

Abstract
Background and Purpose
In the aorta of adult spontaneously hypertensive (SHR), but not in that of normotensive Wistar-Kyoto (WKY), rats, previous exposure to phenylephrine inhibits subsequent contractions to PGE2. The present experiments were designed to examine the mechanism(s) underlying this inhibition.
Experimental Approach
Isometric tension was measured in isolated rings of SHR and WKY aortae. Gene expression and protein presence were measured by quantitative real-time PCR and Western blotting respectively.
Key Results
In aorta of 18 weeks SHR, but not age-matched WKY, pre-exposure to phenylephrine inhibited subsequent contractions to PGE2 that were mediated by thromboxane prostanoid (TP) receptors. This inhibition was not observed in preparations of pre-hypertensive 5-week-old SHR, and was significantly larger in those of 36- than 18-week-old SHR. Pre-exposure to the PKC activator, phorbol 12,13-dibutyrate, also inhibited subsequent contractions to PGE2 in SHR aortae. The selective inhibitor of PKC-ε, ε-V1-2, abolished the desensitization caused by pre-exposure to phenylephrine. Two molecular PKC bands were detected and their relative intensities differed in 36-week-old WKY and SHR vascular smooth muscle. The mRNA expressions of PKC-α, PKC-ε, PK-N2 and PKC-ζ and of G protein-coupled kinase (GRK)-2, GRK4 and β-arrestin2 were higher in SHR than WKY aortae.
Conclusions and Implications
These experiments suggest that in the SHR but not the WKY aorta, α1-adrenoceptor activation desensitizes TP receptors through activation of PKC-ε. This heterologous desensitization is a consequence of the chronic exposure to high arterial pressure.
Tables of Links
| LIGANDS |
|---|
| Calphostin C |
| Go6976 |
| Noradrenaline |
| PGE2 |
| Phenoxybenzamine |
| Phenylephrine |
| S-18886, terutroban |
| U46619 |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (a,bAlexander et al., 2013a, b).
Introduction
Arachidonic acid is transformed to the endoperoxide PGH2 by COX-1 or COX-2 and different synthases then convert PGH2 to one of five primary PGs- PGD2, PGE2, PGF2α, prostacyclin (PGI2) or thromboxane A2 (TXA2; Feletou et al., 2010a)]. PGs are involved in a number of processes, which contribute to hypertension and atherosclerosis (Feletou et al., 2010b,c; Giannarelli et al., 2010; Vanhoutte, 2011). PGs and TXA2 can activate specific GPCRs – the prostanoid DP, EP, FP, IP and TP receptors respectively. However, in isolated arteries of the spontaneously hypertensive rat (SHR), PGs cause vasoconstriction mainly through activation of TP receptors (Tang et al., 2005; 2008).
The TP receptor agonist U46619 elicits comparable contractions in isolated arteries of SHR and Wistar-Kyoto normotensive (WKY) rats (Ge et al., 1995). However, the former preparations are more responsive than the latter to endogenous TP receptor agonists such as endoperoxides (Ge et al., 1995) or prostacyclin (Gluais et al., 2005). The present study was initiated originally to compare contractions with other PGs in WKY and SHR aortae. Serendipitously, we observed that contractions to PGE2 were depressed by previous exposure (pre-exposure) to phenylephrine in SHR but not in WKY aortae (Figure 1). Further experiments reported here were designed to investigate the mechanism(s) underlying this unexpected difference These experiments suggest that activation of α1-adrenoceptors causes heterologous desensitization of TP receptors through activation of PKC-ε in the aorta from adult SHR but not in that from WKY rats.
Figure 1.
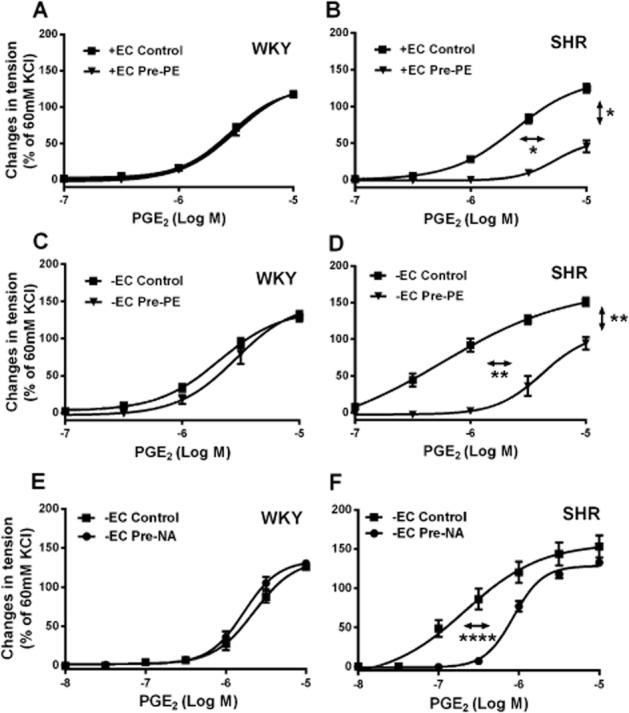
(A–D) Contractions to increasing concentrations of PGE2 in rings [with (+EC) or without (−EC) endothelium] of 36-week-old WKY or SHR aortae with or without previous incubation (40 min) with phenylephrine (2 × 10−7 M; Pre-PE). (E and F) Contractions to increasing concentrations of PGE2 in rings of 18-week-old WKY or SHR aortae (without endothelium) with or without previous incubation (40 min) with noradrenaline (2 × 10−7 M; Pre-NA). Increases in tension above basal are expressed as percentage of the reference contraction to KCl (60 mM) obtained at the beginning of the experiment and shown as means ± SEM; vertical arrows indicate statistical comparisons between responses to the highest concentration tested, whereas horizontal arrows indicate comparisons between −logEC50 values (estimated by regression analysis); (A–D) n = 6, *P ≤ 0.05, **P ≤ 0.01 versus no previous exposure to the α1-adrenoceptor agonist; (E and F) n = 5, ****P ≤ 0.0001 versus without previous incubation with noradrenaline.
Methods
All animal care and experimental procedures were approved by the Committee on the Use of Live Animals in Teaching and Research of The University of Hong Kong. Studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010). A total of 129 animals were used in the present study.
Tissue preparation
The SHR and WKY male rats (5-, 18- or 36-weeks old) were purchased from the Laboratory Animal Services Centre, The Chinese University of Hong Kong, Hong Kong SAR. They were maintained under a 12:12 h light/dark cycle at 21 ± 1°C and fed with standard laboratory chow and water. They were killed with an overdose of pentobarbital sodium (140 mg·kg−1) and exsanguinated. Their thoracic aorta was quickly dissected free, and placed in cold Krebs-Ringer bicarbonate buffer of the following composition (mmol·L−1): NaCl (118), KCl (4.7), CaCl2 (2.5), MgSO4 (1.2), KH2PO4 (1.2), NaHCO3 (25) and glucose (5.5) (control solution). The aortae were cleaned of adherent connective tissue and cut into rings (3∼4 mm in length). In certain preparations, the endothelium was removed mechanically by inserting the tip of a syringe needle into the ring and rolling them back and forth in a container filled with control solution (Furchgott and Zawadzki, 1980).
Isometric tension recording
Rings (with or without endothelium) of SHR or WKY aortae were suspended in organ chambers filled with 5 mL of warmed control solution (37°C) and aerated with 95% O2 and 5% CO2. They were connected to force transducers (FT03; Grass Instrument, Quincy, MA, USA) for isometric tension recording (PowerLab ADInstruments, Sydney, Australia). They were allowed to equilibrate for 1.5 h at optimal resting tension [2.5 g; determined in preliminary experiments (data not shown)]. Then, they were exposed twice to 60 mM KCl in order to obtain a reference contraction. Further increases in tension above the basal level were expressed in percentage of the response to 60 mM KCl, to compensate for differences in the amount of vascular smooth muscle cells between arterial rings.
Protein extraction and Western blotting
Rings (without endothelium) of SHR and WKY aortae were incubated with phenylephrine (2 × 10−7 M) or phorbol 12,13-dibutyrate (PDBu; 10−7 M) for 40 min then immediately frozen in liquid nitrogen for protein extraction. These samples were used to measure protein presence of TP receptors. Untreated preparations without endothelium were used to detect PKC protein expression. WKY and SHR aortic rings were cut into very small pieces using clean scissors and collected in microcentrifuge tubes filled with ice-cold lysis buffer. The samples were sonicated and then centrifuged (2500× g) for 3 min at 4°C. The supernatant fluid was collected into new microcentrifuge tubes for Western blotting analysis. Polyclonal antibodies were used to detect the presence of TP receptors (catalogue number: 10004452, Cayman, Ann Arbor, MI, USA) or total PKC (catalogue number: ab59363, Abcam, Cambridge, UK). Images were processed by ImageJ (NIH, Bethesda, MD, USA).
Reverse transcription and quantitative real-time PCR (qRT-PCR)
RNA extraction was performed using TRIzol reagent (Invitrogen, Carlsbad, CA, USA) following the manufacturer's instructions. Aortic rings of WKY and SHR (without endothelium) were immersed in 1 mL ice-cold TRIzol and homogenized using Tissuelyser LT (Qiagen, Hilden, Germany) for 5 min (twice) and kept at room temperature for 5 min. Then, 200 μL of chloroform were added to the homogenates and the samples were incubated at room temperature for 5 min. All homogenates were centrifuged (12 000× g) for 15 min at 4°C and the upper aqueous phases were transferred to new microcentrifuge tubes. The RNA was precipitated (with 500 μL isopropanol for 2 days at −20°C), collected by centrifugation (12 000× g for 15 min at 4°C), washed with 70% ethanol and suspended in 20 μL RNase-free water (Qiagen). The RNA concentration was determined by absorbance at 260/280 nm using Biotek Epoch (Biotek Instruments, Winooski, VT, USA).
Complementary DNA (cDNA) was synthesized from RNA using an Omniscript Reverse Transcription kit (Qiagen) following the manufacturer's instructions. First, two micrograms of RNA were mixed with Master Mix in microcentrifuge tubes and incubated at 37°C for 60 min. Then, the tubes were placed on ice to inactivate the enzyme and cDNA was stored at −80°C for qRT-PCR; qRT-PCR was performed using SYBR Green Supermix (Bio-Rad, Hercules, CA, USA) following the manufacturer's instructions. The reaction was performed on a StepOnePlus Real-Time PCR System (Applied Biosystem, Foster City, CA, USA). The qRT-PCR data were analysed using the comparative CT (2−ΔΔCT) method (Schmittgen and Livak, 2008). The internal reference gene was the housekeeping gene GAPDH. Primers were designed using NCBI/Primer-BLAST. The primers sequences used are listed in Table 1.
Table 1.
Primers used for quantitative real-time PCR analysis
| Gene name | Gene symbol | Accession ID | Primers |
|---|---|---|---|
| PKC, alpha (PKC-α) | Prkca | NM_001105713.1 | Forward: 5′ CGGATAAGGGACCTGACACT 3′ |
| Reverse: 5′ ACGCACTGCTTGTGAACATT 3′ | |||
| PKC, beta (PKC-β) | Prkcb | NM_001172305.1 | Forward: 5′ ATGACCAAACACCCAGGCAA 3′ |
| Reverse: 5′ GTGTCTCGCTTGTCTCTAGCTT 3′ | |||
| PKC, gamma (PKC-γ) | Prkcg | NM_012628.1 | Forward: 5′ TTTAGACCACGTCCGTGTGG 3′ |
| Reverse: 5′ CACGAAGTCCGGGTTCACAT 3′ | |||
| PKC, delta (PKC-δ) | Prkcd | NM_133307.1 | Forward: 5′ CTACCAATAGCCGGGACACC 3′ |
| Reverse: 5′ ATGCCGCAGTCTTCACACTT 3′ | |||
| PKD1 (PKC-δ1) | Prkd1 | NM_001276715.1 | Forward: 5′ TGGACCAGAAGTTCCCTGAAT 3′ |
| Reverse: 5′ GAAGCTGACAGGACCACTTCA 3′ | |||
| PKD2 (PKC-δ2) | Prkd2 | NM_001013895.1 | Forward: 5′ GTCCGTGGGTGTGATCATGT 3′ |
| Reverse: 5′ GGTCGATGGCTCCAGATGAG 3′ | |||
| PKD3 (PKC-δ3) | Prkd3 | NM_001024263.2 | Forward: 5′ CCGCGTCACTCACAAACTCT 3′ |
| Reverse: 5′ TCCACACTCTGGAAACTTCTGAT 3′ | |||
| PKC, epsilon (PKC-ε) | Prkce | NM_017171.1 | Forward: 5′ CAGGGATTTGAAACTGGACA 3′ |
| Reverse: 5′ CTGCAGGATCTCTGGAGCTA 3′ | |||
| PKC, eta (PKC-η) | Prkch | NM_031085.2 | Forward: 5′ CCCACCTACAACGAGGAGTTC 3′ |
| Reverse: 5′ AGATCCACCCAGCCCTCGAA 3′ | |||
| PKC, theta (PKC-θ) | Prkcq | NM_001276721.1 | Forward: 5′ CGAGGCAAGGTCTCAAGTGT 3′ |
| Reverse: 5′ TAAGGTGCGAGCCTGTTGAG 3′ | |||
| PKC, iota (PKC-ι) | Prkci | NM_032059.1 | Forward: 5′ AAACGCTTTAACAGGCGTGC 3′ |
| Reverse: 5′ CGCCCACACTCAATTGTGAC 3′ | |||
| PKC, zeta (PKC-ζ) | Prkcz | NM_022507.1 | Forward: 5′ AACACGCCAGGTTCTATGCT 3′ |
| Reverse: 5′ TTCCTTGCACATGCCGTAGT 3′ | |||
| PKN1 (PK-N1) | Pkn1 | NM_017175.2 | Forward: 5′ AGGACATCCCTTCCTGGTGA 3′ |
| Reverse: 5′ AGGCCGAATAGAAGACAGCC 3′ | |||
| PKN2 (PK-N2) | Pkn2 | NM_001105755.2 | Forward: 5′ CGTTTTTCCGGCTAACCGAC 3′ |
| Reverse: 5′ TATCCTCGGTTCTCGAGGGG 3′ | |||
| PKN3 (PK-N3) | Pkn3 | NM_001047861.1 | Forward: 5′ GCCGAAGGGAGCACTTTTCT 3′ |
| Reverse: 5′ TACAGACCCAAGCAAGCCAC 3′ | |||
| GPCR kinase 1 (GRK1) | Grk1 | NM_031096.1 | Forward: 5′ AAGACCAAGGGCTATGCAGG 3′ |
| Reverse: 5′ TCTCCACCTTCTCTCCTCGG 3′ | |||
| Adrenergic, beta, receptor kinase 1 (GRK2) | Adrbk1 | NM_012776.1 | Forward: 5′ TCCTGCAGTGTGATAGTGACC 3′ |
| Reverse: 5′ TGAATCAGTGGCACCTTGCT 3′ | |||
| Adrenergic, beta, receptor kinase 2 (GRK3) | Adrbk2 | NM_012897.2 | Forward: 5′ GCATTAAGCTGTTGGACTGC 3′ |
| Reverse: 5′ CTTCTTCCTGGCCTCAATTT 3′ | |||
| GPCR kinase 4 (GRK4) | Grk4 | NM_022928.1 | Forward: 5′ GCTGAAGGCTCGTCAAGGATT 3′ |
| Reverse: 5′ TTCCGATTGGCTGTTGGTCA 3′ | |||
| GPCR kinase 5 (GRK5) | Grk5 | NM_030829.1 | Forward: 5′ GCAACATGCTGCTCACCAAA 3′ |
| Reverse: 5′ CGAAGGGAGGGTCCAACATC 3′ | |||
| GPCR kinase 6 (GRK6) | Grk6 | NM_031657.3 | Forward: 5′ CAGAACGAGATGGTGGAGACTGAG 3′ |
| Reverse: 5′ GCAGTTCCCACAGCAATCCTTT 3′ | |||
| GPCR kinase 7 (GRK7)a | Grk7 | NM_139209.2 | Forward: 5′ CACCTCCATGAACTCGGCAT 3′ |
| Reverse: 5′ ACCATTGGTTCCAGCCCTCT 3′ | |||
| Retina and pineal gland (S-antigen) | Arrestin, Sag | NM_013023.2 | Forward: 5′ GCTAGCTGCTCACCATCTGAA 3′ |
| Reverse: 5′ TAGATGGTCACCGACTTGTCC 3′ | |||
| Arrestin, beta 1 (β-arrestin 1) | Arrb1 | NM_012910.2 | Forward: 5′ CGAGTGTTCAAGAAGGCAAGC 3′ |
| Reverse: 5′ TCAGTGTCACGTAGACTCGCC 3′ | |||
| Arrestin, beta 2 (β-arrestin 2) | Arrb2 | NM_012911.1 | Forward: 5′ CACAAAAGGAACTCCGTGCG 3′ |
| Reverse: 5′ AGCTCTTTGTCCAGGGAAGC 3′ | |||
| Arrestin 3, retinal (arrestin-4) | X-arrestin, Arr3 | NM_001190993.1 | Forward: 5′ CTGGGGATCCTGGTGTCCTA 3′ |
| Reverse: 5′ GTAGCCACTGCTCTCTCACC 3′ | |||
| GAPDH | Gapdh | NM_017008.4 | Forward: 5′ TCATCAACGGGAAACCCATCAC 3′ |
| Reverse: 5′ ACGCCAGTAGACTCCACGACAT 3′ |
All primers are based on gene sequence of Rattus norvegicus, except GRK7, which is based on Homo sapiens.
Data analysis
Results are presented as means ± SEM; n refers to the number of individual experimental animals in each group. For each agonist, the concentration that caused 50% of maximal contraction (EC50) as well as its negative logarithm (−log EC50), and the AUC were calculated by regression analysis. The concentration-contraction curves were analysed by comparing the contraction level reached with the highest concentration of agonist tested, the −log EC50 values (calculated by regression analysis), and/or the AUC. The data, which passed the Kolmogorov–Smirnov test, were analysed using Student's t-test for paired or unpaired observations; otherwise, the Mann–Whitney or Wilcoxon tests were used for statistical comparisons. When more than two means were compared, one-way anova was used for data that passed the Kolmogorov–Smirnov test; otherwise, the Kruskal–Wallis test was used for unpaired and the Friedman test for paired observations; ordinary one-way anova was used for the comparisons of −log EC50. If a significant F value was found, then Tukey's, Dunnett's or Dunn's multiple comparison test were employed to identify differences among groups. Regression analysis (to calculate −log EC50 values and AUC) and statistical analysis were performed using GraphPad Prism version 6 (San Diego, CA, USA). The Western blotting and qRT-PCR data are normalized to the protein presence of β-actin and the mRNA expression of GAPDH respectively. The normalized qRT-PCR data of the SHR group are expressed as the fold change relative to the WKY group (Schmittgen and Livak, 2008); ‘n’ refers to the number of individual rats in each group (two samples were taken from each rat aortic tissue for measurements and the mean of the duplicated values was taken as n = 1). P-values equal or less than 0.05 were considered to indicate statistically significant differences.
Materials
Phenylephrine, noradrenaline, sodium nitroprusside were purchased from Sigma-Aldrich® Chemical Company (St Louis, MO, USA). PGE2 was purchased from Cayman Chemical. U46619 {(Z)-7-[(1S,4R,5R,6S)-5-[(E,3S)-3-hydroxyoct-1-enyl]-3-oxabicyclo[2.2.1]heptan-6-yl]hept-5-enoic acid}} was purchased from Biomol (Plymouth Meeting, PA, USA). Epsilon-V1-2 was purchased from ANASPEC (Fremont, CA, USA). Calphostin C was purchased from Merck (Whitehouse Station, NJ, USA). PDBu and Go6976 were purchased from Tocris Bioscience (Bristol, UK). Phenoxybenzamine was purchased from Santa Cruz (Dallas, TX, USA). S-18886 {3-[(6-amino-4-chlorobenzensulfonyl-2-methyl-5,6,7,8-tetrahydronaphth)-1-yl] propionic acid} was a kind gift from the Institut de Recherches Servier (Suresnes, France).
Results
Pre-incubation with phenylephrine and noradrenaline
To demonstrate that pre-exposure with phenylephrine affects the reactivity of SHR but not WKY aortae, rings (with and without endothelium, from 36-week-old rats) of the same arteries were incubated for 40 min either in control solution or in the presence of phenylephrine (2 × 10−7 M; ‘pre-exposed’ preparations). After repeated washing with control solution and return to basal tension, they were exposed, after waiting a further for 15 min, to cumulatively increasing concentrations of PGE2. The concentration-dependent contractions to the prostanoid were comparable in control and pre-exposed WKY preparations with and without endothelium (Figure 1A and C). By contrast, the contractions to PGE2 were significantly depressed in pre-exposed SHR aortae; this difference persisted in the absence of endothelium (Figure 1B and D). Removal of the endothelium significantly amplified the contractions to PGE2 in SHR, but not in WKY aortae, and these contractions were significantly larger in SHR preparations compared to WKY. All further functional studies were performed on preparations without endothelium.
To verify whether or not a physiological concentration of a natural α-adrenoceptor ligand would exert the same effect as phenylephrine, SHR and WKY aortae (from 18-week-old rats) were pre-incubated for 40 min with noradrenaline (2 × 10−7 M; Cui and Guo, 2002; Di et al., 2011). The pre-exposure to the natural catecholamine also inhibited the subsequent contractions to PGE2 in rings of SHR but not in those of WKY aorta (Figure 1E and F).
To determine whether or not the inhibition by pre-exposure occurs in preparations of pre-hypertensive SHR (Sankhanava Kundu, 2008), aortae from 5-week-old WKY and SHR were incubated with phenylephrine for 40 min. They then were contracted with cumulatively increasing concentrations of PGE2. The contractions to PGE2 were not significantly affected by pre-exposure to phenylephrine in rings of 5-week-old WKY or SHR (Figure 2, upper). Compared with 18-week-old rats, pre-exposure to phenylephrine caused significantly greater reduction of PGE2-induced contractions in preparations from 36-week-old SHR (Figure 2, lower). PGE2 produced larger contractions at low concentrations (3 × 10−8 to 3 × 10−7 M, significantly larger at 10−7 M) in aortae of 18-week than 36-week-old SHR (Figure 2C). As the inhibition of PGE2-induced contraction by phenylephrine pre-exposure was more prominent in aortae of 36-week-old SHR, subsequent experiments were performed in rats at this age, unless specified otherwise.
Figure 2.
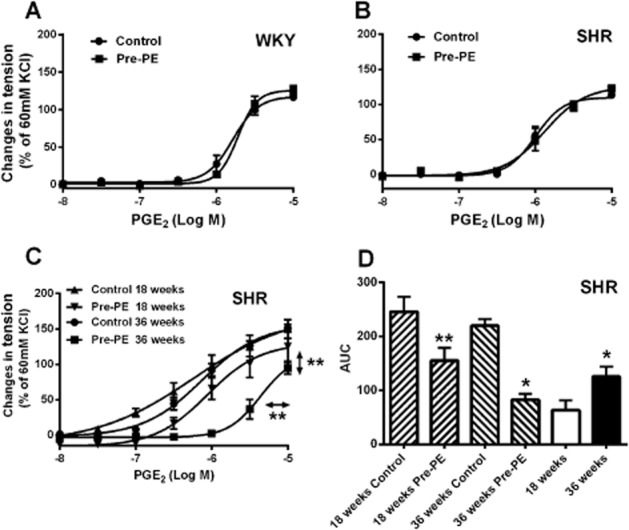
(A and B) Contractions to increasing concentrations of PGE2 in rings of 5-week-old WKY or SHR aortae (without endothelium) with or without previous incubation (40 min) with phenylephrine (2 × 10−7 M; Pre-PE); (C) contractions to increasing concentration of PGE2 shown as concentration-response curves (18 and 36 weeks old); (D) the AUC of Figure 2C, and the differences in PGE2-induced contractions expressed as area between the concentration-response curves between aortic rings of SHR at the same age (18 and 36 weeks old) with or without pre-exposure to phenylephrine (2 × 10−7 M; Pre-PE). Increases in tension above basal are expressed as percentage of the reference contraction to KCl (60 mM) obtained at the beginning of the experiment and data are shown as means ± SEM; vertical arrows indicate statistical comparisons between responses to the highest concentration tested, whereas horizontal arrows indicate comparisons between −logEC50 values (estimated by regression analysis); n = 6, (C) **P ≤ 0.01 versus without pre-exposure to phenylephrine. (D) **P ≤ 0.01 versus 18 weeks control, *P ≤ 0.05 versus 36 weeks control, *P ≤ 0.05 versus 18 weeks (D).
To determine whether or not the inhibitory effect of pre-incubation was long lasting, SHR rings were pre-incubated with phenylephrine. After washout, they were incubated in control solution for either 15 or 60 min before exposing them to cumulatively increasing concentration of PGE2. The subsequent contractions to PGE2 were impaired significantly by pre-exposure to phenylephrine only after the 15 min delay (Figure 3A).
Figure 3.
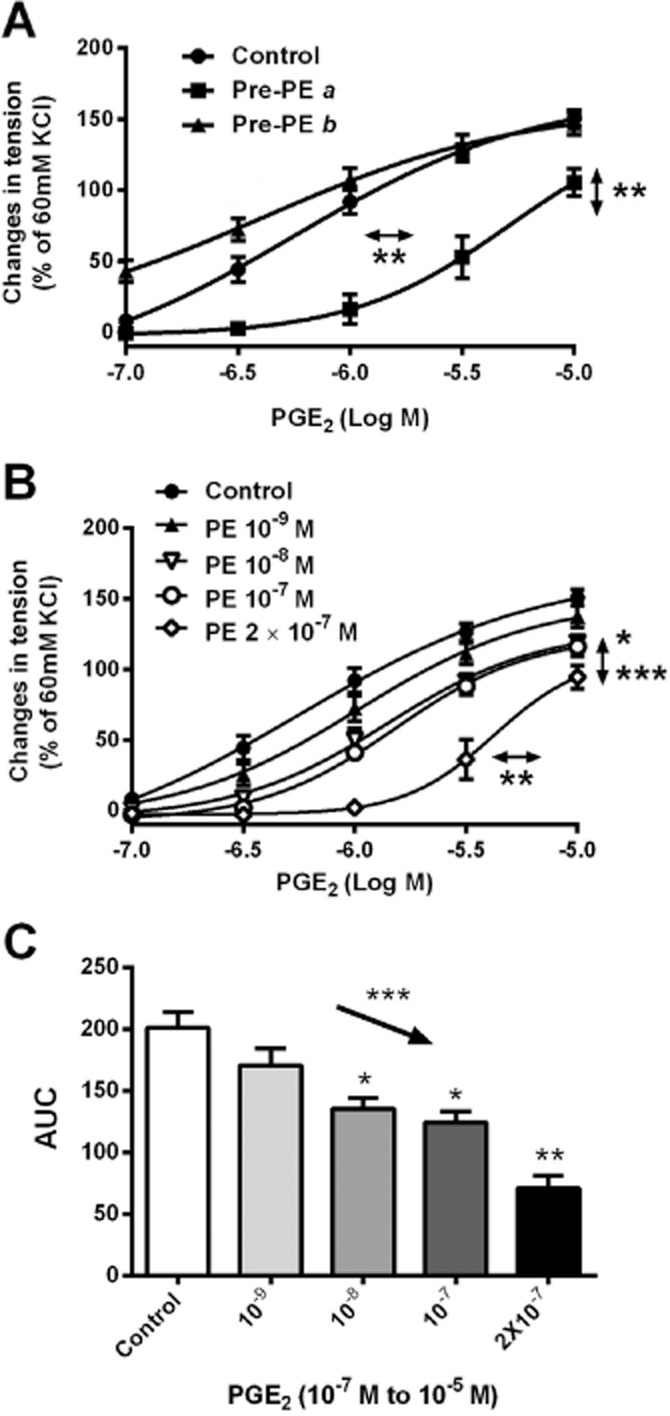
(A) Contractions to increasing concentrations of PGE2 in rings of SHR aortae (without endothelium); Pre-PE a, contractions obtained 15 min after pre-incubation with phenylephrine, Pre-PE b, contractions obtained 60 min after pre-incubation with phenylephrine (n = 4 ∼ 8). (B) Contractions to increasing concentrations of PGE2 in rings of SHR aortae (without endothelium) with or without previous incubation (40 min) with increasing concentrations of phenylephrine (PE, concentration; n = 6). (C) AUC of Figure 3B. Increases in tension above basal are expressed as percentage of the reference contraction to KCl (60 mM) obtained at the beginning of the experiment and shown as means ± SEM; vertical arrows indicate statistical comparisons between responses to the highest concentration tested, whereas horizontal arrows indicate comparisons between −logEC50 values (estimated by regression analysis); (A, B) *P ≤ 0.05 (PE 10−7 M), **P ≤ 0.01, ***P ≤ 0.001 (PE 2 × 10−7 M) versus without pre-exposure to PE. (C) The downward arrow indicates a linear decreasing trend (a systematically decreasing trend, which is statistically distinguished from random behaviour) between columns moving from left to right (***P ≤ 0.001); *P ≤ 0.05, **P ≤ 0.01 versus no previous incubation with PE.
To determine the concentration dependency of the inhibitory effect on the subsequent contractions to PGE2, SHR aortae were incubated for 40 min with increasing concentrations (10−9 to 2 × 10−7 M) of phenylephrine (Figure 3B and C). The statistical analysis revealed a significant linear trend with decreasing responses to PGE2 in function of pre-exposure to increasing amounts of phenylephrine (Figure 3C). Thus, previous exposure to the α1-adrenoceptor agonist inhibited subsequent contractions to vasoconstrictor prostanoid in a concentration-dependent manner.
Role of TP receptors
Aortae of 18-week-old SHR or WKY were incubated with the antagonist of TP receptors, S18886 (10−6 M; Tang et al., 2005) for 40 min before obtaining contractions to increasing concentrations (10−8 to 10−5 M) of PGE2. S18886 abolished the response to the PG in either WKY or SHR preparations (Figure 4A and B). These experiments confirm (Tang et al., 2008) that in the SHR aorta, PGE2 acts as a TP receptor agonist.
Figure 4.
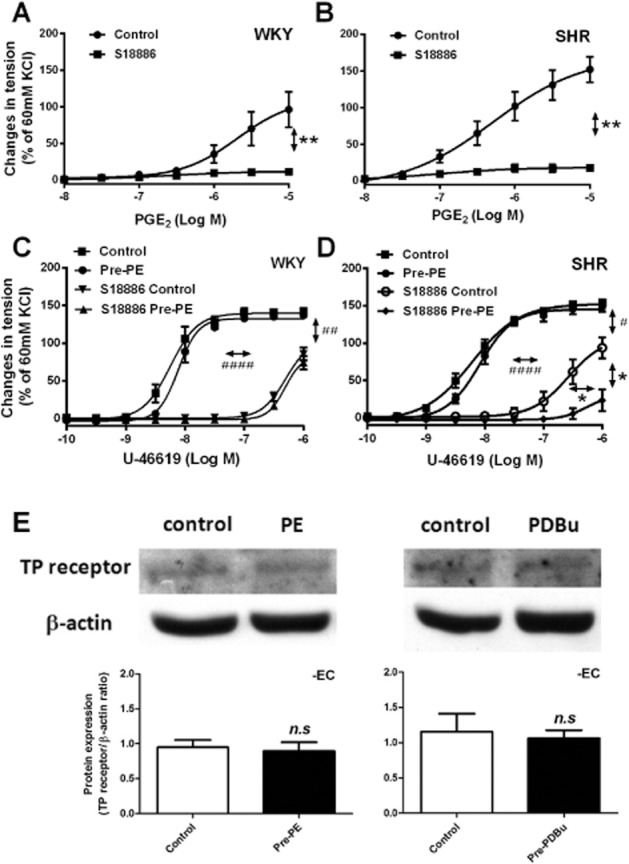
(A, B) Contractions to increasing concentration of PGE2 in rings of WKY or SHR aortae (without endothelium) with or without incubation with S18886 (10−6 M, 40 min); (C, D) contractions to increasing concentrations of U46619 in the absence and presence of S18886 (10−7 M, 20 min) in rings of WKY or SHR aortae (without endothelium), with or without previous incubation with phenylephrine (Pre-PE, 2 × 10−7 M, 40 min). Increases in tension are expressed as percentage of the reference contraction to KCl (60 mM) obtained at the beginning of the experiment and shown as means ± SEM. (E) Western blotting shows TP receptor protein expression in SHR aortae without endothelium, with or without previous incubation (40 min) with phenylephrine (PE, 2 × 10−7 M, left, n = 4, the Mann–Whitney test was used for statistical analysis) or PDBu (10−7 M, right, n = 5); the images were analysed by ImageJ, and the expressions are expressed as the ratio of TP receptor/β-actin shown as means ± SEM. Vertical arrows indicate statistical comparisons between responses to the highest concentration used, whereas horizontal arrows indicate comparisons between −log EC50 values (estimated by regression analysis); (A, B) n = 6, **P ≤ 0.01 versus no incubation with S18886; (C, D) n = 6, #P ≤ 0.05, ##P ≤ 0.01, ####P ≤ 0.0001 incubation with versus without S18886 (no pre-exposure to phenylephrine group); *P ≤ 0.05 versus no previous incubation with phenylephrine.
To confirm the importance of agonist efficacy at TP receptors, the effect of pre-incubation with phenylephrine was compared on subsequent contractions of SHR aortae to U46619 (before and after partial inhibition with 10−7 M S18886). Contractions to U46619 were comparable in untreated WKY and SHR aortae, confirming earlier observations (Ge et al., 1995). Previous exposure to phenylephrine did not inhibit contractions to U46619 under control conditions, or after partial TP receptor blockade with S18886 in WKY preparations (Figure 4C) but did significantly so after partial TP blockade in SHR aortae (Figure 4D).
To assess the effects of pre-exposure on the presence of TP receptors, SHR aortic rings without endothelium were incubated with or without phenylephrine (2 × 10−7 M) for 40 min and washed three times with control solution. They then were frozen in liquid nitrogen. The protein expression of TP receptors was determined by Western blotting. The specific binding to TP receptor was determined using blocking peptide of the TP receptor antibody. Binding of TP receptor antibody was detected near 55 kD in SHR aortae, and was eliminated by co-incubation with blocking peptide; the other protein bands in the blots were similar under the two conditions (data not shown). Thus, the binding near 55 kD was accepted as detection of the protein presence of TP receptors. The protein expression level of TP receptor was not different in smooth muscle of SHR aorta pre-exposed to phenylephrine compared to that of control preparations (Figure 4E, left).
Role of PKC
Next, the role of PKC in the inhibitory effect of previous exposure to α1-adrenoceptor agonists was investigated. Previous incubation with an activator of PKC, PDBu (10−7 M; Hypolite et al., 2013) mimicked the effect of pre-exposure to phenylephrine in SHR (Figure 5B), but not in WKY aortae (Figure 5A) preparations. Administration of the non-selective PKC inhibitor calphostin C (10−6 M, 30 min before incubation with phenylephrine and re-added after washout; Inoue et al., 2011) significantly reduced contractions to PGE2 and abolished the inhibitory effect of pre-exposure to phenylephrine (Figure 5C). These findings suggest that activation of PKC contributes to TP receptor desensitization.
Figure 5.
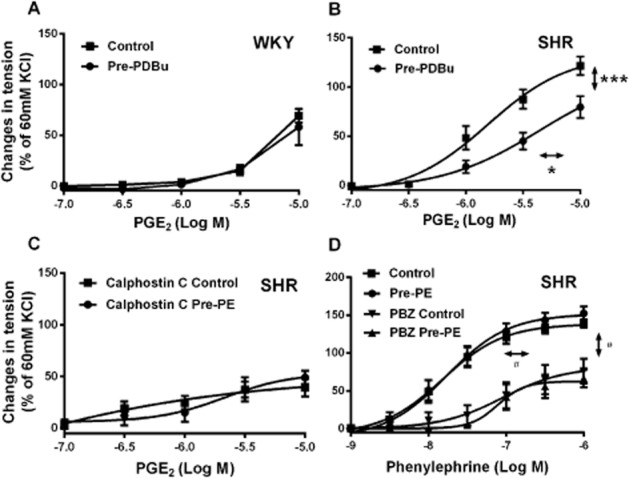
Contractions to increasing concentrations of PGE2 in rings of (A, B) WKY or SHR aortae (without endothelium) with or without previous exposure to PDBu (10−7 M) and (C) SHR aortae (without endothelium) with or without previous exposure to phenylephrine (2 × 10−7 M; Pre-PE) in the presence of calphostin C (10−6 M); (D) Contractions to increasing concentrations of phenylephrine in the absence and presence of 5 × 10−9 M phenoxybenzamine (PBZ) in rings of SHR aortae (without endothelium), with or without previous incubation with phenylephrine (Pre-PE, 2 × 10−7 M, 40 min). Increases in tension above basal are expressed as percentage of the reference contraction to KCl (60 mM) obtained at the beginning of the experiment and shown as means ± SEM; vertical arrows indicate statistical comparisons between responses to the highest concentration tested, whereas horizontal arrows indicate comparisons between −log EC50 values (estimated by regression analysis); n = 6 (B) *P ≤ 0.05, ***P ≤ 0.001 versus no previous incubation with PDBu; (D) #P ≤ 0.05 incubation with versus without PBZ (no pre-exposure to phenylephrine group).
To determine whether or not the effect of pre-exposure was due to the contraction of the preparation to phenylephrine, the following experiments were performed: (i) the pre-exposure did not affect the second contractions evoked by phenylephrine itself with or without co-incubation with an antagonist of α1-adrenoceptors, phenoxybenzamine (5 × 10−9 M, Figure 5D); and (ii) incubation for 40 min with 2 × 10−7 M phenylephrine in the presence or absence of 10−4 M sodium nitroprusside (to maximally stimulate the production of cyclic GMP; Papapetropoulos et al., 1996) and limit the contraction evoked by phenylephrine during the pre-incubation] significantly limited the contraction evoked by PGE2; after repeated washout, the basal tension of the rings was the same, regardless of the presence or absence of sodium nitroprusside (1.93 ± 0.06 and, 2.06 ± 0.05; P > 0.05). The co-incubation with sodium nitroprusside did not prevent the inhibitory effect of the pre-exposure to phenylephrine on subsequent contractions to PGE2 (data not shown). Thus, the contraction produced by phenylephrine during the pre-exposure is unlikely to cause the desensitization.
To confirm the effect of PKC activation on the presence of TP receptors, SHR aortic rings were incubated with PDBu (10−7 M for 40 min) and processed for Western blotting. The protein expression of TP receptors, after incubation with the compound was comparable to that of the control preparations (Figure 4E, right).
The protein presence of total PKC was compared in aortic smooth muscle of WKY and SHR (5 and 36 weeks old) using Western blotting. In preparations of 5-week-old rats, the blots revealed two bands of different molecular mass, suggesting the presence of different PKC isoforms, and the expression of PKC was comparable in the two strains (Figure 6A). However, in aortae of 36-week-old rats, the two bands showed different intensity (Figure 6B). The expression of the band near 95 kD was significantly decreased but that of the band near 72 kD significantly augmented in SHR compared with WKY preparations. The total PKC protein expression was higher in WKY preparations.
Figure 6.
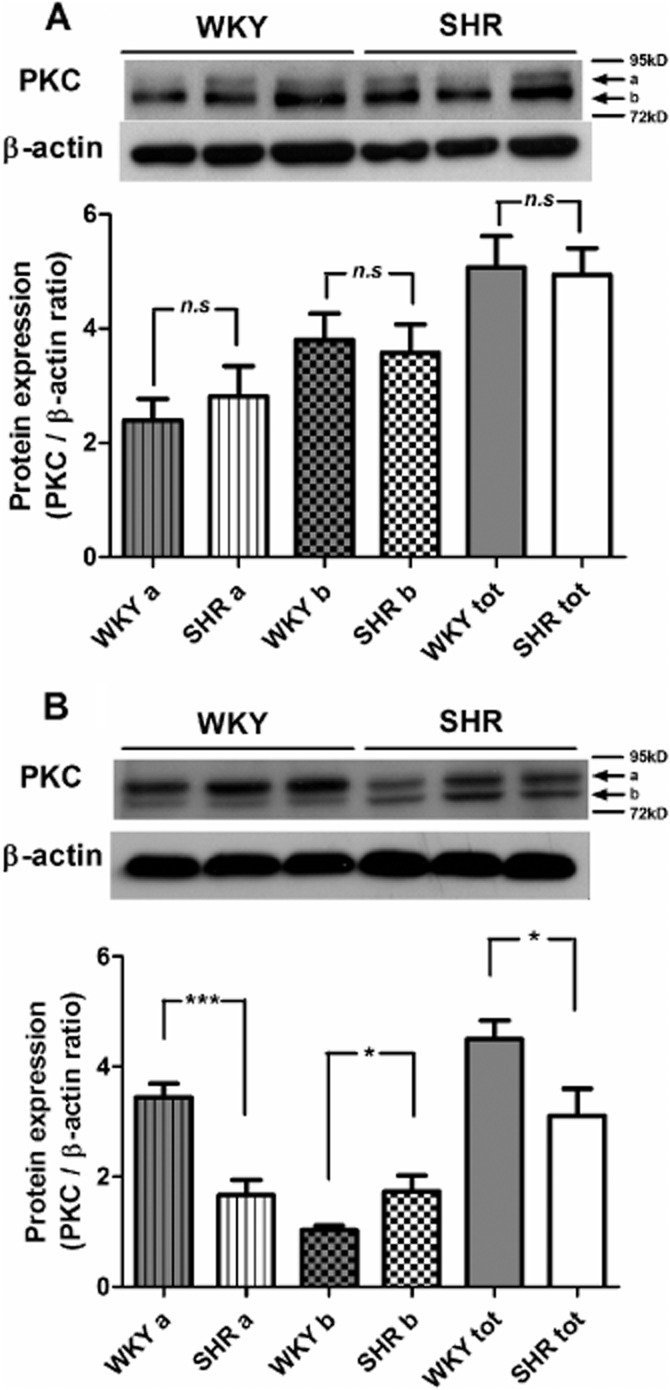
Western blotting shows the total PKC protein expression in aortic smooth muscle cells of (A) 5-week-old WKY or SHR, and (B) 36-week-old WKY or SHR. a, near 95 kD; b, near 72 kD; tot, total expression. The images were analysed by ImageJ; the expressions are expressed as the ratio of PKC/β-actin and shown as means ± SEM, 5 weeks, n = 6, 36 weeks, n = 9; *P ≤ 0.05, ***P ≤ 0.001 versus WKY; n.s., not significant.
Then, the mRNA expressions of 15 different isoforms of the PKC in smooth muscle of WKY and SHR aortae were detected using qRT-PCR. Within the subfamily of conventional PKCs (Webb et al., 2000), only the mRNA expression of PKC-α could be detected and was significantly greater in SHR than in WKY preparations (Figure 7). Within the subfamily of novel PKCs (Webb et al., 2000), the mRNA expressions of PKC-δ, PKC-δ1, PKC-δ2 and PKC-δ3 were comparable in WKY and SHR aortic smooth muscle (data not shown). However, expression of mRNA for PKC-ε was significantly higher in SHR than in WKY preparations (Figure 7). By contrast, the mRNA expression of PKC-η was significantly greater in WKY than in SHR preparations (Figure 7). PKC-θ could not be detected in either WKY or SHR aortae (data not shown). In the subfamily of atypical PKCs (Webb et al., 2000), the mRNA expressions of PK-N2 and PKC-ζ were significantly greater in SHR than WKY aortae (Figure 7), while PK-N3 was not expressed in either (data not shown).
Figure 7.
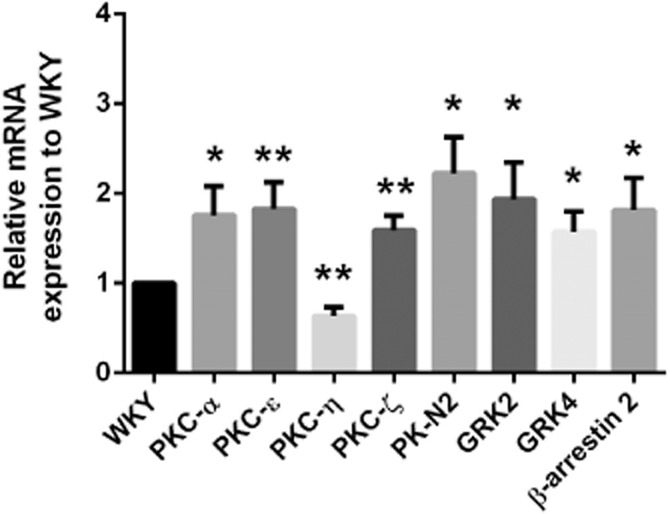
Quantitative real-time PCR results showing mRNA expressions relative to WKY (see Methods) of PKC, GRK isoforms and β-arrestin2 in aortic smooth muscle cells of SHR. The black bar represents the mRNA expression in WKY as 1; the other bars represent the expression, relative to those of WKY, of different mRNAs in SHR. Data shown as means ± SEM. PKC-α, n = 11; other PKC, GRK isoforms and β-arrestin 2, n = 6 ∼ 9. *P ≤ 0.05, **P ≤ 0.01 versus WKY.
G protein-coupled kinase (GRK) and arrestin
It seemed logical to also compare the mRNA expression of different GRK isoforms in aortic smooth muscle of normotensive and hypertensive animals, as PKC may play a role as a GRK regulator (Pronin and Benovic, 1997; Kohout and Lefkowitz, 2003; Lorenz et al., 2003; Gildea et al., 2013). Of the seven known isoforms of GRK reported (Gurevich et al., 2012), the mRNA expressions of GRK1 and GRK7 could not be detected in either WKY or SHR aortic smooth muscle (data not shown). Those of GRK2 and GRK4 were increased significantly in SHR preparations (Figure 7). The mRNA expressions of GRK3, GRK5 and GRK6 were comparable in the two strains (data not shown). The mRNA expression of β-arrestin 2 was significantly greater in SHR than in WKY aortae (Figure 7); the other arrestin isoforms could not be detected in either WKY or SHR smooth muscle (data not shown).
Role of PKC-ε
Finally, the selective inhibitors of PKC-α (Go6976, 10−6 M; Li et al., 2004) and of PKC-ε (ε-V1-2, 5 × 10−6 M; Lee et al., 1999) were used to examine the role of PKC-α and PKC-ε respectively. SHR aortic rings were incubated with one of these inhibitors for 30 min before the pre-exposure to phenylephrine (2 × 10−7 M). After that incubation, the aortic rings were washed with control solution. Cumulative concentration-response curves to PGE2 were then obtained in these rings. Although Go6976 significantly increased the contractions to PGE2 in SHR aortae, the compound did not abolish the difference between aortae, with or without pre-exposure to phenylephrine (Figure 8B). By contrast, ε-V1-2 did not significantly affect the contractions to PGE2 in control aortic rings but restored the response in SHR preparations despite pre-exposure to phenylephrine (Figure 8A).
Figure 8.
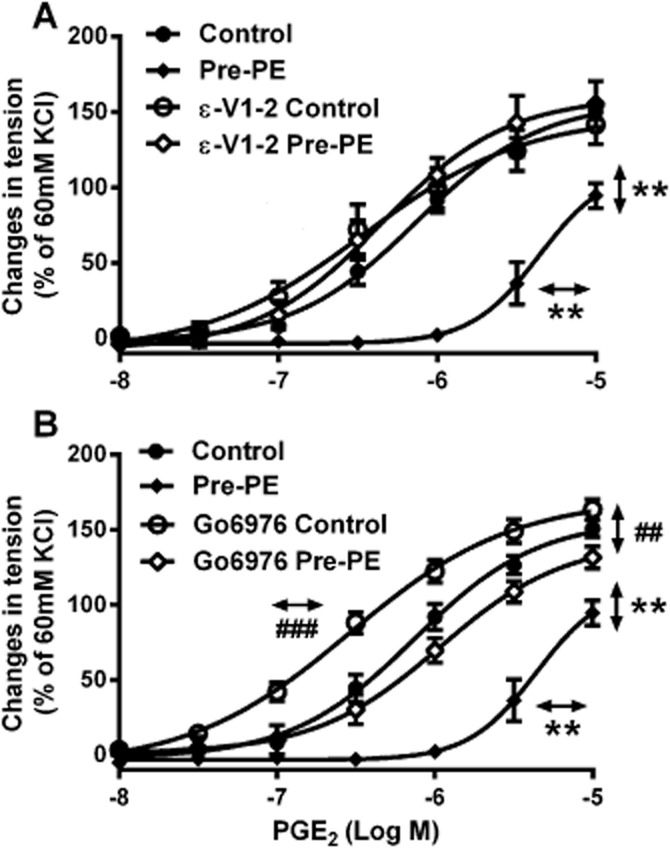
Contractions to increasing concentrations of PGE2 in the absence and presence of (A) ε-V1-2 (5 × 10−6 M) or (B) Go6976 (10−6 M) in rings of SHR aortae (without endothelium), with or without previous incubation with phenylephrine (Pre-PE, 2 × 10−7 M, 40 min). Increases in tension above basal are expressed as percentage of the reference contraction to KCl (60 mM) obtained at the beginning of the experiment and shown as means ± SEM. Vertical arrows indicate statistical comparisons between responses to the highest concentration tested, whereas horizontal arrows indicate comparisons between −log EC50 values (estimated by regression analysis); n = 6, (A, B) **P ≤ 0.01 versus no previous incubation with phenylephrine; (B) ##P ≤ 0.01, ###P ≤ 0.001 versus incubation with Go6976 but without pre-exposure to phenylephrine.
Discussion and conclusions
The main finding of the present study is that, in the aorta of hypertensive rats, previous exposure to synthetic or natural α1-adrenoceptor agonists curtails, in a concentration-dependent manner, the subsequent TP receptor-mediated contractions to PGE2. This curtailment in vascular responsiveness is endothelium-independent and not due to the contraction per se resulting from α1-adrenoceptor activation. Surprisingly, the phenomenon was observed in aortae of adult SHRs but not those of its normotensive counterpart, the WKY or in those of pre-hypertensive SHR (5 weeks old; Sankhanava Kundu, 2008). Furthermore, prolonged hypertension resulted in a more pronounced inhibitory effect of the phenylephrine pre-exposure as it increased with age in the SHR. These findings raise the possibility that the difference between the two strains was due to the chronic exposure of the blood vessels to an increased arterial pressure in the SHR.
Phenylephrine is a selective agonist at α1-adrenoceptors (Godfraind et al., 1982). α1-Adrenoceptors are a subfamily of adrenoceptors, which are targets for adrenaline and noradrenaline, the endogenous mediators of the sympathetic nervous system. Their activation causes vasoconstriction (Civantos Calzada and Aleixandre de Artinano, 2001). The α1-adrenoceptors can be further divided to α1A, α1B and α1D subtypes (Civantos Calzada and Aleixandre de Artinano, 2001). The expression of α1A and α1B-adrenoceptors is comparable in the aorta of WKY and SHR, whereas that of the α1D-subtype is higher in the latter (Godínez-Hernández et al., 2006). α1-adrenoceptors, especially the α1D subtype, play an important role in the homeostatic control of vascular tone, and are involved in sympathetic regulation of arterial BP (Civantos Calzada and Aleixandre de Artinano, 2001; Tanoue et al., 2002). The sympathetic nervous system is hyperactive in the SHR (Head et al., 1985; Scott and Galway, 1985; Mangiarua and Lee, 1990; Ely et al., 1997). Thus, the interaction observed in the present study between the downstream effects of α1-adrenoceptor activation and the response to PGE2 may play a modulating role in reducing prostanoid-induced vasoconstriction in the SHR, in which such responses are prominent (Lüscher and Vanhoutte, 1986; Tang et al., 2005; Tang and Vanhoutte, 2008).
PGE2 is an agonist at both TP and EP receptors causing contraction or relaxation of blood vessels (Tang et al., 2008; Suzuki et al., 2011; Foudi et al., 2012). The contractions it causes in both the SHR and WKY aorta can be attributed to TP receptor activation only, because they were abolished by the selective antagonist S18886. The pre-exposure to the α1-adrenoceptor agonist for 40 min did not modify the protein presence of TP receptors in the vascular smooth muscle cells and its effect was not long lasting, suggesting temporary desensitization. Hence the curtailment observed with noradrenaline and phenylephrine is explained best by the existence of crosstalk between α1-adrenoceptors and TP receptors. A modulatory interaction between the two receptors is supported by the observations that the intensity of the desensitization depends on the concentration of the α1-adrenoceptor agonist used; and that the curtailment was not observed with the more powerful TP receptor agonist U46619, except after partial occupancy of TP receptors with S18886. Hence, desensitization by the pre-exposure to α1-adrenoceptor activation expresses itself when TP receptor stimulation is less efficacious, as seen with other modulating influences (Flavahan et al., 1985; Baretella et al., 2014).
Selective activation of α1-adrenoceptors causes activation of PLC (Wu et al., 1992), which, in turn, activates PKC, an enzyme phosphorylating other proteins to modify cellular activity (Yukawa et al., 1997; Storz et al., 2004; Dorn and Force, 2005; Xiao and Liu, 2013)]. Incubation with calphostin C, a non-selective PKC inhibitor (Inoue et al., 2011) before phenylephrine pre-exposure abolished the curtailment, as did previous exposure to PDBu (activator of PKC; Hypolite et al., 2013). PDBu can down-regulate PKC (Franchi-Gazzola et al., 1996) and may reduce the efficacy of PGE2 rather than mimic phenylephrine desensitization. However, the present results with the selective PKC-ε inhibitor ε-V1-2 (Lee et al., 1999) make the latter interpretation unlikely but suggest that PKC activation contributes to the TP receptor desensitization in the SHR aorta. Transfection studies on cell cultures also suggest that PKC mediates modifications of TP receptors causing their desensitization (Spurney and Coffman, 1997; Yukawa et al., 1997; Walsh and Kinsella, 2000; Yan et al., 2002; Kelley-Hickie and Kinsella, 2004). The present findings indicate that the kinase indeed contributes to TP receptor desensitization in aortae isolated from live animals and that such heterologous desensitization results from crosstalk with α1-adrenoceptors.
Fifteen human PKC isozymes have been identified (Webb et al., 2000), which have been further subdivided into three subfamilies (conventional, novel, and atypical) based on second messenger requirements (Webb et al., 2000). Different expressions of PKC isoforms between SHR and WKY smooth muscle may explain TP receptor desensitization caused by α1-adrenoceptor activation in the former (Magnusson et al., 1998; Castellano and Santos, 2011; Dalton et al., 2013). The PKC expression was comparable in aortae of 5-week-old WKY and pre-hypertensive SHR (Sankhanava Kundu, 2008), in line with the observation that previous activation of α1-adrenoceptors did not cause desensitization at that young age in the latter. The two bands identified by Western blotting in both SHR and WKY aortae suggest the presence of at least two isozymes. The variation in relative intensity of these two molecular weight bands in 36 weeks WKY and SHR implies the presence of mixtures of isoforms in their aortic smooth muscle. Indeed, several of the PKC isoforms have similar molecular weights (Ohanian et al., 1996; Shin et al., 2000; Sampson et al., 2007). The present results thus support the interpretation that variations in PKC isozymes underlie the desensitization of TP receptors observed in the SHR aorta. A similar conclusion was reached in TP receptor desensitization studies on cell cultures (Spurney and Coffman, 1997; Yukawa et al., 1997; Spurney, 1998; Walsh and Kinsella, 2000; Kelley-Hickie and Kinsella, 2004).
The above conclusion is also compatible with the present qRT-PCR results demonstrating differences in genomic expression of PKC isoforms between 36 weeks WKY and SHR aortae. Only conventional (α, β and γ) and novel (δ, δ1, δ2, δ3, ε, η and θ) but not atypical (PK-N1, PK-N2, PK-N3, PKC-ι and PKC-ζ) PKC isoforms can be activated through the same signal transduction pathway as PLC (Breitkreutz et al., 2007). Pre-incubation with PDBu mimicked the desensitization exerted by phenylephrine, an effect that can be attributed to activation of PKC (Way et al., 2000). Since atypical PKCs cannot be activated by phorbol esters or inhibited by calphostin C (Kobayashi et al., 1989; Bruns et al., 1991; Boehm et al., 1996), the increases in PK-N2 and PKC-ζ mRNA expression observed in the present study are not likely to be responsible for TP receptor desensitization. As regards conventional PKCs, the mRNA expression of PKC-α is higher in SHR than WKY aorta, as it is in the heart where it may be related to hypertrophy (Dorn and Force, 2005). The expression of PKC-ε, a novel PKC isoform mediating cardioprotection (Ooie et al., 2003), was greater in SHR than in WKY aortae. This may seem counter-intuitive because the former strain is the one exhibiting cardiovascular dysfunction. In fact, the chronic activation of PKC-ε causes malignant tumors and diabetes (Akita, 2002). Thus, at the early stage of hypertension in the SHR, the elevated BP may trigger an increased PKC-ε expression as a protective response in the vascular system.
Based on the above analysis, PKC-α and PKC-ε were chosen as the likely candidates involved in TP receptor desensitization in the SHR. Indeed, both PKC-α and PKC-ε can be stimulated by α1-adrenoceptor activation (Goldberg et al., 1997; Taguchi et al., 1998). The selective inhibitor of PKC-α, Go6976, although it augmented the contractions to PGE2 (a phenomenon for which the present experiments provide no explanation), did not affect desensitization caused by α1-adrenoceptor activation. By contrast, the selective inhibitor of PKC-ε, ε-V1-2, restored the contractions without affecting control preparations. Thus, PKC-ε is likely to be the isoform activated by α1-adrenoceptors contributing to the heterologous desensitization of TP receptors in SHR aortae.
GRKs and arrestins are key components of the desensitization of GPCRs (Kohout and Lefkowitz, 2003). GRKs phosphorylate the latter and increase the high affinity binding of arrestins to the receptors after their phosphorylation, which prevents G protein coupling and interrupts signalling, thus achieving desensitization (Reiter and Lefkowitz, 2006). The present results show that, of the seven isoforms tested, the mRNA expressions of GRK2 and GRK4 are increased in SHR smooth muscle. In the mouse, reduced expression of GRK2 improves bioavailability of NO and protects against angiotensin II-induced hypertension (Avendano et al., 2014). GRK4 has been linked to both genetic and acquired hypertension (Vandell et al., 2012; Yatabe et al., 2012; Chen et al., 2014). The present experiments demonstrate that β-arrestin 2, the only isoform of arrestin expressed in rat aortae, has a higher expression level in SHR preparations, suggesting that receptor desensitization may be more sensitive in hypertensive animals. Thus, the present higher expressions of GRKs and β-arrestin 2 observed in the SHR preparations not only are consistent with PKC-dependent desensitization but may also indicate a more generalized disorder of receptor function contributing to the hypertensive process.
In summary, the present study reveals a crosstalk between α1-adrenoceptors and TP receptors in vascular smooth muscle of hypertensive but not of normotensive rats. This crosstalk was not observed in preparations of pre-hypertensive SHRs, was amplified by chronic exposure to high BP and was mediated by PKC-ε. Τhe resulting heterologous desensitization observed in preparations of adult hypertensive animals is likely due to the over-expressions of PKC-εisoform and β-arrestin 2. This expression increase may result in a protective response in the vasculature of hypertensive animals. Crosstalk between TP, IP, EP, DP and FP receptors in transfected cells has been studied by others (Walsh and Kinsella, 2000; Foley et al., 2001; Kelley-Hickie and Kinsella, 2004). The present study reports a crosstalk between non-prostanoid and prostanoid receptors that occurs ex vivo and is modulated by disease. The observed unequal expression of PKC isoforms, together with the over-expression of certain GRKs and β-arrestin 2, explains the desensitization of TP receptors occurring in hypertensive animals. It indicates a more generalized disorder of receptor function, which contributes to the hypertensive process in vascular smooth muscle cells.
Acknowledgments
The work described in this study was supported in part by RGC GRF Incentive Award and the Small Project Funding of the University of Hong Kong Research Committee.
Glossary
- PDBu
phorbol 12,13-dibutyrate
- SHR
spontaneously hypertensive rat
Author contributions
Yingzi Zhao, Paul M. Vanhoutte and Susan W.S. Leung participated in research design. Yingzi Zhao conducted experiments and performed data analysis. Yingzi Zhao and Paul M. Vanhoutte wrote the manuscript. Susan W.S. Leung contributed to the writing of the manuscript. All authors have read and approved this manuscript.
Conflict of interest
None.
References
- Akita Y. Protein kinase C-epsilon (PKC-epsilon): its unique structure and function. J Biochem. 2002;132:847–852. doi: 10.1093/oxfordjournals.jbchem.a003296. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Enzymes. Br J Pharmacol. 2013a;170:1797–1867. doi: 10.1111/bph.12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: G protein-coupled receptors. Br J Pharmacol. 2013b;170:1459–1581. doi: 10.1111/bph.12445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avendano MS, Lucas E, Jurado-Pueyo M, Martinez-Revelles S, Vila-Bedmar R, Mayor F, Jr, et al. Increased nitric oxide bioavailability in adult GRK2 hemizygous mice protects against angiotensin II-induced hypertension. Hypertension. 2014;63:369–375. doi: 10.1161/HYPERTENSIONAHA.113.01991. [DOI] [PubMed] [Google Scholar]
- Baretella O, Xu A, Vanhoutte PM. Acidosis prevents and alkalosis augments endothelium-dependent contractions in mouse arteries. Pflugers Arch. 2014;466:295–305. doi: 10.1007/s00424-013-1323-z. [DOI] [PubMed] [Google Scholar]
- Boehm S, Huck S, Freissmuth M. Involvement of a phorbol ester-insensitive protein kinase C in the alpha2-adrenergic inhibition of voltage-gated calcium current in chick sympathetic neurons. J Neurosci. 1996;16:4596–4603. doi: 10.1523/JNEUROSCI.16-15-04596.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breitkreutz D, Braiman-Wiksman L, Daum N, Denning MF, Tennenbaum T. Protein kinase C family: on the crossroads of cell signaling in skin and tumor epithelium. J Cancer Res Clin Oncol. 2007;133:793–808. doi: 10.1007/s00432-007-0280-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruns RF, Miller FD, Merriman RL, Howbert JJ, Heath WF, Kobayashi E, et al. Inhibition of protein kinase C by calphostin C is light-dependent. Biochem Biophys Res Commun. 1991;176:288–293. doi: 10.1016/0006-291x(91)90922-t. [DOI] [PubMed] [Google Scholar]
- Castellano E, Santos E. Functional specificity of ras isoforms: so similar but so different. Genes Cancer. 2011;2:216–231. doi: 10.1177/1947601911408081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K, Fu C, Chen C, Liu L, Ren H, Han Y, et al. Role of GRK4 in the regulation of arterial AT1 receptor in hypertension. Hypertension. 2014;63:289–296. doi: 10.1161/HYPERTENSIONAHA.113.01766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Civantos Calzada B, Aleixandre de Artinano A. Alpha-adrenoceptor subtypes. Pharmacol Res. 2001;44:195–208. doi: 10.1006/phrs.2001.0857. [DOI] [PubMed] [Google Scholar]
- Cui ZJ, Guo LL. Assessing physiological concentrations of endogenous substances in situ by inducing calcium oscillations in vitro. Case of liver. Acta Pharmacol Sin. 2002;23:27–32. [PubMed] [Google Scholar]
- Dalton JE, Fear JM, Knott S, Baker BS, McIntyre LM, Arbeitman MN. Male-specific fruitless isoforms have different regulatory roles conferred by distinct zinc finger DNA binding domains. BMC Genomics. 2013;14:659. doi: 10.1186/1471-2164-14-659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di GQ, Zhou B, Li ZG, Lin QL. Aircraft noise exposure affects rat behavior, plasma norepinephrine levels, and cell morphology of the temporal lobe. J Zhejiang Univ Sci B. 2011;12:969–975. doi: 10.1631/jzus.B1000439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorn GW, 2nd, Force T. Protein kinase cascades in the regulation of cardiac hypertrophy. J Clin Invest. 2005;115:527–537. doi: 10.1172/JCI24178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ely D, Caplea A, Dunphy G, Daneshvar H, Turner M, Milsted A, et al. Spontaneously hypertensive rat Y chromosome increases indexes of sympathetic nervous system activity. Hypertension. 1997;29:613–618. doi: 10.1161/01.hyp.29.2.613. [DOI] [PubMed] [Google Scholar]
- Feletou M, Huang Y, Vanhoutte PM. Vasoconstrictor prostanoids. Pflugers Arch. 2010a;459:941–950. doi: 10.1007/s00424-010-0812-6. [DOI] [PubMed] [Google Scholar]
- Feletou M, Kohler R, Vanhoutte PM. Endothelium-derived vasoactive factors and hypertension: possible roles in pathogenesis and as treatment targets. Curr Hypertens Rep. 2010b;12:267–275. doi: 10.1007/s11906-010-0118-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feletou M, Vanhoutte PM, Verbeuren TJ. The thromboxane/endoperoxide receptor (TP): the common villain. J Cardiovasc Pharmacol. 2010c;55:317–332. doi: 10.1097/fjc.0b013e3181d8bc8a. [DOI] [PubMed] [Google Scholar]
- Flavahan NA, Lindblad LE, Verbeuren TJ, Shepherd JT, Vanhoutte PM. Cooling and alpha 1- and alpha 2-adrenergic responses in cutaneous veins: role of receptor reserve. Am J Physiol. 1985;249:H950–H955. doi: 10.1152/ajpheart.1985.249.5.H950. [DOI] [PubMed] [Google Scholar]
- Foley JF, Kelley LP, Kinsella BT. Prostaglandin D(2) receptor-mediated desensitization of the alpha isoform of the human thromboxane A(2) receptor. Biochem Pharmacol. 2001;62:229–239. doi: 10.1016/s0006-2952(01)00661-x. [DOI] [PubMed] [Google Scholar]
- Foudi N, Gomez I, Benyahia C, Longrois D, Norel X. Prostaglandin E2 receptor subtypes in human blood and vascular cells. Eur J Pharmacol. 2012;695:1–6. doi: 10.1016/j.ejphar.2012.08.009. [DOI] [PubMed] [Google Scholar]
- Franchi-Gazzola R, Visigalli R, Bussolati O, Gazzola GC. Involvement of protein kinase C∊ in the stimulation of anionic amino acid transport in cultured human fibroblasts. J Biol Chem. 1996;271:26124–26130. doi: 10.1074/jbc.271.42.26124. [DOI] [PubMed] [Google Scholar]
- Furchgott RF, Zawadzki JV. The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature. 1980;288:373–376. doi: 10.1038/288373a0. [DOI] [PubMed] [Google Scholar]
- Ge T, Hughes H, Junquero DC, Wu KK, Vanhoutte PM, Boulanger CM. Endothelium-dependent contractions are associated with both augmented expression of prostaglandin H synthase-1 and hypersensitivity to prostaglandin H2 in the SHR aorta. Circ Res. 1995;76:1003–1010. doi: 10.1161/01.res.76.6.1003. [DOI] [PubMed] [Google Scholar]
- Giannarelli C, Zafar MU, Badimon JJ. Prostanoid and TP-receptors in atherothrombosis: is there a role for their antagonism? Thromb Haemost. 2010;104:949–954. doi: 10.1160/TH10-03-0195. [DOI] [PubMed] [Google Scholar]
- Gildea JJ, Tran HT, Van Sciver RE, Bigler Wang D, Carlson JM, Felder RA. A novel role for c-Myc in G protein-coupled receptor kinase 4 (GRK4) transcriptional regulation in human kidney proximal tubule cells. Hypertension. 2013;61:1021–1027. doi: 10.1161/HYPERTENSIONAHA.111.00321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gluais P, Lonchampt M, Morrow JD, Vanhoutte PM, Feletou M. Acetylcholine-induced endothelium-dependent contractions in the SHR aorta: the Janus face of prostacyclin. Br J Pharmacol. 2005;146:834–845. doi: 10.1038/sj.bjp.0706390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godfraind T, Miller RC, Lima JS. Selective alpha 1- and alpha 2-adrenoceptor agonist-induced contractions and 45Ca fluxes in the rat isolated aorta. Br J Pharmacol. 1982;77:597–604. doi: 10.1111/j.1476-5381.1982.tb09337.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godínez-Hernández D, Gallardo-Ortíz I, López-Sánchez P, Villalobos-Molina R. Captopril therapy decreases both expression and function of α1D-adrenoceptors in pre-hypertensive rat aorta. Auton Autacoid Pharmacol. 2006;26:21–29. doi: 10.1111/j.1474-8673.2005.00358.x. [DOI] [PubMed] [Google Scholar]
- Goldberg M, Zhang HL, Steinberg SF. Hypoxia alters the subcellular distribution of protein kinase C isoforms in neonatal rat ventricular myocytes. J Clin Invest. 1997;99:55. doi: 10.1172/JCI119133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurevich EV, Tesmer JJ, Mushegian A, Gurevich VV. G protein-coupled receptor kinases: more than just kinases and not only for GPCRs. Pharmacol Ther. 2012;133:40–69. doi: 10.1016/j.pharmthera.2011.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Head R, Cassis L, Robinson R, Westfall D, Stitzel R. Altered catecholamine contents in vascular and nonvascular tissues in genetically hypertensive rats. J Vasc Res. 1985;22:196–204. doi: 10.1159/000158601. [DOI] [PubMed] [Google Scholar]
- Hypolite JA, Lei Q, Chang S, Zderic SA, Butler S, Wein AJ, et al. Spontaneous and evoked contractions are regulated by PKC-mediated signaling in detrusor smooth muscle: involvement of BK channels. Am J Physiol Renal Physiol. 2013;304:F451–F462. doi: 10.1152/ajprenal.00639.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue T, Kobayashi K, Inoguchi T, Sonoda N, Fujii M, Maeda Y, et al. Reduced expression of adipose triglyceride lipase enhances tumor necrosis factor alpha-induced intercellular adhesion molecule-1 expression in human aortic endothelial cells via protein kinase C-dependent activation of nuclear factor-kappaB. J Biol Chem. 2011;286:32045–32053. doi: 10.1074/jbc.M111.285650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. Animal research: Reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley-Hickie LP, Kinsella BT. EP1- and FP-mediated cross-desensitization of the alpha (alpha) and beta (beta) isoforms of the human thromboxane A2 receptor. Br J Pharmacol. 2004;142:203–221. doi: 10.1038/sj.bjp.0705695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi E, Ando K, Nakano H, Iida T, Ohno H, Morimoto M, et al. Calphostins (UCN-1028), novel and specific inhibitors of protein kinase CI fermentation, isolation, physico-chemical properties and biological activities. J Antibiot. 1989;42:1470–1474. doi: 10.7164/antibiotics.42.1470. [DOI] [PubMed] [Google Scholar]
- Kohout TA, Lefkowitz RJ. Regulation of G protein-coupled receptor kinases and arrestins during receptor desensitization. Mol Pharmacol. 2003;63:9–18. doi: 10.1124/mol.63.1.9. [DOI] [PubMed] [Google Scholar]
- Lee YH, Kim I, Laporte R, Walsh MP, Morgan KG. Isozyme-specific inhibitors of protein kinase C translocation: effects on contractility of single permeabilized vascular muscle cells of the ferret. J Physiol. 1999;517(Pt 3):709–720. doi: 10.1111/j.1469-7793.1999.0709s.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Li W, Liu W, Altura BT, Altura BM. Mechanisms of hydroxyl radical-induced contraction of rat aorta. Eur J Pharmacol. 2004;499:171–178. doi: 10.1016/j.ejphar.2004.07.094. [DOI] [PubMed] [Google Scholar]
- Lorenz K, Lohse MJ, Quitterer U. Protein kinase C switches the Raf kinase inhibitor from Raf-1 to GRK-2. Nature. 2003;426:574–579. doi: 10.1038/nature02158. [DOI] [PubMed] [Google Scholar]
- Lüscher T, Vanhoutte PM. Endothelium-dependent contractions to acetylcholine in the aorta of the spontaneously hypertensive rat. Hypertension. 1986;8:344–348. doi: 10.1161/01.hyp.8.4.344. [DOI] [PubMed] [Google Scholar]
- Magnusson P, Larsson L, Englund G, Larsson B, Strang P, Selin-Sjogren L. Differences of bone alkaline phosphatase isoforms in metastatic bone disease and discrepant effects of clodronate on different skeletal sites indicated by the location of pain. Clin Chem. 1998;44:1621–1628. [PubMed] [Google Scholar]
- Mangiarua EI, Lee RM. Increased sympathetic innervation in the cerebral and mesenteric arteries of hypertensive rats. Can J Physiol Pharm. 1990;68:492–499. doi: 10.1139/y90-070. [DOI] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohanian V, Ohanian J, Shaw L, Scarth S, Parker PJ, Heagerty AM. Identification of protein kinase C isoforms in rat mesenteric small arteries and their possible role in agonist-induced contraction. Circ Res. 1996;78:806–812. doi: 10.1161/01.res.78.5.806. [DOI] [PubMed] [Google Scholar]
- Ooie T, Takahashi N, Nawata T, Arikawa M, Yamanaka K, Kajimoto M, et al. Ischemia-induced translocation of protein kinase C-epsilon mediates cardioprotection in the streptozotocin-induced diabetic rat. Circ J. 2003;67:955–961. doi: 10.1253/circj.67.955. [DOI] [PubMed] [Google Scholar]
- Papapetropoulos A, Cziraki A, Rubin JW, Stone CD, Catravas JD. cGMP accumulation and gene expression of soluble guanylate cyclase in human vascular tissue. J Cell Physiol. 1996;167:213–221. doi: 10.1002/(SICI)1097-4652(199605)167:2<213::AID-JCP4>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, et al. NC-IUPHAR. The IUPHAR/BPS Guide to PHARMACOLOGY: an expert-driven knowledge base of drug targets and their ligands. Nucl Acids Res. 2014;42(Database Issue):D1098–D1106. doi: 10.1093/nar/gkt1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pronin AN, Benovic JL. Regulation of the G protein-coupled receptor kinase GRK5 by protein kinase C. J Biol Chem. 1997;272:3806–3812. doi: 10.1074/jbc.272.6.3806. [DOI] [PubMed] [Google Scholar]
- Reiter E, Lefkowitz RJ. GRKs and beta-arrestins: roles in receptor silencing, trafficking and signaling. Trends Endocrinol Metab. 2006;17:159–165. doi: 10.1016/j.tem.2006.03.008. [DOI] [PubMed] [Google Scholar]
- Sampson LJ, Davies LM, Barrett-Jolley R, Standen NB, Dart C. Angiotensin II-activated protein kinase C targets caveolae to inhibit aortic ATP-sensitive potassium channels. Cardiovasc Res. 2007;76:61–70. doi: 10.1016/j.cardiores.2007.05.020. [DOI] [PubMed] [Google Scholar]
- Sankhanava Kundu JPR. The story of spontaneously hypertensive rat (SHR): a review. Al Ameen J Med Sci. 2008;1:65–66. [Google Scholar]
- Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative CT method. Nat Protoc. 2008;3:1101–1108. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- Scott T, Galway G. The relationship between altered blood vessel structure, hypertension, and the sympathetic nervous system. Can J Physiol Pharm. 1985;63:387–391. doi: 10.1139/y85-069. [DOI] [PubMed] [Google Scholar]
- Shin HG, Barnett JV, Chang P, Reddy S, Drinkwater DC, Pierson RN, et al. Molecular heterogeneity of protein kinase C expression in human ventricle. Cardiovasc Res. 2000;48:285–299. doi: 10.1016/s0008-6363(00)00185-1. [DOI] [PubMed] [Google Scholar]
- Spurney RF. Role of C-terminal serines in desensitization and phosphorylation of the mouse thromboxane receptor. J Biol Chem. 1998;273:28496–28503. doi: 10.1074/jbc.273.43.28496. [DOI] [PubMed] [Google Scholar]
- Spurney RF, Coffman TM. The C-terminus of the thromboxane receptor contributes to coupling and desensitization in a mouse mesangial cell line. J Pharmacol Exp Ther. 1997;283:207–215. [PubMed] [Google Scholar]
- Storz P, Doppler H, Toker A. Protein kinase Cdelta selectively regulates protein kinase D-dependent activation of NF-kappaB in oxidative stress signaling. Mol Cell Biol. 2004;24:2614–2626. doi: 10.1128/MCB.24.7.2614-2626.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki J, Ogawa M, Watanabe R, Takayama K, Hirata Y, Nagai R, et al. Roles of prostaglandin E2 in cardiovascular diseases. Int Heart J. 2011;52:266–269. doi: 10.1536/ihj.52.266. [DOI] [PubMed] [Google Scholar]
- Taguchi K, Yang M, Goepel M, Michel MC. Comparison of human α1-adrenoceptor subtype coupling to protein kinase C activation and related signalling pathways. Naunyn Schmiedebergs Arch Pharmacol. 1998;357:100–110. doi: 10.1007/pl00005143. [DOI] [PubMed] [Google Scholar]
- Tang EH, Vanhoutte PM. Gene expression changes of prostanoid synthases in endothelial cells and prostanoid receptors in vascular smooth muscle cells caused by aging and hypertension. Physiol Genomics. 2008;32:409–418. doi: 10.1152/physiolgenomics.00136.2007. [DOI] [PubMed] [Google Scholar]
- Tang EH, Feletou M, Huang Y, Man RY, Vanhoutte PM. Acetylcholine and sodium nitroprusside cause long-term inhibition of EDCF-mediated contractions. Am J Physiol Heart Circ Physiol. 2005;289:H2434–H2440. doi: 10.1152/ajpheart.00568.2005. [DOI] [PubMed] [Google Scholar]
- Tang EH, Jensen BL, Skott O, Leung GP, Feletou M, Man RY, et al. The role of prostaglandin E and thromboxane-prostanoid receptors in the response to prostaglandin E2 in the aorta of Wistar Kyoto rats and spontaneously hypertensive rats. Cardiovasc Res. 2008;78:130–138. doi: 10.1093/cvr/cvm112. [DOI] [PubMed] [Google Scholar]
- Tanoue A, Nasa Y, Koshimizu T, Shinoura H, Oshikawa S, Kawai T, et al. The α 1D-adrenergic receptor directly regulates arterial blood pressure via vasoconstriction. J Clin Invest. 2002;109:765–775. doi: 10.1172/JCI14001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandell AG, Lobmeyer MT, Gawronski BE, Langaee TY, Gong Y, Gums JG, et al. G protein receptor kinase 4 polymorphisms: beta-blocker pharmacogenetics and treatment-related outcomes in hypertension. Hypertension. 2012;60:957–964. doi: 10.1161/HYPERTENSIONAHA.112.198721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanhoutte PM. Endothelium-dependent contractions in hypertension when prostacyclin becomes ugly. Hypertension. 2011;57:526–531. doi: 10.1161/HYPERTENSIONAHA.110.165100. [DOI] [PubMed] [Google Scholar]
- Walsh MT, Kinsella BT. Regulation of the human prostanoid TPalpha and TPbeta receptor isoforms mediated through activation of the EP(1) and IP receptors. Br J Pharmacol. 2000;131:601–609. doi: 10.1038/sj.bjp.0703624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Way KJ, Chou E, King GL. Identification of PKC-isoform-specific biological actions using pharmacological approaches. Trends Pharmacol Sci. 2000;21:181–187. doi: 10.1016/s0165-6147(00)01468-1. [DOI] [PubMed] [Google Scholar]
- Webb BL, Hirst SJ, Giembycz MA. Protein kinase C isoenzymes: a review of their structure, regulation and role in regulating airways smooth muscle tone and mitogenesis. Br J Pharmacol. 2000;130:1433–1452. doi: 10.1038/sj.bjp.0703452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu D, Katz A, Lee CH, Simon MI. Activation of phospholipase C by alpha 1-adrenergic receptors is mediated by the alpha subunits of Gq family. J Biol Chem. 1992;267:25798–25802. [PubMed] [Google Scholar]
- Xiao H, Liu M. Atypical protein kinase C in cell motility. Cell Mol Life Sci. 2013;70:3057–3066. doi: 10.1007/s00018-012-1192-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan FX, Yamamoto S, Zhou HP, Tai HH, Liao DF. Serine 331 is major site of phosphorylation and desensitization induced by protein kinase C in thromboxane receptor alpha. Acta Pharmacol Sin. 2002;23:952–960. [PubMed] [Google Scholar]
- Yatabe J, Yatabe MS, Yoneda M, Felder RA, Jose PA, Sanada H. Hypertension-Related Gene Polymorphisms of G-Protein-Coupled Receptor Kinase 4 Are Associated with NT-proBNP Concentration in Normotensive Healthy Adults. Int J Hypertens. 2012;2012:806810. doi: 10.1155/2012/806810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yukawa M, Yokota R, Eberhardt RT, von Andrian L, Ware JA. Differential desensitization of thromboxane A2 receptor subtypes. Circ Res. 1997;80:551–556. doi: 10.1161/01.res.80.4.551. [DOI] [PubMed] [Google Scholar]