

Abstract
Background and purpose
Using an innovative chemical approach, peptide welding technology (PWT), a tetrabranched derivative of nociceptin/orphanin FQ (N/OFQ) has been generated and pharmacologically characterized. Both in vitro and in vivo PWT2-N/OFQ displayed the same pharmacological profile to the natural ligand. It was more potent and produced longer-lasting effects. The aim of the present study was to investigate the spinal effects of PWT2-N/OFQ in nociceptive and neuropathic pain models in mice and non-human primates.
Experimental Approach
Tail withdrawal assay in mice and monkeys was used as a nociceptive pain model and mechanical threshold in mice subjected to chronic constriction injury was used as a neuropathic pain model. The antinociceptive effects of spinally administered N/OFQ and PWT2-N/OFQ were assessed in these models.
Key Results
PWT2-N/OFQ mimicked the spinal antinociceptive effects of N/OFQ both in nociceptive and neuropathic pain models in mice as well as in non-human primates displaying 40-fold higher potency and a markedly prolonged duration of action. The effects of N/OFQ and PWT2-N/OFQ were sensitive to the N/OFQ receptor (NOP) antagonist SB-612111, but not to opioid receptor antagonists.
Conclusions and Implications
The present study has demonstrated that PWT2-N/OFQ mimicked the antinociceptive effects of the natural peptide in rodents and non-human primates acting as a potent and longer-lasting NOP-selective agonist. More generally, PWT derivatives of biologically active peptides can be viewed as innovative pharmacological tools for investigating those conditions and states in which selective and prolonged receptor stimulation promotes beneficial effects.
Tables of Links
| TARGETS |
|---|
| GPCRs |
| NOP receptor |
| μ opioid receptor |
| LIGANDS |
|---|
| N/OFQ, nociceptin/orphanin FQ |
| Naltrexone |
| SB-612111 |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guideto PHARMACOLOGY 2013/14 (Alexander et al., 2013).
Introduction
Peptide welding technology (PWT) is a novel chemical strategy that allows the synthesis of multi-branched peptides with extraordinarily high yield, purity and reproducibility (Guerrini et al., 2014). Using this approach, different tetrabranched derivatives of endogenous peptides, including nociceptin/orphanin FQ (N/OFQ), substance P, neurokinins A and B, and more recently neuropeptide S, have been synthesized and characterized. In vitro these compounds maintained the biological activity, potency and selectivity of action of the natural peptide. In vivo PWT derivatives showed higher potency and a marked prolongation of action compared with the natural sequences (Guerrini et al., 2014; Rizzi et al., 2014; Ruzza et al., 2014; 2015).
In particular, three different tetrabranched derivatives of the neuropeptide N/OFQ have been synthesized and characterized using in vitro and in vivo experiments. In vitro at the human N/OFQ peptide receptor (NOP; receptor binding, [35S]GTPγS binding, and calcium mobilization studies) PWT-N/OFQ derivatives behaved as high-affinity potent and selective full agonists (Rizzi et al., 2014). In the electrically stimulated mouse vas deferens, these compounds mimicked the inhibitory action of the natural sequence with similar maximal effects and threefold higher potencies. The NOP-selective antagonist SB-612111 antagonized the effects of N/OFQ and PWT derivatives with similar pKB values (8.02–8.48) (Guerrini et al., 2014). In vivo after supraspinal administration, PWT2-N/OFQ stimulated food intake in mice (Guerrini et al., 2014) and mimicked the inhibitory effects exerted by N/OFQ on locomotor activity (Rizzi et al., 2014). In these assays, PWT2-N/OFQ was 40-fold more potent, and elicited a more intense and extremely long-lasting action compared with the natural peptide (Guerrini et al., 2014; Rizzi et al., 2014).
In rodents, N/OFQ and NOP receptor agonists have been reported to produced variable effects on pain transmission depending on the route of administration as well as pain modality (Zeilhofer and Calo, 2003; Lambert, 2008; Schroder et al., 2014). At the spinal level N/OFQ consistently produces antinociceptive effects. Indeed the antinociceptive action of N/OFQ injected intrathecally (i.t.) initially reported by Xu et al. (1996) has been later confirmed in several laboratories. This action likely derives from presynaptic inhibition of excitatory synaptic transmission in the superficial layers of the dorsal horn (Liebel et al., 1997). The importance of this biological action of N/OFQ is corroborated by the finding that animals knockout for the NOP or the ppN/OFQ genes display a robust pronociceptive phenotype in the formalin test (Depner et al., 2003; Rizzi et al., 2007; 2014). The antinociceptive action of spinally administered N/OFQ and NOP receptor agonists is evident in models of acute pain, and is even more pronounced in models of inflammatory and neuropathic pain (see Schroder et al., 2014). The antinociceptive properties of NOP agonists given i.t. have been confirmed in non-human primate studies using N/OFQ (Ko et al., 2006; Ko and Naughton, 2009) as well as NOP-selective (Hu et al., 2010) or non-selective (Molinari et al., 2013) compounds. Interestingly, spinal NOP-mediated antinociception is not associated with typical μ-opioid (MOP) receptor side effects such as itch (Lin and Ko, 2013). Overall, these studies provide functional evidence for NOP receptor agonists as analgesics.
In the present work, the effects of the novel NOP receptor-ligand PWT2-N/OFQ have been investigated in nociceptive (tail withdrawal) and neuropathic (chronic constriction injury) pain models, after spinal administration in mice. In addition, any possible effects on locomotor performance in mice were evaluated using the rotarod test. The effects of i.t. PWT2-N/OFQ were also investigated in non-human primates by measuring tail withdrawal latencies and scratching behaviour.
Methods
Animals
All animal care and experimental procedures were conducted as humanely as possible and complied with the Guide for the Care and Use of Laboratory Animals as adopted and promulgated by the U.S. National Institutes of Health (Bethesda, MD, USA). The animal studies were approved by the Ethical Committee for the Use of Laboratory Animals (CEASA) of the University of Ferrara and by the Italian Ministry of Health (authorization number 316/2013-B; protocol number A13-053 for NHP) and by the Institutional Animal Care and Use Committee in Wake Forest University School of Medicine (Winston-Salem, NC, USA). All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010).
Male CD-1 mice were used in tail withdrawal and rotarod experiments (20–25 g, Harlan, Udine, Italy; total number of 140) and in chronic constriction injury (25–30 g, Harlan Industries, Indianapolis, IN, USA; total number of 64) studies. Mice were housed five per cage with food and water ad libitum and a 12 h light/dark cycle under standard laboratory conditions. Three adult male and female rhesus monkeys (Macaca mulatta, aged 10–17 years old) weighing between 8.1 and 14.6 kg were used. Monkeys were individually housed and their daily diet consisted of approximately 25–30 biscuits (Purina Monkey Chow; Ralston Purina Co., St. Louis, MO, USA), fresh fruit and water ad libitum. For 1 month before the present study, the monkeys were not exposed to any pharmacological treatment. All monkeys had previously been trained in the warm water tail withdrawal assay and they were housed in facilities accredited by the American Association for the Accreditation of Laboratory Animal Care.
Mouse studies
All tail withdrawal assay experiments began at 10:00 h and were performed according to the procedure described by Nazzaro et al. (2007). Briefly, the animals were placed in a holder and the distal half of the tail was immersed in water at 48°C. A cut-off time of 20 s was chosen to avoid tissue damage. If mice did not remove their tails within 20 s the tail was removed from the water and a maximum time of 20 s was recorded. Baseline values for tail withdrawal latency were determined before drug administration in each animal. Subsequent tail withdrawal latencies were measured at multiple time points after i.t. administration of N/OFQ or PWT2-N/OFQ.
The Rotarod test was used to investigate motor performance of the same mice used in the tail withdrawal assay following i.t. injection of N/OFQ or PWT2-N/OFQ. The day before the experiment, each mouse was tested on the rotarod (rota-rod for mice Ugo Basile, Varese, Italy) at a speed of 15 r.p.m. for 2 min. The day of the test, control and treated mice were positioned on the rotarod and the time they spent on the apparatus was measured (120 s cut-off).
Nerve injury was induced by ligation of the sciatic nerve as described previously (Bennett and Xie, 1988) with modification for mice. Briefly, mice were anaesthetized with i.p. injection of 80 mg·kg−1 ketamine and 12 mg·kg−1 xylazine. An incision was made between the gluteus superficialis and biceps femoris muscles in the right leg to expose the sciatic nerve. Four chromic gut ligatures (5-0 gut suture) were tied loosely around the sciatic nerve at 1 mm intervals in such a way that the epineural blood flow was occluded, but not arrested. The wound was closed by suturing the muscles and the skin.
Tactile allodynia was measured in mice with chronic constriction injury as paw withdrawal thresholds in response to probing the ipsilateral paw with calibrated von Frey filaments as described previously (Sukhtankar et al., 2013). No changes were observed in withdrawal thresholds of the contralateral paw. Mice were habituated for 45 min in suspended cages designed with wire mesh floors. Von Frey filaments with buckling weights ranging from 0.04 to 2.0 g were then applied perpendicularly to the plantar surface of the ipsilateral paw and held for 5 s. A positive response was indicated by a sharp withdrawal of the paw. For each mouse, testing began with a von Frey filament corresponding to the weight of 0.4 g. If the animal made a positive response, the next filament with lower force was applied. If the response was negative, the next filament with higher force was used. The experiment ended once the animal had made four responses after the first positive response. Paw withdrawal threshold was determined using the Dixon non-parametric method (Dixon, 1980). Baseline values for paw withdrawal thresholds were determined before chronic constriction injury as well as before drug administration. Changes in tactile allodynia in response to drug treatment were determined 2 weeks after the nerve injury was induced. Separate groups of mice (n = 8 per group) were used to study each dosing condition.
All behavioural tests were conducted by trained experimenters who were unaware of experimental conditions.
Monkey studies
Surgical implantation of i.t. catheters was performed according to the detailed procedures described by Ding et al. (2015). Briefly, animals were anaesthetized with ketamine (10 mg·kg−1, i.m.), intubated and maintained under isoflurane anaesthesia. Strict aseptic surgery procedures were followed. Hemilaminectomy was performed in the lateral aspect of the L4 or L5 vertebral body. The catheter was inserted into the i.t. space and advanced rostrally to place the catheter tip in the L1–L2 lumbar region. The catheter was then routed s.c. to the vascular access port site and was attached to the port. Animals received buprenorphine (0.02 mg·kg−1, i.m.) and meloxicam (0.15 mg·kg−1, s.c.) as post-operative analgesics and Ceftiofur (2.2 mg·kg−1, i.m.) as a post-operative antibiotic. Post-operative care and incision site observations were performed daily for 14 days or until fully healed. Compared with a conventional method using anaesthesia to deliver drugs in the CNS, monkeys with implanted catheters allow easier i.t. drug delivery and immediate measurement of early drug effects. The warm water tail withdrawal assay was used to evaluate thermal antinociceptive effects of PWT2-N/OFQ (Ko and Naughton, 2009). Monkeys were seated in primate restraint chairs, and the lower part of their shaved tail (approximately 15 cm) was immersed into a thermal flask containing water maintained at either 42, 46 or 50°C. Water at 42 and 46°C was used as non-noxious stimuli, and 50°C water was used as an acute noxious stimulus. Tail withdrawal latencies were measured at each temperature using a computerized timer by experimenters who were unaware of experimental conditions. If monkeys did not remove their tails within 20 s (cut-off), the flask was removed and a maximum time of 20 s was recorded. Test sessions began with baseline measurements at each temperature. Subsequent tail withdrawal latencies were measured at multiple time points after i.t. administration of PWT2-N/OFQ.
Behavioural activities of monkeys were recorded in their home cages for scoring scratching behaviour, which is associated with itch sensation (Ko et al., 2004). Each 15 min recording session was conducted at 1, 3, 6 and 24 h after i.t. PWT2-N/OFQ. A scratch was defined as one brief (<1 s) episode of scraping contact of the forepaw or hind paw on the skin surface. Total scratches were counted for each 15 min session by experimenters who were blinded to experimental conditions.
Drug administration
N/OFQ and PWT2-N/OFQ (synthesized in house as previously described Guerrini et al., 1997; Guerrini et al., 2014) and naltrexone (National Institute on Drug Abuse, Bethesda, MD, USA) were dissolved in sterile water. SB-612111 hydrochloride (Tocris Bioscience, Bristol, UK) was dissolved in 1:1:8 ratio of DMSO, Tween 80 and sterile water.
In mice, drugs were administered i.t. in the volume of 5 μL as previously described (Sukhtankar et al., 2013). Briefly, the mouse was secured by a firm grip on the pelvic girdle, and the test drug was delivered by lumbar puncture between the L5/L6 vertebrae using a 30 G needle attached to a 10 μL Hamilton syringe. Mice in the control group received an i.t. administration of sterile water. In monkeys implanted with i.t. catheters (Ding et al., 2015), drugs were administered i.t. in a volume of 1 mL through the subcutaneous access port. There was a minimum of 1 week interval between drug administrations.
Experimental design
The first part of the study was to characterize the effectiveness and duration of PWT2-N/OFQ in comparison with N/OFQ in mouse tail withdrawal assay with or without chronic constriction injury.
Withdrawal latency time was measured by an experienced observer unaware of drug treatmenst. Tail withdrawal latency was determined immediately before and at several time points after i.t. injection of 5 μL of vehicle (control), N/OFQ (0.1–10.0 nmol) or PWT2-N/OFQ (2.5–250.0 pmol). In addition the selective NOP antagonist SB-612111 (1 mg·kg−1) was administered i.v. (100 μL·mouse−1 into the caudal tail vein using a 30 G needle attached to a sterile syringe) 30 min before spinal injection of PWT2-N/OFQ (25 pmol).
The rotator test was performed on the same mice used in the tail withdrawal assay 20 and 60 min after i.t. injection of N/OFQ and PWT2-N/OFQ respectively.
Mice with nerve injury-induced tactile allodynia received an i.t. administration of PWT2-N/OFQ (2.5–250 pmol), N/OFQ (0.1–10.0 nmol), or vehicle. Paw withdrawal thresholds were then measured at several time points after i.t. PWT2-N/OFQ or N/OFQ. In addition, antagonist studies were conducted to determine the involvement of spinal NOP and opioid receptors in the anti-allodynic effects of i.t. PWT2-N/OFQ. The selective NOP antagonist SB-612111 (1 or 3 μg), opioid antagonist naltrexone (3 μg) or vehicle was administered i.t. 10 min before i.t. PWT2-N/OFQ (25 pmol). Paw withdrawal thresholds were measured 0.5 h after PWT2-N/OFQ treatment to determine antagonist effects.
The second part of the study was to characterize the effectiveness and duration of PWT2-N/OFQ against thermal nociception in monkeys. Monkeys received i.t. PWT2-N/OFQ (0.3–3.0 nmol) or vehicle and their nociceptive thresholds were measured at several time points after administration. To determine the effect of PWT2-N/OFQ in eliciting itch sensation, monkeys received i.t. PWT2-N/OFQ (3 nmol) or vehicle and their behavioural activities were scored for scratching responses at 1, 3, 6 and 24 h after administration.
Data analysis
Tail withdrawal latency (s) or paw withdrawal latency (g) data are shown in time course experiments. If the animal did not respond to any von Frey filament or did not withdraw their tail, the cut-off value was assigned. Curve fitting was performed using PRISM 4.0 (GraphPad Software Inc., San Diego, CA, USA). Mean values (mean ± SEM) were calculated from individual animals for all behavioural endpoints. All data were analysed by two-way anova (drug vs. time in dose–response curve experiments, antagonist vs. agonist in antagonist experiments) with repeated measures followed by Bonferroni's multiple comparisons test except the rotarod test data that were analysed by one-way anova followed by Dunnett's test for multiple comparisons. The criterion for significance for all tests was set at P < 0.05.
Results
In the mouse tail withdrawal assay, i.t. injection of N/OFQ in the range 0.1–10 nmol produced a dose- and time-dependent antinociceptive effect (Figure 1A). This effect peaked 5 min post-injection and lasted (with the highest dose of N/OFQ, i.e. 10 nmol) for 1.5 h. However, after i.t. injection of 10 nmol of N/OFQ, all mice exhibited hind limb flaccidity and a marked decrease in spontaneous locomotor activity. Thus, we investigated the effects of spinal N/OFQ in the rotarod test. As reported in Figure 2A, all mice treated with saline reached the cut-off time. N/OFQ at 0.1 nmol did not modify animal performance on the rotarod while in the 1–10 nmol range it dose-dependently impaired performance producing statistically significant effects at 10 nmol.
Figure 1.
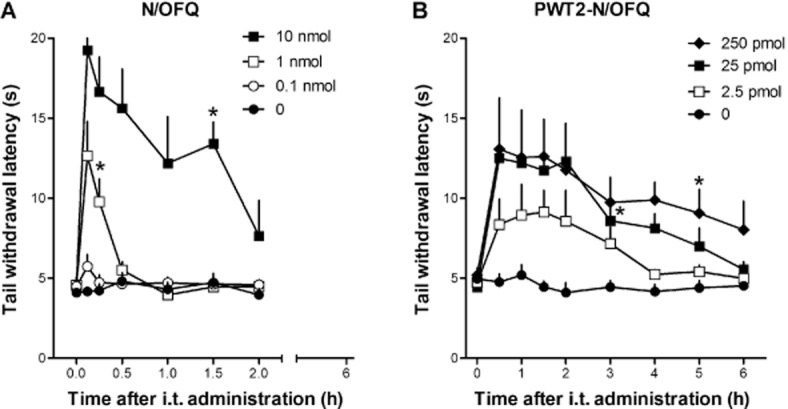
Dose–response curves to i.t. N/OFQ (0.1–10 nmol, panel A) and PWT2-N/OFQ (2.5–250 pmol, panel B) in the mouse tail withdrawal assay. N/OFQ and PWT2-N/OFQ-induced dose-dependent (F(3,182) = 82.4; P < 0.05; F(4,178) = 24.9; P < 0.05) and time-dependent (F(6,182) = 14.2; P < 0.05; (F(8,178) = 7.94; P < 0.05) antinociceptive effects. Each data point represents mean ± SEM (n = 12). *P < 0.05, significantly different from vehicle controls, from the first to the corresponding time point).
Figure 2.
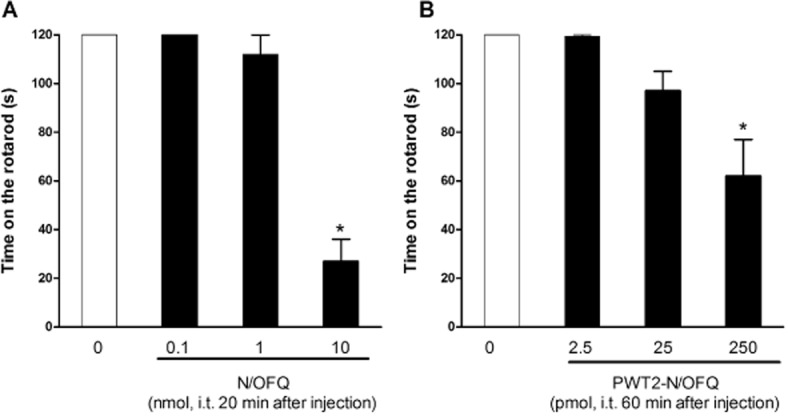
Dose–response curves to i.t. N/OFQ (0.1–10 nmol, panel A) and PWT2-N/OFQ (2.5–250 pmol, panel B) in the rotarod test performed in the same mice used in the tail withdrawal assay. Data are time on the rotarod (s) 20 min or 60 min following i.t. injection of N/OFQ or PWT2-N/OFQ respectively. Each data point represents mean ± SEM (n = 12). *P < 0.05, significantly different from vehicle.
As shown in Figure 1B, PWT2-N/OFQ given i.t., in the range 2.5–250.0 pmol, elicited dose- and time-dependent antinociceptive effects. Multiple comparisons test indicated that 25 pmol of PWT-N/OFQ produced statistically significant antinociception between 30 and 180 min and that 250 pmol of PWT2-N/OFQ produced statistically significant antinociception between 30 and 300 min time points. In comparison with N/OFQ, the tetrabranched peptide elicited slightly lower antinociceptive effects, but was approximately 40-fold more potent, and proded longer-lasting effects. In particular the increase in tail withdrawal latency induced by PWT2-N/OFQ peaked at 30 min, remained stable for 3 h, and then slowly returned to basal levels. The same animals were evaluated in the rotarod assay 60 min after injection. PWT2-N/OFQ at 2.5 and 25.0 pmol did not modify animal performance on the rod while at 250 pmol the tetrapeptide caused a statistically significant impairment of animal performance (Figure 2B).
As shown in Figure 3, i.v. administration of the NOP antagonist, SB-612111 (1 mg·kg−1, 30 min before i.t. injection) did not modify, per se, tail withdrawal latencies, but was able to fully prevent the antinociceptive effect induced by 25 pmol PWT2-N/OFQ. Of note, this dose of antagonist has previously been shown to prevent the antinociceptive action of i.t. N/OFQ (Rizzi et al., 2007).
Figure 3.
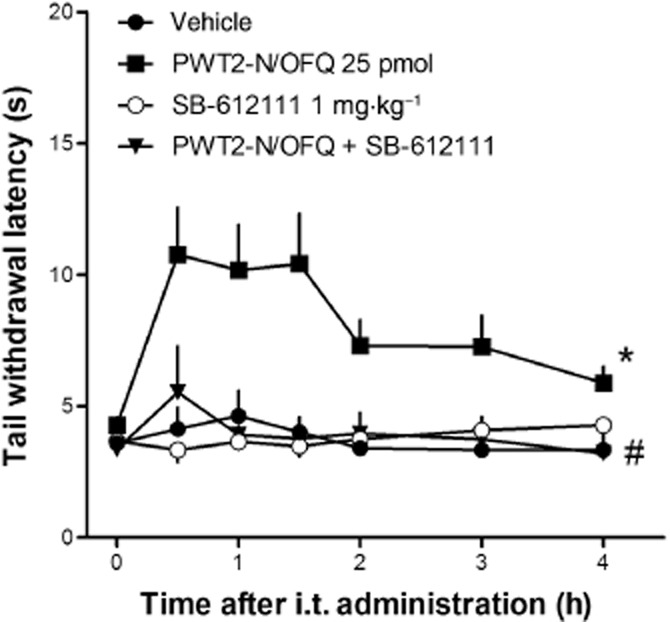
Effects of SB-612111 (1 mg·kg−1, i.v.) on i.t. PWT2-N/OFQ in the mouse tail withdrawal assay. Each data point represents mean ± SEM (n = 8). Raw data from tail withdrawal experiments were converted to the area under the time (period 0–120 min) × withdrawal latency curve (AUC). AUC data are as follows: vehicle: 690 ± 55; PWT2-N/OFQ 25 pmol: 1555 ± 219; SB-612111: 660 ± 35; PWT2-N/OFQ + SB-612111: 737 ± 139. *P < 0.05, significantly different from vehicle; #P > 0.05, significantly different from PWT2-N/OFQ; two-way anova.
Figure 4 compares the effects of i.t. N/OFQ (0.1–10.0 nmol) and PWT2-N/OFQ (2.5–250.0 pmol) on tactile allodynia in mice with chronic constriction injury. Before injury, all mice maintained paw withdrawal thresholds above the cut-off value (2 g). Two weeks after the nerve injury, the paw withdrawal thresholds were significantly reduced (0.04 g). Paw withdrawal thresholds did not change in mice injected i.t. with vehicle. I.t. N/OFQ produced anti-allodynic effects in both dose- and time-dependent manners (Figure 4A). N/OFQ 0.1 nmol elicited statistically significant, but short-lasting anti-allodynic effects; 1 and 10 nmol N/OFQ produced greater effects that lasted up to 3 h from injection.
Figure 4.
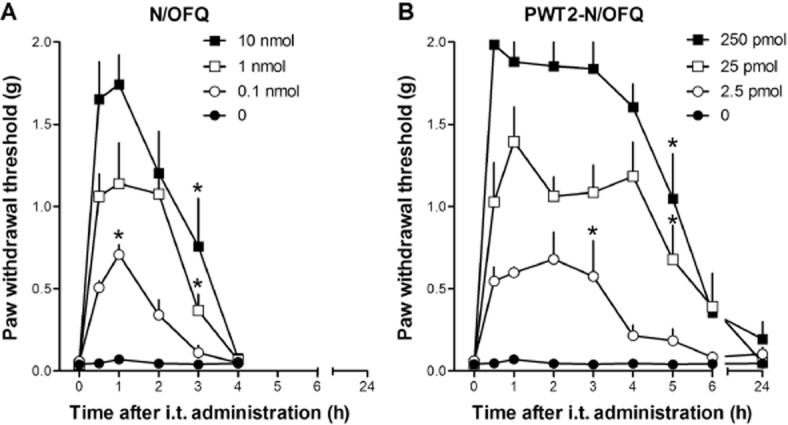
Dose–response curves to i.t. N/OFQ (0.1–10 nmol, panel A) and PWT2-N/OFQ (2.5–250 pmol, panel B) in mice with neuropathic pain. N/OFQ and PWT2-N/OFQ N/OFQ induced a dose-dependent (F(3,28) = 24.8; P < 0.05; F(3,28) = 52.5; P < 0.05) and time-dependent (F(4,112) = 42; P < 0.05; F(7,196) = 36.8; P < 0.05) anti-allodynic effect. Each data point represents mean ± SEM (n = 8). *P < 0.05, significantly different from vehicle controls, from the first to the corresponding time point. Note that the zero-time point is a pretest value (see Methods).
PWT2-N/OFQ mimicked the anti-allodynic effects of N/OFQ (Figure 4B). Multiple comparisons test indicated that 2.5 pmol PWT-N/OFQ produced anti-allodynic effects between 0.5 and 3.0 h time points. Both 25 and 250 pmol i.t. PWT2-N/OFQ produced anti-allodynic effects between 0.5 and 5.0 h time points.
Spinal injection of the NOP antagonist SB-612111, attenuated the anti-allodynic effects of PWT2-N/OFQ (25 pmol) in a dose-dependent (1 and 3 μg) manner. In contrast, the opioid receptor antagonist naltrexone (3 μg) did not affect the action of PWT2-N/OFQ (Figure 5).
Figure 5.
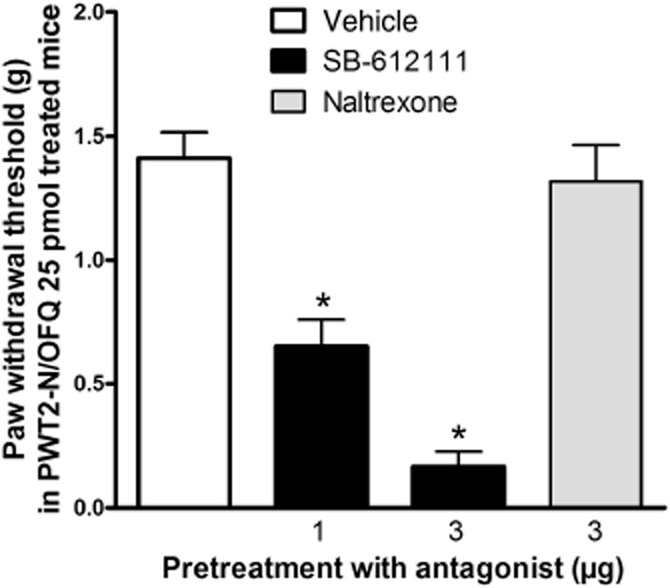
Effects of SB-612111 (1 or 3 μg, i.t.) and naltrexone (3 μg, i.t.) on i.t. PWT2-N/OFQ in mice with neuropathic pain. Antagonists were given10 min before PWT2-N/OFQ. Behavioural responses were measured 0.5 h after administration of PWT2-N/OFQ. SB-612111 attenuated the anti-allodynic effects of i.t. PWT2-N/OFQ in a dose-dependent (F(2, 21) = 45.5; P < 0.05) manner. Each data point represents mean ± SEM (n = 8). *P < 0.05, significantly different from vehicle controls.
In monkeys, i.t. PWT2-N/OFQ produced antinociception against an acute nociceptive stimulus (50°C water) in both dose- and time-dependent manners. Multiple comparisons test indicated that 0.3 nmol i.t. PWT2-N/OFQ produced antinociception between 0.5 and 2.5 h time points while the antinociceptive effects elicited by 1 and 3 nmol PWT2-N/OFQ were still statistically significant 24 h from injection (Figure 6A). The higher dose of PWT2-N/OFQ 3 nmol did not significantly elicit scratching responses at any time point (Figure 6B). I.t. PWT2-N/OFQ in monkeys at the three doses tested did not cause any overt side effects including sedation or motor impairment.
Figure 6.
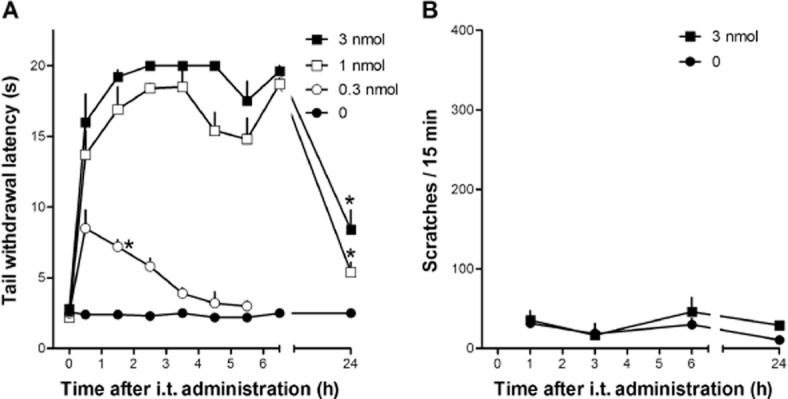
Dose–response curves to i.t. PWT2-N/OFQ (0.3–3 nmol, panel A) in the monkey tail withdrawal assay. PWT2-N/OFQ induced a dose-dependent (F(3,6) = 382; P < 0.05) and time-dependent (F(8,16) = 55; P < 0.05) antinociceptive effect. (B) Data shown are scratching responses in a 15 min observation period after i.t. PWT2-N/OFQ. Each data point represents mean ± SEM (n = 3). *P < 0.05, significantly different from vehicle control, from the first to the corresponding time point. Note that the zero- time point is a pretest value (see Methods).
Discussion and conclusions
In this study, we investigated the antinociceptive effects of the novel NOP receptor agonist PWT2-N/OFQ in mice and monkeys. I.t. PWT2-N/OFQ elicited robust antinociceptive effects similar to N/OFQ in both nociceptive and neuropathic pain models in mice as well as in non-human primates. PWT2-N/OFQ displayed higher potency and a markedly prolonged duration of action.
In the mouse tail withdrawal assay, i.t. N/OFQ produced a dose- and time-dependent antinociceptive effect that peaked at 5 min post-injection and lasted (with the highest dose) for 1.5 h. However after i.t. injection of 10 nmol N/OFQ, all mice exhibited hind limb flaccidity, decreased spontaneous locomotor activity and a profound impairment of performance on the rotarod. Therefore, 1 nmol is the highest dose of N/OFQ that produced an unbiased antinociceptive action but this effect was short-lasting (15 min). In mice with chronic constriction injury, N/OFQ elicited anti-allodynic effects in both dose- and time-dependently. In these mice, N/OFQ showed increased potency and duration of action compared with mice in the tail withdrawal assay. These results suggest that the antinociceptive action of spinal NOP activation was more efficient against neuropathic than nociceptive pain. This proposal is supported by earlier studies demonstrating that spinal N/OFQ is more effective in neuropathic than nociceptive pain models (Kamei et al., 1999; Courteix et al., 2004) and its electrophysiological actions are more pronounced in the spinal cord (Sotgiu et al., 2004) and dorsal root ganglia (Abdulla and Smith, 1998) of the former than the latter models. Although the mechanisms of N/OFQ spinal antinociceptive effects are not fully understood, the evidence that both NOP receptor and N/OFQ expression are increased in pain-relevant areas in models of neuropathic pain (Rosen et al., 2000; Briscini et al., 2002; Ma et al., 2005; Chen and Sommer, 2006) may, at least in part, explain the higher effectiveness of the peptide against neuropathic pain. These topics are discussed in detail by Schroder et al. (2014).
Similar to N/OFQ, PWT2-N/OFQ produced antinociceptive effects both in mice in the tail withdrawal assay and in those with chronic constriction injury. The novel peptide was approximately 40-fold more potent and produced longer-lasting effects. These results perfectly match previous findings obtained comparing the supraspinal effects of N/OFQ and PWT2-N/OFQ on food intake (Guerrini et al., 2014) and locomotor activity (Rizzi et al., 2014). The increase in agonist potency and longer-lasting effects of PWT2-N/OFQ may likely derive from the lower susceptibility to peptidase action that characterizes multi-branched peptides, compared with free peptide sequences, as previously demonstrated by Bracci et al. (2003). Another difference between N/OFQ and PWT2-N/OFQ was the kinetics of action. The onset of action of PWT2-N/OFQ was delayed compared with that of N/OFQ, but the duration of action was longer. Interestingly these in vivo differences parallel those obtained in vitro when studying the action of the two compounds in tissues where PWT2-N/OFQ mimicked N/OFQ effects with slower kinetics and wash-resistant effects (Guerrini et al., 2014). It is worthy to note that similar results were obtained comparing the effects of substance P and PWT2-substance P in bioassay studies (Ruzza et al., 2014). Thus, slow kinetics of action and reduced sensitivity to wash-out seems to be a common feature of PWT derivatives that can possibly be related to their multivalent ligand nature. Indeed, multivalent ligands may promote longer-lasting target binding via different mechanisms (Gestwicki et al., 2002) and this phenomenon can be important for the prolongation of in vivo drug action (Vauquelin and Charlton, 2010). Thus, lower susceptibility to peptidase action associated with longer-lasting target binding may synergize and explain the prolonged duration of action of PWT derivatives, compared with free peptide sequences.
Results of the antagonist studies with i.t. PWT2-N/OFQ matched those previously obtained with N/OFQ (Rizzi et al., 2007). Given i.t., PWT2-N/OFQ induced antinociception which was fully blocked by the NOP receptor-selective antagonist SB-612111 (Zaratin et al., 2004) while the classical opioid receptor antagonist naltrexone was inactive. These findings demonstrated that the antinociceptive effects of PWT2-N/OFQ are due to selective activation of the NOP receptor. This is corroborated by previous in vitro studies that demonstrated high selectivity of PWT2-N/OFQ for NOP over classical opioid receptors (Guerrini et al., 2014; Rizzi et al., 2014) and, most importantly, by in vivo studies where i.c.v. PWT2-N/OFQ inhibited motor activity in wild-type, but not NOP receptor knockout mice (Rizzi et al., 2014).
Finally, i.t. N/OFQ produced robust antinociceptive effects in non-human primates in the tail withdrawal assay, in the dose range 10–100 nmol, and these effects lasted for 2.5 h (Ko et al., 2006; Ko and Naughton, 2009). Under the same experimental conditions, PWT2-N/OFQ mimicked the action of the natural NOP receptor agonist, but was approximately 30-fold more potent. This ratio of potency is in line with the present and the previous (Guerrini et al., 2014; Rizzi et al., 2014) rodent studies. Moreover, statistically significant antinociceptive effects were measured in response to i.t. PWT2-N/OFQ even 24 h after injection. Thus, the duration of action of the PWT derivative is approximately 10-fold longer than that of the natural peptide (i.e. 24 vs. 2.5 h). To our knowledge, PWT2-N/OFQ produces the longest duration of antinociceptive effects in the non-human primate model, as compared with the clinically used analgesic morphine (∼4–6 h) or with experimentally developed compounds (∼4–6 h) (Ko et al., 2006; Hu et al., 2010; Molinari et al., 2013).
The increase in duration of action provided by PWT was greater in monkeys than in mice. It is known that peptide degradation and peptidase activity is different between rodents and primates (Liederer and Borchardt, 2005), which may contribute to much longer duration of action for N/OFQ and PWT2-N/OFQ in primates. In addition, future studies can be integrated with MS to quantify the amount of PWT2-N/OFQ at the 24 h time point following administration. Although the analgesic effectiveness of NOP receptor agonists in patients with neuropathic pain is unknown, these agonists fully attenuated capsaicin-induced allodynia in monkeys (Ko and Naughton, 2009; Hu et al., 2010). Capsaicin evokes pain by activating the transient receptor potential vanilloid type 1, which has been implicated in the transduction of diverse pain modalities including diabetic neuropathy (Szolcsanyi and Sandor, 2012). Given their full antiallodynic effects against capsaicin (Hu et al., 2010), NOP receptor agonists possess a promising clinical profile. More importantly, NOP receptor agonists produce morphine-comparable antinociception across different pain models in monkeys without eliciting severe side effects (Lin and Ko, 2013). Given that itch is a long-standing side effect associated with the use of i.t. morphine (Cousins and Mather, 1984; Ganesh and Maxwell, 2007), lack of scratching in response to i.t. PWT2-NOFQ in monkeys suggests that this compound as well as other NOP receptor-selective agonists (Hu et al., 2010) may have therapeutic potential as novel analgesics.
In summary, this study demonstrated that spinally administered PWT2-N/OFQ inhibited nociceptive and neuropathic pain in mice and monkeys. The PWT derivative was more potent than the natural peptide and elicited prolonged effects that lasted for more than 24 h in non-human primates. In general, the present findings confirm and extend previous results obtained with N/OFQ (Guerrini et al., 2014; Rizzi et al., 2014) and other peptides (Ruzza et al., 2014; 2015) suggesting that the PWT approach can be successfully applied to different peptides to generate novel ligands displaying the same pharmacological activity and selectivity of action of the natural sequence. In vivo, these display higher potency and prolonged duration of action.
Acknowledgments
The present study was supported by the University of Ferrara (FAR grants to G. C. and R. G.), UFPeptides s.r.l. and the U.S. Public Health Service (R01-DA032568, R01-AR059193 and W81XWH-13-2-0045 to M. C. K.).
Glossary
- i.t
intrathecally
- MOP
μ-opioid peptide receptor
- N/OFQ
nociceptin/orphanin FQ
- NOP
N/OFQ peptide receptor
- PWT
peptide welding technology
Author contributions
A. R., D. D. S., H. D., R. G., G. C. and M. C. K. participated in research design. A. R., D. D. S., H. D., K. H. and C. R. conducted experiments and performed data analysis. R. G. designed, synthesized, and purified PWT2-N/OFQ and N/OFQ. A. R., D. D. S. and H. D. wrote the first draft of the paper. The final version of the paper was revised by R. G., G. C. and M. C. K.
Statement of conflicts of interest
Girolamo Calò and Remo Guerrini are inventors of the EP13162532.9 patent application focused on PWT technology and are among the founders of the University of Ferrara spin-off company UFPeptides s.r.l., the assignee of this patent application.
References
- Abdulla FA, Smith PA. Axotomy reduces the effect of analgesic opioids yet increases the effect of nociceptin on dorsal root ganglion neurons. J Neurosci. 1998;18:9685–9694. doi: 10.1523/JNEUROSCI.18-23-09685.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SP, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: G protein-coupled receptors. Br J Pharmacol. 2013;170:1459–1581. doi: 10.1111/bph.12445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett GJ, Xie YK. A peripheral mononeuropathy in rat that produces disorders of pain sensation like those seen in man. Pain. 1988;33:87–107. doi: 10.1016/0304-3959(88)90209-6. [DOI] [PubMed] [Google Scholar]
- Bracci L, Falciani C, Lelli B, Lozzi L, Runci Y, Pini A, et al. Synthetic peptides in the form of dendrimers become resistant to protease activity. J Biol Chem. 2003;278:46590–46595. doi: 10.1074/jbc.M308615200. [DOI] [PubMed] [Google Scholar]
- Briscini L, Corradini L, Ongini E, Bertorelli R. Up-regulation of ORL-1 receptors in spinal tissue of allodynic rats after sciatic nerve injury. Eur J Pharmacol. 2002;447:59–65. doi: 10.1016/s0014-2999(02)01833-2. [DOI] [PubMed] [Google Scholar]
- Chen Y, Sommer C. Nociceptin and its receptor in rat dorsal root ganglion neurons in neuropathic and inflammatory pain models: implications on pain processing. J Peripher Nerv Syst. 2006;11:232–240. doi: 10.1111/j.1529-8027.2006.0093.x. [DOI] [PubMed] [Google Scholar]
- Courteix C, Coudore-Civiale MA, Privat AM, Pelissier T, Eschalier A, Fialip J. Evidence for an exclusive antinociceptive effect of nociceptin/orphanin FQ, an endogenous ligand for the ORL1 receptor, in two animal models of neuropathic pain. Pain. 2004;110:236–245. doi: 10.1016/j.pain.2004.03.037. [DOI] [PubMed] [Google Scholar]
- Cousins MJ, Mather LE. Intrathecal and epidural administration of opioids. Anesthesiology. 1984;61:276–310. [PubMed] [Google Scholar]
- Depner UB, Reinscheid RK, Takeshima H, Brune K, Zeilhofer HU. Normal sensitivity to acute pain, but increased inflammatory hyperalgesia in mice lacking the nociceptin precursor polypeptide or the nociceptin receptor. Eur J Neurosci. 2003;17:2381–2387. doi: 10.1046/j.1460-9568.2003.02676.x. [DOI] [PubMed] [Google Scholar]
- Ding H, Hayashida K, Suto T, Sukhtankar DD, Kimura M, Mendenhall V, et al. Supraspinal actions of N/OFQ, morphine and substance P in regulating pain and itch in nonhuman primates. Br J Pharmacol. 2015 doi: 10.1111/bph.13124. doi: 10.1111/bph.13124. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon WJ. Efficient analysis of experimental observations. Annu Rev Pharmacol Toxicol. 1980;20:441–462. doi: 10.1146/annurev.pa.20.040180.002301. [DOI] [PubMed] [Google Scholar]
- Ganesh A, Maxwell LG. Pathophysiology and management of opioid-induced pruritus. Drugs. 2007;67:2323–2333. doi: 10.2165/00003495-200767160-00003. [DOI] [PubMed] [Google Scholar]
- Gestwicki JE, Cairo CW, Strong LE, Oetjen KA, Kiessling LL. Influencing receptor-ligand binding mechanisms with multivalent ligand architecture. J Am Chem Soc. 2002;124:14922–14933. doi: 10.1021/ja027184x. [DOI] [PubMed] [Google Scholar]
- Guerrini R, Calo G, Rizzi A, Bianchi C, Lazarus LH, Salvadori S, et al. Address and message sequences for the nociceptin receptor: a structure–activity study of nociceptin-(1–13)-peptide amide. J Med Chem. 1997;40:1789–1793. doi: 10.1021/jm970011b. [DOI] [PubMed] [Google Scholar]
- Guerrini R, Marzola E, Trapella C, Pela M, Molinari S, Cerlesi MC, et al. A novel and facile synthesis of tetra branched derivatives of nociceptin/orphanin FQ. Bioorg Med Chem. 2014;22:3703–3712. doi: 10.1016/j.bmc.2014.05.005. [DOI] [PubMed] [Google Scholar]
- Hu E, Calo G, Guerrini R, Ko M. Long lasting antinociceptive spinal effects in primates of the novel nociceptin/orphanin FQ receptor agonist UFP-112. Pain. 2010;148:107–113. doi: 10.1016/j.pain.2009.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamei J, Ohsawa M, Kashiwazaki T, Nagase H. Antinociceptive effects of the ORL1 receptor agonist nociceptin/orphanin FQ in diabetic mice. Eur J Pharmacol. 1999;370:109–116. doi: 10.1016/s0014-2999(99)00112-0. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko MC, Naughton NN. Antinociceptive effects of nociceptin/orphanin FQ administered intrathecally in monkeys. J Pain. 2009;10:509–516. doi: 10.1016/j.jpain.2008.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko MC, Song MS, Edwards T, Lee H, Naughton NN. The role of central mu opioid receptors in opioid-induced itch in primates. J Pharmacol Exp Ther. 2004;310:169–176. doi: 10.1124/jpet.103.061101. [DOI] [PubMed] [Google Scholar]
- Ko MC, Wei H, Woods JH, Kennedy RT. Effects of intrathecally administered nociceptin/orphanin FQ in monkeys: behavioral and mass spectrometric studies. J Pharmacol Exp Ther. 2006;318:1257–1264. doi: 10.1124/jpet.106.106120. [DOI] [PubMed] [Google Scholar]
- Lambert DG. The nociceptin/orphanin FQ receptor: a target with broad therapeutic potential. Nat Rev Drug Discov. 2008;7:694–710. doi: 10.1038/nrd2572. [DOI] [PubMed] [Google Scholar]
- Liebel JT, Swandulla D, Zeilhofer HU. Modulation of excitatory synaptic transmission by nociceptin in superficial dorsal horn neurones of the neonatal rat spinal cord. Br J Pharmacol. 1997;121:425–432. doi: 10.1038/sj.bjp.0701149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liederer BM, Borchardt RT. Stability of oxymethyl-modified coumarinic acid cyclic prodrugs of diastereomeric opioid peptides in biological media from various animal species including human. J Pharm Sci. 2005;94:2198–2206. doi: 10.1002/jps.20452. [DOI] [PubMed] [Google Scholar]
- Lin AP, Ko MC. The therapeutic potential of nociceptin/orphanin FQ receptor agonists as analgesics without abuse liability. ACS Chem Neurosci. 2013;4:214–224. doi: 10.1021/cn300124f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma F, Xie H, Dong ZQ, Wang YQ, Wu GC. Expression of ORL1 mRNA in some brain nuclei in neuropathic pain rats. Brain Res. 2005;1043:214–217. doi: 10.1016/j.brainres.2005.01.037. [DOI] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molinari S, Camarda V, Rizzi A, Marzola G, Salvadori S, Marzola E, et al. Dmt1]N/OFQ(1–13)-NH2: a potent nociceptin/orphanin FQ and opioid receptor universal agonist. Br J Pharmacol. 2013;168:151–162. doi: 10.1111/j.1476-5381.2012.02115.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nazzaro C, Rizzi A, Salvadori S, Guerrini R, Regoli D, Zeilhofer HU, et al. UFP-101 antagonizes the spinal antinociceptive effects of nociceptin/orphanin FQ: behavioral and electrophysiological studies in mice. Peptides. 2007;28:663–669. doi: 10.1016/j.peptides.2006.11.004. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, et al. NC-IUPHAR. The IUPHAR/BPS Guide to PHARMACOLOGY: an expert-driven knowledge base of drug targets and their ligands. Nucl Acids Res. 2014;42:D1098–D1106. doi: 10.1093/nar/gkt1143. (Database Issue): [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzi A, Gavioli EC, Marzola G, Spagnolo B, Zucchini S, Ciccocioppo R, et al. Pharmacological characterization of the nociceptin/orphanin FQ receptor antagonist SB-612111 [(-)-cis-1-methyl-7-[[4-(2,6-dichlorophenyl)piperidin-1-yl]methyl]-6,7,8,9 -tetrahydro-5H-benzocyclohepten-5-ol]: in vivo studies. J Pharmacol Exp Ther. 2007;321:968–974. doi: 10.1124/jpet.106.116780. [DOI] [PubMed] [Google Scholar]
- Rizzi A, Malfacini D, Cerlesi MC, Ruzza C, Marzola E, Bird MF, et al. In vitro and in vivo pharmacological characterization of nociceptin/orphanin FQ tetrabranched derivatives. Br J Pharmacol. 2014;171:4138–4153. doi: 10.1111/bph.12799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen A, Lundeberg T, Bytner B, Nylander I. Central changes in nociceptin dynorphin B and Met-enkephalin-Arg-Phe in different models of nociception. Brain Res. 2000;857:212–218. doi: 10.1016/s0006-8993(99)02432-4. [DOI] [PubMed] [Google Scholar]
- Ruzza C, Rizzi A, Malfacini D, Cerlesi MC, Ferrari F, Marzola E, et al. Pharmacological characterization of tachykinin tetrabranched derivatives. Br J Pharmacol. 2014;171:4125–4137. doi: 10.1111/bph.12727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruzza C, Rizzi A, Malfacini D, Pulga A, Pacifico S, Salvadori S, et al. In vitro and in vivo pharmacological characterization of a neuropeptide S tetrabranched derivative. Pharma Res Per. 2015;3:e00108. doi: 10.1002/prp2.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroder W, Lambert DG, Ko MC, Koch T. Functional plasticity of the N/OFQ-NOP receptor system determines analgesic properties of NOP receptor agonists. Br J Pharmacol. 2014;171:3777–3780. doi: 10.1111/bph.12744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotgiu ML, Bellomi P, Biella GE. Efficacy of nociceptin inhibition on WDR neuron activity is enhanced in mononeuropathic rats. Brain Res. 2004;998:251–254. doi: 10.1016/j.brainres.2003.11.025. [DOI] [PubMed] [Google Scholar]
- Sukhtankar DD, Zaveri NT, Husbands SM, Ko MC. Effects of spinally administered bifunctional nociceptin/orphanin FQ peptide receptor/mu-opioid receptor ligands in mouse models of neuropathic and inflammatory pain. J Pharmacol Exp Ther. 2013;346:11–22. doi: 10.1124/jpet.113.203984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szolcsanyi J, Sandor Z. Multisteric TRPV1 nocisensor: a target for analgesics. Trends Pharmacol Sci. 2012;33:646–655. doi: 10.1016/j.tips.2012.09.002. [DOI] [PubMed] [Google Scholar]
- Vauquelin G, Charlton SJ. Long-lasting target binding and rebinding as mechanisms to prolong in vivo drug action. Br J Pharmacol. 2010;161:488–508. doi: 10.1111/j.1476-5381.2010.00936.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu XJ, Hao JX, Wiesenfeld-Hallin Z. Nociceptin or antinociceptin: potent spinal antinociceptive effect of orphanin FQ/nociceptin in the rat. Neuroreport. 1996;7:2092–2094. [PubMed] [Google Scholar]
- Zaratin PF, Petrone G, Sbacchi M, Garnier M, Fossati C, Petrillo P, et al. Modification of nociception and morphine tolerance by the selective opiate receptor-like orphan receptor antagonist (-)-cis-1-methyl-7-[[4-(2,6-dichlorophenyl)piperidin-1-yl]methyl]-6,7,8,9- tetrahydro-5H-benzocyclohepten-5-ol (SB-612111) J Pharmacol Exp Ther. 2004;308:454–461. doi: 10.1124/jpet.103.055848. [DOI] [PubMed] [Google Scholar]
- Zeilhofer HU, Calo G. Nociceptin/orphanin FQ and its receptor–potential targets for pain therapy? J Pharmacol Exp Ther. 2003;306:423–429. doi: 10.1124/jpet.102.046979. [DOI] [PubMed] [Google Scholar]