application of extracellular solution. Representative traces are shown. The relative inhibition of TRPC6 (B), TRPC3 (D) and TRPC7 channels (F) was calculated from the AUC for each concentration SAR7334 and IC50s were derived from the best fit of the data (solid lines) with a logistic model. Data are given as means ± SEM (n = 5–10).
By contrast, the compound did not have appreciable effects on TRPC4 or TRPC5 channels (IC50 > 10 μM, see Supporting Information Fig. S3). To verify the channel-inhibiting activity of SAR7334, we performed patch-clamp experiments on TRPC6-HEK-FITR cells. Application of 50 μM OAG stimulated typical non-selective cation currents through TRPC6 channels in these cells. SAR7334 dose-dependently reduced TRPC6 currents with an IC50 of 7.9 nM (Figure 3).
Figure 3.
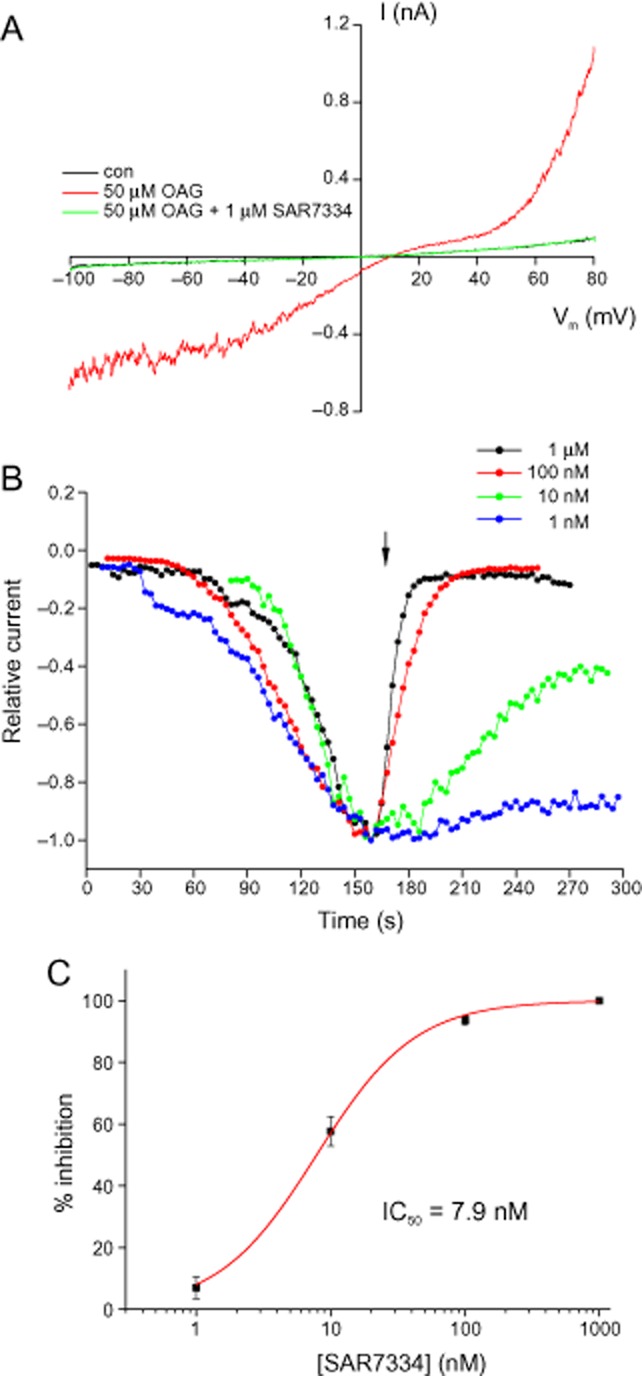
SAR7334 potently inhibits TRPC6 whole-cell currents. TRPC6 currents were elicited in TRPC6-HEK-FITR cells by application of 50 μM OAG. (A) Current traces measured during voltage ramps before and after application of the TRPC6 channel activator OAG and after consecutive additions of SAR7334. (B) Time course and dose dependence of SAR7334-induced TRPC6 inhibition. Whole-cell currents were measured at −70 mV during voltage ramps and plotted versus time. For better comparison of responses, current amplitudes were normalized to the maximal current after addition of OAG and maximal currents were superimposed. The arrow indicates application of SAR7334. (C) TRPC6 currents were measured as in (B) and inhibition was determined in steady state after application of the respective concentration of SAR7334. The dose–response relationship was fitted to a logistic function. Data are given as means ± SEM (n = 3–12).
Thus, SAR7334 blocked TRPC6-mediated ion currents and Ca2+ influx with similar potency. Likewise, recordings from TRPC3- and TRPC5-expressing cells (Supporting Information Fig. S4) confirmed the substantially lower potency of the compound towards these ion channels, as seen in the FLIPR experiments.
As physiological activation of TRPC6 channels is triggered by PLC-coupled receptors, we further investigated how receptor-stimulated TRPC6 responses were affected by SAR7334. For this purpose, we challenged TRPC6-HEK-FITR cells with trypsin which had been shown to activate PLC-linked proteinase-activated receptors 2 and to robustly induce intracellular Ca2+ release in HEK cells (Kawabata et al., 1999). In most TRPC6-HEK-FITR cells, trypsin evoked large, rapidly declining TRPC6 currents that reached an almost steady-state level within 30–60 s. This current was substantially reduced by 100 nM SAR7334 (Figure 4), whereas Ca2+ release and, hence, the underlying PLC activity was not attenuated by up to 10 μM of the inhibitor (Supporting Information Fig. S5).
Figure 4.
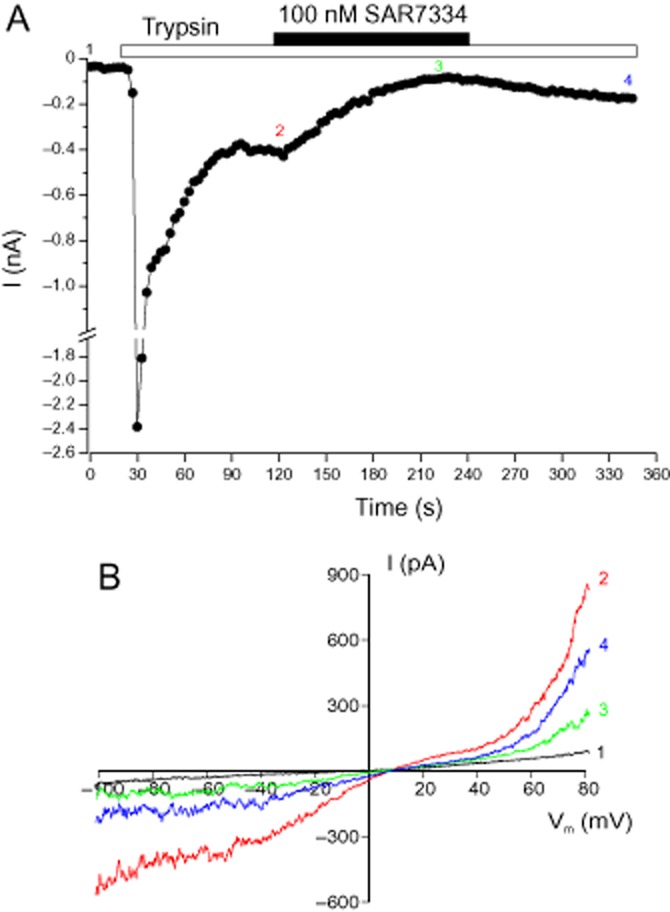
Inhibition of receptor-stimulated TRPC6 currents by of SAR7334. Whole-cell currents were measured in TRPC6-HEK-FITR cells. (A) The time course of TRPC6 currents at −70 mV during application of 200 nM trypsin and 100 nM SAR7334 is depicted. Trypsin and SAR7334 were added to the extracellular solution during the time indicated by the bars. (B) Current responses measured during voltage ramps at the time points indicated in A are shown.
The reduction of trypsin-induced TRPC6 currents by 100 nM SAR7334 amounted to 88 ± 3% (n = 5) which was not different from the corresponding effect on OAG-induced current responses (see Figure 3C).
Having observed inhibition of both OAG- and receptor-induced TRPC6 currents by SAR7334, we next wanted to test the activity of the compound in a relevant physiological model. Acute HPV has been shown to exclusively depend on the activity of TRPC6 channels (Weissmann et al., 2006). We, therefore, investigated the effect of SAR7334 on hypoxic vasoconstriction in perfused isolated lungs from mice and found that this compound abolished hypoxia-induced increases in pulmonary arterial pressure. Half maximal inhibition was achieved at about 100 nM indicating that SAR7334 is able to efficiently block native TRPC6 channels in situ (Figure 5).
Figure 5.
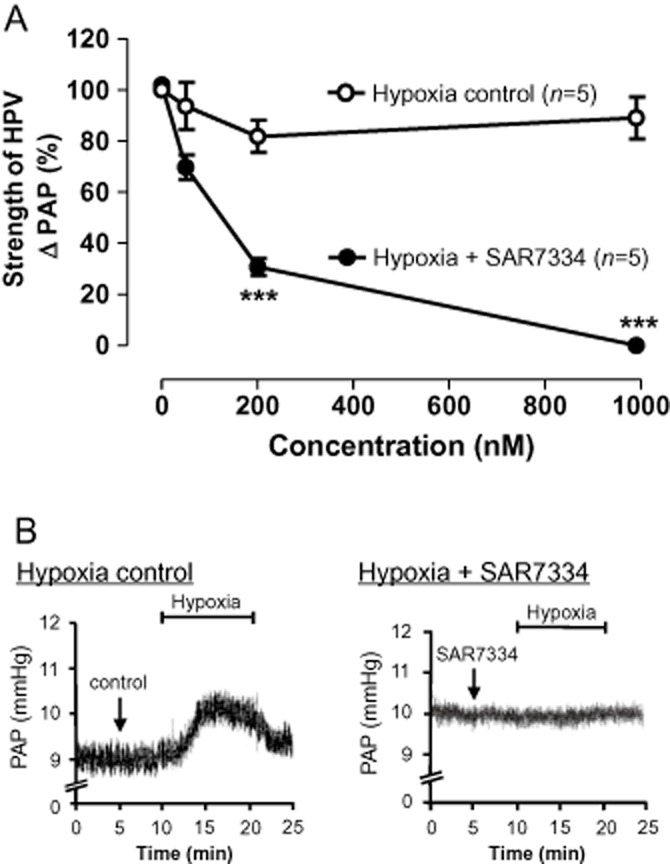
Effect of SAR7334 on hypoxia-induced vasoconstriction in isolated perfused and ventilated lungs. (A) Acute hypoxic pulmonary vasoconstriction (HPV) was assessed as the increase in pulmonary artery pressure (ΔPAP) upon hypoxic ventilation in isolated perfused mouse lungs. Data were normalized to the reference pressure response in the absence of SAR7334 for each experiment. SAR7334 dose-dependently reduced the strength of HPV. SAR7334 had no significant effect on normoxic vascular tone (not shown). Data shown are means ± SEM (n = 5). ***P < 0.001, significantly different from control response. (B) Original recordings of PAP in the absence (left) or presence (right) of 1 μM SAR7334. Hypoxic challenge is indicated by the bars.
Selectivity testing showed that the compound had little effect on other ion channels or receptors (Supporting Information Fig. S3). Notably, store-operated Ca2+ entry driven by ORAI1/STIM1 was also largely resistant to SAR7334 (Supporting Information Fig. S6). Thus, despite the reported interaction of ORAI1 and STIM1 with TRPC6 channels (Liao et al., 2007; Jardin et al., 2009), it seems unlikely that they directly confer sensitivity to SAR7334. In addition to compound selectivity, we were interested in the pharmacokinetic properties of SAR7334. Measuring plasma concentrations in rats revealed that pharmacologically effective concentrations of the substance were reached and maintained for several hours after oral administration (Figure 6).
Taken together, the high potency, good selectivity profile and appropriate pharmacokinetic properties suggest that SAR7334 is a suitable molecule for the investigation of TRPC6-mediated processes in vivo. Therefore, we decided to use the compound to study the effect of inhibiting TRPC6 channels on BP in SHR. For this purpose, BP was monitored telemetrically in conscious animals. Our results illustrated in Figure 7 did not show an effect of SAR7334 on arterial pressure, questioning the role of TRPC6 channels in regulation of systemic vascular tone in rodents.
Figure 7.
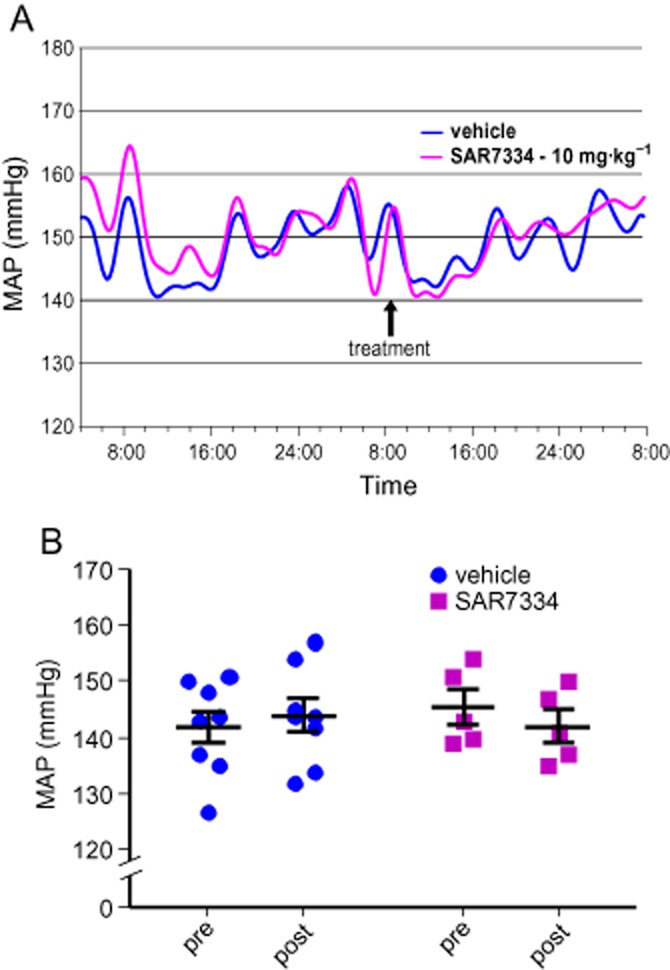
SAR7334 does not affect BP in conscious SHR. Mean arterial pressure (MAP) was measured telemetrically in SHR. The time course of average MAP in vehicle-treated (n = 8) and SAR7334-treated (n = 5) groups is illustrated (A). Statistical comparison of MAP in vehicle- and SAR7334-treated animals 24 h before compound application (pre) and after treatment (post) did not show a time-dependent or treatment-related effect or a significant interaction of both factors (repeated measures two-way anova) (B).
Discussion and conclusions
In this work, we used a rational drug design approach to synthesize libraries of novel small-molecule inhibitors of TRPC6 channels. The aminoindanol derivative SAR7334 was among the most potent TRPC6 channel blockers identified by FLIPR screening. Patch-clamp experiments confirmed that SAR7334 suppressed TRPC6 currents with an IC50 < 10 nM. Thus, the potency of the compound is comparable with that of the recently identified anilino-thiazole series of TRPC6/3 channel inhibitors (Washburn et al., 2013).
Previous studies in TRPC6−/− animals, as well as pharmacological studies, have demonstrated that acute HPV in mice is mediated specifically by TRPC6 channels (Weissmann et al., 2006; Urban et al., 2012). The results of our experiments in the isolated perfused lung model were consistent with these data. Importantly, these results also demonstrated that SAR7334 was able to suppress native TRPC6 channel activity. It is not yet clear why inhibition of HPV required significantly higher concentrations of the compound than blockade of recombinant TRPC6 channel responses. An explanation for the shift in dose dependence often seen in intact lung experiments compared with cellular assays might be that compound access to the target arterial muscle cells is limited by the endothelial barrier. However, it is equally possible that native TRPC6 channel complexes incorporate other TRPC subunits or auxiliary proteins such as ORAI1 and STIM1 that modify channel pharmacology. The TRPC1 channel is co-expressed with TRPC6 channels in precapillary pulmonary artery smooth muscle cells, whereas all other TRPC channel isoforms including TRPC3 and TRPC7 are only present at very low levels (Weissmann et al., 2006). Although the biochemical evidence for the formation of TRPC1/TRPC6 channel complexes is contradictory (Xu et al., 1997; Hofmann et al., 2002; Strubing et al., 2003), functional TRPC1/TRPC6 heteromers have been described in heterologous systems (Storch et al., 2012). Therefore, such channels could potentially exist in pulmonary arteries and account for the observed attenuated sensitivity to SAR7334. In any case, it will be interesting to examine the effect of SAR7334 in models that co-express TRPC6 channels together with putative binding partners in order to compare the pharmacology of such heteromeric assemblies with that of native TRPC6 channel complexes.
The expression of TRPC6 channels in vascular myocytes and its demonstrated role in receptor and pressure-induced Ca2+ influx support the idea that the channel may be involved in BP regulation. Consistent with this proposal, up-regulation of TRPC6 channels has been detected in several hypertensive animal models (Bae et al., 2007; Mita et al., 2010; Zulian et al., 2010). However, the study of vascular effects of these channels in TRPC6−/− mice is obscured by compensatory up-regulation of TRPC3 channels in some, but not all, vascular beds (Dietrich et al., 2005; Weissmann et al., 2006). The hypertensive phenotype of TRPC6−/− animals, caused by exaggerated TRPC3-mediated Ca2+ entry into smooth muscle cells, does not allow us to draw conclusions about the vascular function of TRPC6 channels in wild-type animals. Therefore, we set out to clarify the effect of acute inhibition of TRPC6 channels in SHR, an established rodent hypertension model. Telemetric assessment of BP did not show any effect of SAR7334 application despite the fact that circulating compound levels were sufficient to achieve complete inhibition of TRPC6 channels. This result indicates that TRPC6 channels do not play a major role in systemic BP regulation in SHR. Nevertheless, it remains to be seen whether the same holds true for other hypertensive models, for example those with increased levels of vascular TRPC6 channels, and whether local haemodynamics are modulated by TRPC6 channel inhibition.
SAR7334 combines high potency with a good oral pharmacokinetic profile and reasonable selectivity versus TRPC3 and TRPC7 channels which make this compound a valuable tool for further evaluation of TRPC channel pharmacology in vivo. Clearly, more work is needed to fully explore the potential therapeutic utility of SAR7334 and other TRPC channel blockers. Given the compelling data associating TRPC6 channels with diseases such as FSGS or lung ischaemia reperfusion-induced oedema (Reiser et al., 2005; Winn et al., 2005; Weissmann et al., 2012), we believe that such efforts are worthwhile pursuing.
Acknowledgments
We would like to thank Andrea Leonhardt, Stefan Müller, Tim Koschella and Karin Quanz for expert technical assistance as well as Markus Rupp for performing isolated lung experiments. We also thank Dr. Werngard Czechtizky and Dr. Klaus Steinmeyer for critical reading of the manuscript.
Glossary
- [Ca2+]i
intracellular calcium concentration
- FITR
Flp-In T-Rex
- FLIPR
fluorometric imaging plate reader
- FSGS
focal segmental glomerulosclerosis
- HPV
hypoxic pulmonary vasoconstriction
- OAG
1-oleoyl-2-acetyl-sn-glycerol
- SHR
spontaneously hypertensive rats
- THF
tetrahydrofuran
- TRP
transient receptor potential
- TRPC
transient receptor potential canonical
Author contributions
T. M., M. F., G. H., H.-W. K., S. H., B. F., N. W., W. L., T. S., M. L., K. S., L. W. and C. S. performed the research and analysed the data. T. M., M. F., G. H., H.-W. K., S. H., N. W., W. L., T. S., M. L., K. S., L. W., H. R. and C. S. designed the research. T. M. and C. S. wrote the manuscript.
Conflicts of interests
T Maier, M Follmann, G Hessler, H-W Kleemann, S Hachtel, W Linz, T Schmidt, M Löhn, K Schroeter, L Wang and C Strübing were employees of Sanofi-Aventis Deutschland GmbH at the time this study was conducted. T Maier, M Follmann, G Hessler, H-W Kleemann, S Hachtel, L Wang and C Strübing are inventors on a patent application (WO2011107474 A1) related to this work. T Schmidt is a contributor to WO2011107474 A1 according to applicable German law. N Weissmann was funded by a grant from Sanofi-Aventis Deutschland GmbH.
Supporting Information
Figure S1 Examples of chemical structures used to derive the pharmacophore model for TRPC6 channel inhibitors.
Figure S2 Stereoselective synthesis of SAR7334.
Figure S3 Selectivity profile of SAR7334.
Figure S4 Effect of SAR7334 on heterologously expressed TRPC3 and TRPC5 channels.
Figure S5 SAR7334 does not inhibit trypsin-induced Ca2+ release in TRPC6- HEK-FITR cells.
Figure S6 Intact store-operated Ca2+ entry in SAR7334-treated cells.
References
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Catterall WA, et al. The Concise Guide to PHARMACOLOGY 2013/14: Ion Channels. Br J Pharmacol. 2013a;170:1607–1651. doi: 10.1111/bph.12447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL. Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Enzymes. Br J Pharmacol. 2013b;170:1797–1867. doi: 10.1111/bph.12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson M, Roshanravan H, Khine J, Dryer SE. Angiotensin II activation of TRPC6 channels in rat podocytes requires generation of reactive oxygen species. J Cell Physiol. 2014;229:434–442. doi: 10.1002/jcp.24461. [DOI] [PubMed] [Google Scholar]
- Bae YM, Kim A, Lee YJ, Lim W, Noh YH, Kim EJ, et al. Enhancement of receptor-operated cation current and TRPC6 expression in arterial smooth muscle cells of deoxycorticosterone acetate-salt hypertensive rats. J Hypertens. 2007;25:809–817. doi: 10.1097/HJH.0b013e3280148312. [DOI] [PubMed] [Google Scholar]
- Barrera NP, Shaifta Y, McFadzean I, Ward JP, Henderson RM, Edwardson JM. AFM imaging reveals the tetrameric structure of the TRPC1 channel. Biochem Biophys Res Commun. 2007;358:1086–1090. doi: 10.1016/j.bbrc.2007.05.039. [DOI] [PubMed] [Google Scholar]
- Bon RS, Beech DJ. In pursuit of small molecule chemistry for calcium-permeable non-selective TRPC channels – mirage or pot of gold? Br J Pharmacol. 2013;170:459–474. doi: 10.1111/bph.12274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietrich A, Mederos YS, Gollasch M, Gross V, Storch U, Dubrovska G, et al. Increased vascular smooth muscle contractility in TRPC6-/- mice. Mol Cell Biol. 2005;25:6980–6989. doi: 10.1128/MCB.25.16.6980-6989.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Y, Winters A, Ding M, Graham S, Akopova I, Muallem S, et al. Reactive oxygen species-mediated TRPC6 protein activation in vascular myocytes, a mechanism for vasoconstrictor-regulated vascular tone. J Biol Chem. 2011;286:31799–31809. doi: 10.1074/jbc.M111.248344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming I, Rueben A, Popp R, Fisslthaler B, Schrodt S, Sander A, et al. Epoxyeicosatrienoic acids regulate Trp channel dependent Ca2+ signaling and hyperpolarization in endothelial cells. Arterioscler Thromb Vasc Biol. 2007;27:2612–2618. doi: 10.1161/ATVBAHA.107.152074. [DOI] [PubMed] [Google Scholar]
- Freedman J, Vaal MJ, Huber EW. The Mitsunobu reaction of some indan amino alcohols. J Org Chem. 1991;56:670–672. [Google Scholar]
- Fuchs B, Rupp M, Ghofrani HA, Schermuly RT, Seeger W, Grimminger F, et al. Diacylglycerol regulates acute hypoxic pulmonary vasoconstriction via TRPC6. Respir Res. 2011;12:20. doi: 10.1186/1465-9921-12-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guner O, Clement O, Kurogi Y. Pharmacophore modeling and three dimensional database searching for drug design using catalyst: recent advances. Curr Med Chem. 2004;11:2991–3005. doi: 10.2174/0929867043364036. [DOI] [PubMed] [Google Scholar]
- Harper MT, Londono JE, Quick K, Londono JC, Flockerzi V, Philipp SE, et al. Transient receptor potential channels function as a coincidence signal detector mediating phosphatidylserine exposure. Sci Signal. 2013;6:ra50. doi: 10.1126/scisignal.2003701. [DOI] [PubMed] [Google Scholar]
- Harteneck C, Gollasch M. Pharmacological modulation of diacylglycerol-sensitive TRPC3/6/7 channels. Curr Pharm Biotechnol. 2011;12:35–41. doi: 10.2174/138920111793937943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann T, Obukhov AG, Schaefer M, Harteneck C, Gudermann T, Schultz G. Direct activation of human TRPC6 and TRPC3 channels by diacylglycerol. Nature. 1999;397:259–263. doi: 10.1038/16711. [DOI] [PubMed] [Google Scholar]
- Hofmann T, Schaefer M, Schultz G, Gudermann T. Subunit composition of mammalian transient receptor potential channels in living cells. Proc Natl Acad Sci U S A. 2002;99:7461–7466. doi: 10.1073/pnas.102596199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jardin I, Gomez LJ, Salido GM, Rosado JA. Dynamic interaction of hTRPC6 with the Orai1-STIM1 complex or hTRPC3 mediates its role in capacitative or non-capacitative Ca2+ entry pathways. Biochem J. 2009;420:267–276. doi: 10.1042/BJ20082179. [DOI] [PubMed] [Google Scholar]
- Kawabata A, Saifeddine M, Al-Ani B, Leblond L, Hollenberg MD. Evaluation of proteinase-activated receptor-1 (PAR1) agonists and antagonists using a cultured cell receptor desensitization assay: activation of PAR2 by PAR1-targeted ligands. J Pharmacol Exp Ther. 1999;288:358–370. [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. Animal research: Reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurogi Y, Guner OF. Pharmacophore modeling and three-dimensional database searching for drug design using catalyst. Curr Med Chem. 2001;8:1035–1055. doi: 10.2174/0929867013372481. [DOI] [PubMed] [Google Scholar]
- Kwon Y, Hofmann T, Montell C. Integration of phosphoinositide- and calmodulin-mediated regulation of TRPC6. Mol Cell. 2007;25:491–503. doi: 10.1016/j.molcel.2007.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larrow JF, Roberts E, Verhoeven TR, Ryan KM, Senanayake CH, Reider PJ, et al. (1S,2R)-1-aminoindan-2-ol [1H-inden-2-ol, 1-amino-2,3-dihydro-(1S-cis)-] Org Synth. 1999;76:46–56. [Google Scholar]
- Liao Y, Erxleben C, Yildirim E, Abramowitz J, Armstrong DL, Birnbaumer L. Orai proteins interact with TRPC channels and confer responsiveness to store depletion. Proc Natl Acad Sci U S A. 2007;104:4682–4687. doi: 10.1073/pnas.0611692104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohn M, Plettenburg O, Ivashchenko Y, Kannt A, Hofmeister A, Kadereit D, et al. Pharmacological characterization of SAR407899, a novel rho-kinase inhibitor. Hypertension. 2009;54:676–683. doi: 10.1161/HYPERTENSIONAHA.109.134353. [DOI] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miehe S, Crause P, Schmidt T, Lohn M, Kleemann HW, Licher T, et al. Inhibition of diacylglycerol-sensitive TRPC channels by synthetic and natural steroids. PLoS ONE. 2012;7:e35393. doi: 10.1371/journal.pone.0035393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mio K, Ogura T, Hara Y, Mori Y, Sato C. The non-selective cation-permeable channel TRPC3 is a tetrahedron with a cap on the large cytoplasmic end. Biochem Biophys Res Commun. 2005;333:768–777. doi: 10.1016/j.bbrc.2005.05.181. [DOI] [PubMed] [Google Scholar]
- Mita M, Ito K, Taira K, Nakagawa J, Walsh MP, Shoji M. Attenuation of store-operated Ca2+ entry and enhanced expression of TRPC channels in caudal artery smooth muscle from Type 2 diabetic Goto-Kakizaki rats. Clin Exp Pharmacol Physiol. 2010;37:670–678. doi: 10.1111/j.1440-1681.2010.05373.x. [DOI] [PubMed] [Google Scholar]
- Moran MM, McAlexander MA, Biro T, Szallasi A. Transient receptor potential channels as therapeutic targets. Nat Rev Drug Discov. 2011;10:601–620. doi: 10.1038/nrd3456. [DOI] [PubMed] [Google Scholar]
- Nilius B, Owsianik G. Transient receptor potential channelopathies. Pflugers Arch. 2010;460:437–450. doi: 10.1007/s00424-010-0788-2. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, et al. NC-IUPHAR. The IUPHAR/BPS Guide to PHARMACOLOGY: an expert-driven knowledge base of drug targets and their ligands. Nucl Acids Res. 2014;42(Database Issue):D1098–D1106. doi: 10.1093/nar/gkt1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiser J, Polu KR, Moller CC, Kenlan P, Altintas MM, Wei C, et al. TRPC6 is a glomerular slit diaphragm-associated channel required for normal renal function. Nat Genet. 2005;37:739–744. doi: 10.1038/ng1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo K, Rainer PP, Shalkey Hahn V, Lee DI, Jo SH, Andersen A, et al. Combined TRPC3 and TRPC6 blockade by selective small-molecule or genetic deletion inhibits pathological cardiac hypertrophy. Proc Natl Acad Sci U S A. 2014;111:1551–1556. doi: 10.1073/pnas.1308963111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storch U, Forst AL, Philipp M, Gudermann T, Schnitzler M. Transient receptor potential channel 1 (TRPC1) reduces calcium permeability in heteromeric channel complexes. J Biol Chem. 2012;287:3530–3540. doi: 10.1074/jbc.M111.283218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strubing C, Krapivinsky G, Krapivinsky L, Clapham DE. Formation of novel TRPC channels by complex subunit interactions in embryonic brain. J Biol Chem. 2003;278:39014–39019. doi: 10.1074/jbc.M306705200. [DOI] [PubMed] [Google Scholar]
- Urban N, Hill K, Wang L, Kuebler WM, Schaefer M. Novel pharmacological TRPC inhibitors block hypoxia-induced vasoconstriction. Cell Calcium. 2012;51:194–206. doi: 10.1016/j.ceca.2012.01.001. [DOI] [PubMed] [Google Scholar]
- Washburn DG, Holt DA, Dodson J, McAtee JJ, Terrell LR, Barton L, et al. The discovery of potent blockers of the canonical transient receptor channels, TRPC3 and TRPC6, based on an anilino-thiazole pharmacophore. Bioorg Med Chem Lett. 2013;23:4979–4984. doi: 10.1016/j.bmcl.2013.06.047. [DOI] [PubMed] [Google Scholar]
- Weissmann N, Akkayagil E, Quanz K, Schermuly RT, Ghofrani HA, Fink L, et al. Basic features of hypoxic pulmonary vasoconstriction in mice. Respir Physiol Neurobiol. 2004;139:191–202. doi: 10.1016/j.resp.2003.10.003. [DOI] [PubMed] [Google Scholar]
- Weissmann N, Dietrich A, Fuchs B, Kalwa H, Ay M, Dumitrascu R, et al. Classical transient receptor potential channel 6 (TRPC6) is essential for hypoxic pulmonary vasoconstriction and alveolar gas exchange. Proc Natl Acad Sci U S A. 2006;103:19093–19098. doi: 10.1073/pnas.0606728103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weissmann N, Sydykov A, Kalwa H, Storch U, Fuchs B, Schnitzler M, et al. Activation of TRPC6 channels is essential for lung ischaemia-reperfusion induced oedema in mice. Nat Commun. 2012;3:649. doi: 10.1038/ncomms1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winn MP, Conlon PJ, Lynn KL, Farrington MK, Creazzo T, Hawkins AF, et al. A mutation in the TRPC6 cation channel causes familial focal segmental glomerulosclerosis. Science. 2005;308:1801–1804. doi: 10.1126/science.1106215. [DOI] [PubMed] [Google Scholar]
- Wu LJ, Sweet TB, Clapham DE. International Union of Basic and Clinical Pharmacology. LXXVI. Current progress in the mammalian TRP ion channel family. Pharmacol Rev. 2010a;62:381–404. doi: 10.1124/pr.110.002725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Eder P, Chang B, Molkentin JD. TRPC channels are necessary mediators of pathologic cardiac hypertrophy. Proc Natl Acad Sci U S A. 2010b;107:7000–7005. doi: 10.1073/pnas.1001825107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu XZ, Li HS, Guggino WB, Montell C. Coassembly of TRP and TRPL produces a distinct store-operated conductance. Cell. 1997;89:1155–1164. doi: 10.1016/s0092-8674(00)80302-5. [DOI] [PubMed] [Google Scholar]
- Zulian A, Baryshnikov SG, Linde CI, Hamlyn JM, Ferrari P, Golovina VA. Upregulation of Na+/Ca2+ exchanger and TRPC6 contributes to abnormal Ca2+ homeostasis in arterial smooth muscle cells from Milan hypertensive rats. Am J Physiol Heart Circ Physiol. 2010;299:H624–H633. doi: 10.1152/ajpheart.00356.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Examples of chemical structures used to derive the pharmacophore model for TRPC6 channel inhibitors.
Figure S2 Stereoselective synthesis of SAR7334.
Figure S3 Selectivity profile of SAR7334.
Figure S4 Effect of SAR7334 on heterologously expressed TRPC3 and TRPC5 channels.
Figure S5 SAR7334 does not inhibit trypsin-induced Ca2+ release in TRPC6- HEK-FITR cells.
Figure S6 Intact store-operated Ca2+ entry in SAR7334-treated cells.