

Significance
The significance of the results reported is in two areas. (i) Because the cannabinoid receptor type 2 (CB2) agonists seem to be general protective agents, HU-433, a new specific CB2 agonist, may be of major therapeutic importance. (ii) Enantiomers usually have different activity profiles. We report now that HU-433 and its enantiomer HU-308 are both specific CB2 agonists, but whereas HU-433 is much more potent than HU-308 in the rescue of ovariectomy-induced bone loss and ear inflammation, its binding to the CB2 receptor (through which the activity of both enantiomers takes place) is substantially lower compared with HU-308. This situation questions the usefulness of universal radioligands for comparative binding studies.
Keywords: endocannabinoids, osteoporosis, enantiomers, pose occupancy
Abstract
Activation of the CB2 receptor is apparently an endogenous protective mechanism. Thus, it restrains inflammation and protects the skeleton against age-related bone loss. However, the endogenous cannabinoids, as well as Δ9-tetrahydrocannabinol, the main plant psychoactive constituent, activate both cannabinoid receptors, CB1 and CB2. HU-308 was among the first synthetic, selective CB2 agonists. HU-308 is antiosteoporotic and antiinflammatory. Here we show that the HU-308 enantiomer, designated HU-433, is 3–4 orders of magnitude more potent in osteoblast proliferation and osteoclast differentiation culture systems, as well as in mouse models, for the rescue of ovariectomy-induced bone loss and ear inflammation. HU-433 retains the HU-308 specificity for CB2, as shown by its failure to bind to the CB1 cannabinoid receptor, and has no activity in CB2-deficient cells and animals. Surprisingly, the CB2 binding affinity of HU-433 in terms of [3H]CP55,940 displacement and its effect on [35S]GTPγS accumulation is substantially lower compared with HU-308. A molecular-modeling analysis suggests that HU-433 and -308 have two different binding conformations within CB2, with one of them possibly responsible for the affinity difference, involving [35S]GTPγS and cAMP synthesis. Hence, different ligands may have different orientations relative to the same binding site. This situation questions the usefulness of universal radioligands for comparative binding studies. Moreover, orientation-targeted ligands have promising potential for the pharmacological activation of distinct processes.
The CB2 cannabinoid receptor functions as an endogenous protective entity (1). Thus, it is an important regulator of bone mass and inflammation. It represents a therapeutic target that avoids the undesired psychotropic effects caused by CB1 receptor activation. CB2 is expressed in osteoblasts, the bone-forming cells, and in osteoclasts, the bone-resorbing cells (2). Monocytes/macrophages, B cells, certain T-cell subtypes, and mast cells also express CB2 (3–7). In the skeleton, activation of CB2 favors bone formation over resorption, thus protecting the skeleton against age-related bone loss. Inflammatory responses are restrained by CB2 agonists in several instances such as hepatic ischemia-reperfusion injury (8), uveitis (9), and contact dermatitis (10). With their terpene and resorcinol-derived moieties, some synthetic CB2 agonists, such as HU-308 (10), JWH-133 (11), and HU-910 (12), structurally resemble the phytocannabinoids Δ9-tetrahydrocannabinol and cannabidiol. Other, non-phytocannabinoid-type agonists have been also reported (13). HU-308 was one of the first fully characterized, highly selective, and highly efficacious cannabinoid type-2 agonist (10). It has three chiral centers, namely, at carbon atoms in positions 3, 4, and 6 (Scheme 1). HU-308 has a 3R, 4S, 6S configuration; that of its enantiomer (HU-433) is 3S, 4R, 6R.
Scheme 1.

Structures of HU-433 and -308.
In most experiments, the optimal HU-308 mitogenic activity in osteoblasts, as well as its antiosteoclastogenic effect, was obtained by using concentrations in the nanomolar range (2, 14). Surprisingly, testing a new HU-308 batch, we noticed a 3- to 4-magnitude decrease in the dose that triggers optimal proliferative response in osteoblasts, reasoning that this preparation contained a significant amount of the HU-308 enantiomer, presumably responsible for the enhanced activity. We report here that this enantiomer, designated HU-433, was synthesized and compared with HU-308, and indeed showed markedly enhanced bone-anabolic and antiinflammatory activities, but inferior CB2 receptor binding affinity. Although both enantiomers seem to target the same binding pocket, two different orientations relative to the binding site are possible, leading to different behavior of the two enantiomers due to their different occupancy of these orientations. This observation appears to reflect a situation in which relatively small differences in possible binding conformations of the ligands within the receptor—referred to as poses—lead to significant diminution of receptor-binding properties and a marked increase in biological activity.
Results and Discussion
Selective agonists of enhanced activity and reduced affinity are of interest and importance because they may contribute to the biochemical and pharmacological investigation of individual receptors, as well as serve as drug leads with shorter ligand–receptor interaction and thus more controllable manipulations of the pharmacological action. As a rule, enantiomers do not exhibit the same binding orientation or even bind to the same site. Indeed, one enantiomer of a chiral drug may have a desired beneficial effect, whereas the other may be inactive or cause entirely different effects (15). Hence, it is not entirely surprising that HU-433 and -308 have different potencies. What is surprising is that this difference is inversely related to their binding affinity.
HU-433 was synthesized following essentially the same synthetic route reported for HU-308 (10), except that the starting material for the preparation of HU-308 was (+)-α-pinene, which was converted to (1S)-(+)-myrtenol, whereas the synthesis of HU-433 started directly from (1R)-(−)-myrtenol (Scheme 2). The optical rotations of HU-308 and -433 are [α]D = +124° and [α]D = −120°, respectively. In mouse models, the antiosteoporotic and antiinflammatory activity of HU-433 is 1,000- to 10,000-fold higher than that of HU-308 (see below). However, the affinity that HU-433 displays for the human CB2 (hCB2) receptor is 25.7-fold less than the hCB2 receptor affinity displayed by HU-308, a difference that is statistically significant, as indicated by the lack of overlap of the 95% confidence limits of the hCB2 mean Ki values of these two compounds (Fig. 1A). HU-433 also appeared to be less potent and efficacious than HU-308 at activating the hCB2 receptor in the [35S]GTPγS binding assay (Fig. 1B). However, neither the mean EC50 nor the mean Emax that it displayed in this assay was significantly different from the corresponding EC50 and Emax values of HU-308. Importantly, like HU-308 (10), HU-433 does not bind to CHO cell membranes expressing CB1 (Ki > 10,000 nM). Furthermore, at least in ex vivo osteoblast and in vivo inflammatory models, genetic or pharmacological ablation of CB2 completely blocks the HU-433 activity (Figs. 2 A and D and 3B).
Scheme 2.

Synthesis of HU-433. Reagents and conditions were as follows: (a) (1R)-(-)-myrtenol 95%, pivaloyl chloride, pyridine, 0 °C, then overnight at room temperature (r.t.), 91% isolated. (b) CrO3, CH3CN, 0 °C, then t-BuOOH, 1 h at r.t., 25% isolated. (c) EtOH, NaBH4, 4 h, 91% isolated. (d) CH2Cl2, dry p-TSA, 5-(1,1-dimethylheptyl)resorcinol,1.5 h. at r.t., 43% isolated. (e) K2CO3, DMF, CH3I, overnight at r.t. 67% yield. (f) Et2O, LiAlH4, reflux, 80% isolated.
Fig. 1.

(A) Displacement of [3H]CP55940 by HU-308 and -433 from specific binding sites in membranes obtained from CHO cells transfected with hCB2 receptors. Each symbol represents the mean percent displacement ± SE. Mean Ki values for this displacement with 95% confidence limits shown in parentheses were calculated by the Cheng–Prusoff equation to be 11.5 nM (9.47 and 14.0 nM; n = 6) for HU-308 and 296 nM (242 and 362 nM; n = 6) for HU-433. (B) Mean log concentration–response curves of HU-308, -433, and CP55940 for stimulation of [35S]GTPγS binding to hCB2 CHO cell membranes. Each symbol represents the mean percentage increase in [35S]GTPγS binding ± SE. Mean EC50 values for the production of these increases by HU-308, -433 and CP55940 with their 95% confidence limits shown in parentheses are 6.4 nM (1.8 and 23.3 nM; n = 6), 130 nM (12.2 and 1,386 nM; n = 5), and 3.0 nM (0.24 and 37.6 nM; n = 4), respectively. The corresponding mean Emax values (maximal effects) of these three compounds, with their 95% confidence limits shown in parentheses, are 27.7% (22.0% and 33.5%), 15.6% (8.6% and 22.5%), and 36.8% (24.2% and 49.3%), respectively.
Fig. 2.

Mitogenic signaling of HU-433 in osteoblasts involves CB2 and its downstream signaling pathway. (A and B) Number of wild-type (WT) (♦) and CB2−/− (♢) newborn mouse calvarial osteoblasts by HU-433 (A) and HU-308 (B). (C) DNA synthesis. (D and E) inhibition of HU-433 (D) and HU-308 (E) stimulated cell number by CB2 antagonist SR144528 (SR2). (F and G) Inhibition of HU-433 stimulated cell number by pertussis toxin (PTX) (F) and MEK/Erk1,2 inhibitor PD98059 (G). (H) Stimulation of Mapkapk2 mRNA levels by HU-433. Data are mean ± SE obtained in triplicate culture wells per condition. *P < 0.05 vs. Cntl (control).
Fig. 3.

HU-433 attenuates xylene-induced ear swelling in WT, but not CB2-deficient, mice. (A) WT mice. (B) CB2−/− mice. ■, VEH; ◆, HU-433; ▲, Indomethacin. Data are mean ± SE obtained in six mice per condition. *P < 0.05 vs. VEH-treated mice.
Both HU-433 and -308 stimulate osteoblast proliferation, exhibiting a dose–response biphasic effect. However, the peak effect of HU-433 is at 10−12 M concentration (Fig. 2 A and C), whereas that of HU-308 is at 10−9 M (Fig. 2B) [sometimes 10−8 M (2)], presenting a 103- to 104-fold higher potency for HU-433. The activity of either enantiomer is completely absent in CB2−/− osteoblasts (Fig. 2 A and B). Also, the selective CB2 receptor antagonist SR144528, although having no effect by itself, similarly blocks the HU-433 and -308 proliferative activity (Fig. 2 D and E). These data indicate that, at least in osteoblasts, the HU-433 activity, like that of HU-308, is critically CB2-dependent.
We recently reported a downstream Gi-protein–Erk1,2–Mapkapk2–CREB–cyclin D1 signaling cascade through CB2 (14). To assess whether HU-433 activates the same pathway, we tested the effect of the respective Gi-protein and MEK/Erk1,2 blockers pertussis toxin (PTX) and PD98059 (PD) on HU-433 proliferative activity. As in the case of HU-308 (14), either inhibitor completely blocks the HU-433 effect on osteoblasts (Fig. 2 F and G). Like HU-308, HU-433 also stimulates Mapkapk2 expression (Fig. 2H). Together, these findings confirm that HU-433 not only selectively targets CB2, but also elicits at least some of the same biological responses as HU-308, although with a vastly enhanced potency.
HU-308 mitigates ex vivo osteoclastogenesis dose-dependently with a peak effect of ∼40% at 10−8 M (2). This mitigation occurred consequent to reduced proliferation of osteoclast progenitors and to almost complete inhibition of receptor activated nuclear factor κB ligand (RANKL) expression in bone marrow-derived stromal cells (2). Here we show that in ex vivo osteoclastogenic cultures, incubated in the presence of macrophage colony-stimulating factor and RANKL, HU-433 reduces the number of osteoclasts identified as tartrate resistant acid phosphatase (TRAP)-positive multinucleated cells. Although the magnitude of this reduction is similar to that induced by HU-308 (2), it occurs at a 103-fold lower ligand concentration (Fig. 4 A, C, and D). We further show that HU-433 stimulates apoptosis of these cells, increasing the number of ghosts by ∼40% (Fig. 4 B–D). In an ex vivo assay for degradation of the mineralized extracellular matrix (16), the decrease in osteoclast formation translates into a reduced number of resorption pits (Fig. 4 E, G, and H). Pit area is also reduced (Fig. 4F). Osteoclast activity is cyclic, and the pit size is critically affected by the number of cycles (17). Hence, the present reduction in pit area is consistent with the increase in osteoclast apoptosis, which in turn results in shorter duration of the osteoclast activity and decreased number of resorption cycles. Thus, it seems that the two enantiomers restrain bone resorption via distinct mechanisms.
Fig. 4.

HU-433 inhibits number of intact osteoclasts. (A) Number of intact osteoclasts. (B) Number of apoptotic osteoclasts. (C) HU-433–free control culture stained with TRAP. (D) TRAP staining of cultures challenged with 10−11 M HU-433. (E–H) Pit formation in osteoclastogenic cultures of intact cells grown on dentin slices. (E) Number of pits per area. (F) Average pit area. (G and H) Toluidine blue staining of HU-433–free vehicle-treated control (G) and HU-433–treated cultures (H). Arrows, apoptotic osteoclasts. Cntl, HU-433-free vehicle-treated control. Quantitative data are mean ± SE obtained in four to six culture wells per condition. *P < 0.05 vs. cntl.
Ovariectomy (OVX)-induced bone loss is the most widely used animal model for osteoporosis. To assess rescue of OVX-induced bone loss by HU-433, animals were left untreated for 6 wk after OVX, followed by a 6-wk daily (5 d/wk) treatment with vehicle or 2, 20, or 200 µg/kg HU-433. Animals administered vehicle showed a ∼55% decrease in bone volume density (BV/TV) compared with sham-OVX HU-433–free controls. Because of the potential therapeutic implications of HU-433 as a bone anabolic agent, and because cannabinoid ligands often show biphasic effects (18), we tested three doses. Indeed, the effect of HU-433 on the trabecular bone microstructure is biphasic, reminiscent of its bell shaped dose-related activity in the osteoblast culture (Fig. 2). The 20 µg/kg dose completely reversed the OVX-induced decrease in BV/TV (Fig. 5 A and B) associated primarily with increased trabecular thickness (Fig. 5C), a higher trabecular number (Fig. 5D), and higher connectivity (Fig. 5E). The 2 and 200 µg/kg doses had no effect on the bone microstructural parameters (Fig. 5 B–D). However, all three doses similarly increased the level of the serum bone formation marker osteocalcin (Fig. 5F), but only the 20 and 200 µg/kg doses inhibited the serum bone resorption surrogate Trap5b (Fig. 5G). The incomplete correlation between the bone microstructural parameters and serum remodeling markers may be due to the continuous cumulative effect by HU-433 over the 6-wk treatment period, whereas changes in bone remodeling that we detected may reflect its status at around the time of treatment termination and blood sampling. By comparison, in the same mouse model for osteoporosis, HU-308 at 20 mg/kg per day reversed <50% of the bone loss (19).
Fig. 5.

HU-433 rescues bone loss in distal femoral metaphysis of OVX mice. (A) Representative images from femora with median bone volume density values. (B) Bone volume density (BV/TV). (C) Trabecular thickness (Tb.Th). (D) Trabecular number (Tb.N). (E) Connectivity density (Conn. D). (F) Serum osteocalcin. (G) Serum Trap5b. Data are mean ± SE obtained in 8–12 mice per condition. VEH, vehicle. *P < 0.05 vs. VEH.
Acute edema is one of the inflammatory responses restrained by CB2 activation (20). Therefore, to evaluate whether the markedly enhanced potency of HU-433 vs. HU-308 is shared by tissues other than bone, we tested HU-433 for antiinflammatory activity in an ear-swelling model, a commonly used test for assessing the antiinflammatory activity of cannabinoid receptor agonists (21). By using this model, it has been previously reported that a single 50 mg/kg dose of HU-308 causes a ∼50% inhibition of arachidonic acid-induced ear swelling (10, 22). In the same model, using topically administered xylene as irritant, we show that a single dose of 20 µg/kg HU-433 given before the xylene application inhibits swelling almost to the same extent (Fig. 3A), representing a 2,500-fold higher potency by HU-433 than by HU-308. This effect was completely prevented by genetic ablation of CB2 (Fig. 3B). Osteoblasts originate in the stromal cell system (23), whereas osteoclasts and mast cells are hematopoietic (24, 25). Therefore, the markedly enhanced inhibition of ear swelling, together with the vastly greater effects in osteoblast and osteoclast cultures and on the enhancement of bone mass, portray HU-433 as a highly efficacious and potent CB2 agonist.
We carried out molecular-modeling studies to provide a possible explanation for the discrepancy between the vastly enhanced potency of HU-433 vs. HU-308 and its reduced binding affinity to CB2, in terms of CP55,940 displacement. CB2 belongs to the rhodopsin-like family of seven transmembrane G-protein–coupled receptors (GPCRs) (26, 27). Therefore, the crystal structure of bovine rhodopsin (26) was used as a template to construct a homology model of CB2. We demonstrate that both enantiomers present similar binding poses, almost perpendicular to the receptor z axis, with the aliphatic chain turning deep into the binding pocket (Fig. 6A).
Fig. 6.
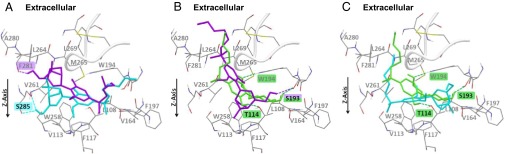
The binding pocket of CB2 with lowest energy poses of HU-308 (cyan) and HU-433 (purple) (A); lowest energy pose of CP 55,940 (green) along with an equivalent pose of HU-433 (purple) (B); and lowest energy pose of CP55,940 (green) along with equivalent pose of HU-308 (cyan) (C). Binding site orientation is identical in all images. Gray fonts represent residues forming hydrophobic interactions with ligands. Residues forming hydrogen bonds with ligands are highlighted by respective ligand color.
The hydrophobic lining of the CB2 binding site usually shares common interactions with both HU-308 and -433. That said, the bicyclic ring of HU-308 is buried deeper in transmembrane domains 3, 6, and 7 compared with the corresponding bicyclic ring of HU-433, which is closer to the extracellular aspect of the cell membrane (Fig. 6A). In this scenario, the HU-433 hydrophobic interactions are indeed weaker than those of HU-308. In addition, HU-433 seems to have a weaker H-bonding network than HU-308. In particular, the hydroxyl group of HU-433 forms only one H-bond with the backbone carbonyl oxygen of F281, whereas the corresponding hydroxyl group of HU-308 forms two H-bonds with both the backbone carbonyl and the hydroxyl group of S285 (Fig. 6A). This binding mode explains the lower affinity of HU-433 compared with that of HU-308.
CP55,940 is a nonselective cannabinoid receptor ligand often used as the radioactive competitor in CB2 binding studies (28). Contrary to the lowest binding pose of HU-433 and -308, the orientation of CP55,940 is almost parallel to the CB2 z axis, with the longest aliphatic chain pointing toward the extracellular aspect and the two rings deeply embedded in the binding pocket (parallel pose). Similar binding modes were found for HU-433 (Fig. 6B) and, to a lesser extent, for HU-308 (Fig. 6C). The parallel pose of HU-433 demonstrates hydrophobic interactions similar to those of CP55,940, but has only one H-bond compared with three for CP55,940 (Fig. 6B). In fact, the score of the parallel pose of HU-433 is only slightly higher than that of its binding pose, and the size of the respective clusters is also similar (Table S1), suggesting that HU-433 may also largely populate the parallel pose. HU-308 cannot occupy the parallel pose due to steric hindrance of the bicyclic ring, but rather a somewhat similar pose (Fig. 6C). This HU-308 parallel pose undergoes hydrophobic interactions but shares no H-bonds with the binding pocket. Actually, HU-308 has a higher score for its parallel pose than for the binding pose, with overall fewer poses in the respective cluster (Fig. 6C and Table S1), suggesting that this pose is considerably less populated by HU-308 than its lowest binding pose. Considering that the parallel pose is mainly adopted by the highly active CP55,940, the difference in occupancy of this pose by the two enantiomers may explain the higher activity of HU-433.
Table S1.
Autodock scores and cluster size of two different poses of the three agonists
| Molecule | Binding pose | Parallel pose |
| HU-308 | −11.04 (32) | −8.70 (15) |
| HU-433 | −9.90 (18) | −9.53 (18) |
| CP55,940 | −10.96 (27) | −11.47 (12) |
Scoring is in kcal/mol. Number of similar poses in each cluster is given in parentheses.
Although considerations based on molecular modeling explain the vastly enhanced biological potency of HU-433 vs. HU-308, they do not resolve the weaker HU-433 stimulation of GTPγS binding to CB2 compared with HU-308. It is reasonable to assume that, although the two enantiomers activate the same mitogenic signaling pathway, GTPγS is not associated with this pathway. Indeed, GTPγS seems not to be involved in Erk1,2 activation by Gαi-dependent GPCRs (29, 30). A corollary of this conclusion is that HU-433 has its binding orientation parallel to the CB2 z axis, which favors Erk1,2.
Another factor that can contribute to the differential in vivo activity of the two ligands is the differences in their possible allosteric interactions with CB2 receptors. Fig. 2B shows a very shallow concentration–response curve for HU-308 (over 5 log units), which strongly suggests negative cooperativity, whereas the analogous curve for HU-433 (Fig. 2A) is much steeper (3 log units) and more compatible with a Hill coefficient of 1.0—i.e., lack of cooperativity. The possibility of allosteric interactions needs to be more thoroughly investigated in further studies.
In conclusion, we present here two enantiomer agonists of the CB2 cannabinoid receptor with different, apparently paradoxical pharmacological properties, one with stronger binding to the receptor and weaker potency and the other with a lower binding affinity and higher potency in several biological assays. Using these enantiomers, the present analyses suggest that the CB2 receptor allows multiple ligand-binding modes—for example, one that favors GTPγS accumulation and another that signals through Erk1,2. This previously unreported situation questions the usefulness of universal radioligands for comparative binding studies because many ligands show inferior binding determinants when tested against structurally nonidentical radioactive competitors. Designing orientation-specific ligands has vast potential for the pharmacological activation of distinct processes.
The high potency of HU-433 in osteoblast proliferation and osteoclast differentiation, as well as its antiinflammatory activity, indicate that it may represent a therapeutically valuable molecule.
Materials and Methods
Synthesis of HU-308 and -433.
HU-308 was synthesized as reported (10). The procedure for preparing HU-433 is summarized in Scheme 2. Myrtenyl pivalate was prepared by reacting pivaloyl chloride with myrtenol in dry pyridine. The product was further reacted with CrO3 in CH3CN and t-BuOOH, and the resulting crude was chromatographed on silica gel to give 4-oxo-myrtenyl pivalate that was reduced by NaBH4, leading to 4-hydroxymyrtenyl pivalate as colorless oil. This oil was added to a solution of dry pTSA (p-toluene sulfonic acid) and 5-(1,1-dimethylheptyl)resorcinol in dry dichloromethane. The resultant 2-(3-myrtenyl pivalate)-5-(1,1-dimethylheptyl)resorcinol was reacted with methyl iodide to give the methylated 2-(3-myrtenyl pivalate)-5-(1,1-dimethylheptyl)resorcinol that was purified by silica gel chromatography and reduced with LiAlH4 to generate HU-433. The detailed synthetic method is reported in SI Materials and Methods.
Binding Assays.
Materials.
The (–)-cis-3-[2-hydroxy-4-(1,1-dimethylheptyl)phenyl]-trans-4-(3-hydroxypropyl)cyclohexanol (CP55940) was purchased from Tocris. For the binding experiments, [3H]CP55940 (174.6 Ci·mmol−1) and [35S]GTPγS (1,250 Ci·mmol−1) were obtained from PerkinElmer Life Sciences; GTPγS was from Roche Diagnostic; and GDP and dimethyl sulfoxide (DMSO) were from Sigma-Aldrich. HU-308, -433, and CP55940 were stored at −20 °C as stock solutions of 10 mM in DMSO, with the concentration of this vehicle in all assay wells being 0.1% DMSO.
CHO cells.
CHO cells stably transfected with cDNA encoding human cannabinoid CB2 receptors (Bmax = 215 pmol·mg−1) were purchased from PerkinElmer Life Sciences. They were maintained at 37 °C in 5% (vol/vol) CO2 in Dulbecco’s modified Eagle medium nutrient mixture F-12 HAM, supplemented with 1 mM l-glutamine, 10% (vol/vol) FBS, 0.6% penicillin–streptomycin, and 400 µg·ml−1 G418. These hCB2 CHO cells were passaged twice weekly by using PBS-EDTA buffer (1 mM EDTA in PBS solution, pH = 7).
Membrane preparation.
Binding assays with [3H]CP55940 or [35S]GTPγS were performed with hCB2 CHO cell membranes, prepared as described (31). Cells were removed from flasks by scraping, centrifuged at 489 × g, and then frozen as a pellet at −20 °C until required. Before use in a radioligand binding assay, cells were defrosted and diluted in Tris binding buffer (radioligand displacement assay, pH = 7) or GTPγS binding buffer ([35S]GTPγS binding assay, pH = 7.4). Protein assays were performed by using a Bio-Rad Dc kit.
Radioligand displacement assay.
The assays were carried out with 0.7 nM [3H]CP55940 and Tris binding buffer (50 mM Tris⋅HCl, 50 mM Tris-base, 0.1% BSA, pH 7.4) in a total assay volume of 500 µL, by using the filtration procedure described (31). Binding was initiated by the addition of hCB2 CHO cell membranes (50 µg of protein per well). All assays were performed at 37 °C for 60 min before termination by the addition of ice-cold Tris binding buffer and vacuum filtration by using a 24-well sampling manifold (Brandel Cell Harvester) and Brandel GF/B filters that had been soaked in wash buffer at 4 °C for at least 24 h (Brandel, pH = 7). Each reaction well was washed six times with a 1.2-mL aliquot of Tris binding buffer, and filters were oven-dried for 60 min and then placed in 5 mL of scintillation fluid (Ultima Gold XR; PerkinElmer). Radioactivity was quantified by a liquid scintillation counter (Tri-Carb 2800TR; PerkinElmer). Specific binding was defined as the difference between the binding that occurred in the presence and absence of 1 µM unlabeled CP55940.
[35S]GTPγS binding assay.
The method used for measuring agonist-stimulated binding of [35S]GTPγS was based on a described protocol (32). The assays were carried out with GTPγS binding buffer (50 mM Tris⋅HCl, 50 mM Tris-base, 5 mM MgCl2, 1 mM EDTA, 100 mM NaCl, 1 mM DTT, 0.1% BSA, pH = 7.4) in the presence of [35S]GTPγS (0.1 nM) and GDP (30 µM) in a final volume of 500 µL. Binding was initiated by the addition of [35S]GTPγS to the wells. Nonspecific binding was measured in the presence of 30 µM GTPγS. All assays were performed at 30 °C for 60 min before termination by addition of ice-cold Tris binding buffer and vacuum filtration as described for the radioligand binding assays. The radioactivity was quantified by liquid scintillation spectrometry. In all of the [35S]GTPγS binding assays, we used membranes obtained from hCB2 CHO cells at a protein concentration of 50 µg per well.
Analysis of Data.
Values are expressed as means and variability as SE or as 95% confidence limits. The concentrations of the compounds under investigation that produced a 50% displacement of radioligand from specific binding sites (IC50 value) were calculated by using GraphPad Prism (Version 5.0), and the corresponding Ki values were calculated by using the equation of Cheng and Prusoff (33). Mean values for EC50 and maximal effect (Emax) and SE or 95% confidence limits of these values were calculated by nonlinear regression analysis using the equation for a sigmoid concentration–response curve (GraphPad Prism; Version 5.0). Bmax and Kd values were calculated from data obtained in saturation binding assays (34), by using a one site-specific binding equation (GraphPad Prism; Version 5.0).
Animals.
C57BL/6J mice were used in all experiments. The experimental protocols were approved by the Institutional Animal Care and Use Committee of the Hebrew University of Jerusalem. CB2−/− mice on C57BL/6J background were kindly provided by Andreas Zimmer (University of Bonn, Bonn) and bred in the Hebrew University animal facility.
Cell Cultures.
Newborn mouse calvarial osteoblasts were prepared from 5-d-old mice by successive collagenase digestion (35). The cells were grown to subconfluence in α-MEM supplemented with 10% (vol/vol) FCS and then serum starved for 2 h. Cell counts and/or BrdU incorporation were determined after additional 48-h incubation in α-MEM supplemented with 0.5% BSA and enantiomers (2). Osteoclastogenic cultures were established from bone marrow-derived monocytes of 10- to 11-wk old mice as reported and grown for 4–5 d in medium containing macrophage–colony-stimulating factor, RANKL (R&D Systems), and enantiomers (36).
Real-Time RT-PCR.
Total RNA was isolated from NeMCO cells and incubated with or without HU-433 by using the TRI Reagent Kit (Molecular Research Center), followed by a phenol-chloroform phase extraction and isopropyl precipitation. The RNeasy kit was used for the purification of the RNA. Real-time RT-PCR analysis for Mapkapk2 was carried out by using RealTime ready Assay ID 3000082 (Mapkapk2) and 307884 (GAPDH) (Roche Diagnostica). Data were normalized to GAPDH and presented as relative quantity vs. 3-h control cultures.
Ear Swelling.
Xylene was pipetted to the inner and outer aspect of one external ear. HU-433 or the ethanol–cremophor–saline (1:1:18) vehicle was injected s.c. 24 h before xylene application. Indomethacin, used as a positive control, was given 30 min before the xylene application. PBS was applied to the other ear. Ear thickness was measured by using a Mitutoyo micrometer immediately before as well as 30, 90, and 150 min after xylene or PBS application. Ear swelling was expressed as the difference between the xylene- and PBS-treated ears. Significance was defined as P < 0.05.
Effect of HU-433 on Bone Loss.
Mice at 10 wk of age were subjected to bilateral OVX or sham-OVX. HU-433 was administered intraperitoneally 5 d/wk for 6 wk as an ethanol:cremophor:saline (1:1:18) solution. To assess in vivo the effect of HU-433 on bone formation, newly formed bone was vitally labeled by the fluorochrome calcein (Sigma) and injected intraperitoneally (15 mg/kg) 4 and 1 d before killing. At death, the femoral bones were separated and preserved, as reported (2). The skeletal microstructure and remodeling was analyzed by a combined microcomputed tomography (μCT)/histomorphometric system (2, 37, 38). Briefly, femoral metaphyses were examined by a μCT system (μCT 40; Scanco Medical AG) at a 20-μm resolution in all three spatial dimensions. After μCT image acquisition, the specimens were embedded undecalcified in Technovit 9100 (Heraeus Kulzer). Longitudinal sections through the midfrontal plane were left unstained for dynamic histomorphometry based on the vital calcein double labeling. To identify osteoclasts, consecutive sections were stained for TRAP. Parameters were determined according to a standardized nomenclature (39).
Serum Markers of Bone Remodeling.
Blood was collected retro-orbitally at the time of killing. Serum osteocalcin was determined by using a two-site enzyme immunoassay (EIA) kit (Biomedical Technologies Inc.). Osteoclast-derived TRAP5b was measured in the same specimens by using an EIA kit (Immunodiagnostic System).
Molecular Modeling.
The sequence and domain information of the hCB2 receptor was obtained from SWISS-PROT (accession no. P34972) (40). The model was generated by using bovine rhodopsin crystal structure as a template, and further refinement of the loops was carried out iteratively by using Modeler (Version 9v9) (41). The best model chosen had <3.4% residues outside the allowed region of the Ramachandran plot. Validation of the model was done by using available experimental data and previously derived models of CB2 (27, 42).
Optimization of HU-308 (3R, 4S, 6S), HU-433 (3S, 4R, 6R), and CP55,940 was done at B3LYP/6-311G** by using the Gaussian09 software package (43). These optimized geometries were used as the initial conformations for the docking within Autodock (Version 4) (44). Respective partial charges for the ligands and receptor were calculated according to Gasteiger (45) and Kollman (46). Charges of nonpolar hydrogen atoms were merged into heavy atoms. The binding site of the protein was detected by the Computed Atlas of Surface Topography of Proteins server (47) and validated with available experimental and theoretical data (26, 27, 48).
Binding poses were identified by using the Lamarckian Genetic Algorithm. The receptor was treated as rigid, whereas the ligand was allowed torsional flexibility. The centroid of three residues reported important for binding, F117, W258, and W194 (49), was defined as the grid center that was constructed of 60 × 60 × 60 grid points with a 0.375-Å spacing. The system parameters were set as default, but with 107 energy evaluations. Using these parameters, we generated 100 poses with lowest energies. The best scoring and lowest root-mean-square deviation (rmsd) solutions from the resulting 100 poses were clustered into groups, with rmsd values <1.0 Å and considered the predicted binding conformers.
Statistical Analysis.
Multiple groups were compared by using ANOVA. When significant differences were indicated by ANOVA, differences between group means were analyzed by using the Student–Newman–Keuls test for pairwise comparisons. A t test was used when two samples were compared.
SI Materials and Methods
Synthesis of HU-433.
Preparation of myrtenyl pivalate.
Pivaloyl chloride (8 g, 67 mmol) was added slowly to a solution of myrtenol (commercial myrtenol; Aldrich; CAS 19894-97-4), [α]D= −51°) (5 g, 33 mmol) in dry pyridine (25 mL), with cooling in ice bath. The mixture was stirred overnight at room temperature. Then the mixture was diluted with ether (25 mL), ice water was added, the organic layer was collected and washed with 10% HCl several times and then with NaHCO3, dried over Na2SO4, and evaporated to give 7.17 g (91%) of colorless oil.
Preparation of 4-oxomyrtenyl pivalate.
For the preparation of 4-oxomyrtenyl pivalate, CrO3 (1.06 g, 10.6 mmol) was dissolved in CH3CN (35 mL) and stirred at 0 °C. Then t-BuOOH (17.5 mL 70%/H2O, 126 mmol) was added, followed by the immediate addition of a solution of myrtenyl pivalate (5 g, 20 mmol) in CH3CN (38 mL). The reaction mixture was brought to room temperature and was stirred for 1 h. The reaction mixture was diluted with ether, and 10% Na2SO3 in water (130 mL) was added. The mixture was extracted several times with ether, dried, and evaporated. The crude mixture was purified by silica gel column chromatography (0–10% ether in petroleum ether) to give an oil that crystallized from pentane to give 4-oxo-myrtenyl pivalate (0.9 g, 19%) melting point 35 °C, [α]D= −162°.
Preparation of 4-hydroxymyrtenyl pivalate.
The 4-oxo-myrtenyl pivalate (1 g, 4 mmol) was dissolved in ethanol (15 mL). NaBH4 (0.2 g, 5.3 mmol) was added slowly, and the resulting suspension was stirred at room temperature for 15 min. The ethanol was evaporated, and then ether and water were added. The organic layer was separated, dried, and evaporated to give a colorless oil (0.92 g, 91%).
Condensation of 4-hydroxymyrtenyl pivalate with 5-(1,1-dimethylheptyl)resorcinol.
To a solution of dry p-toluenesulfonic acid (0.16 g, 0.96 mmol) and 5-(1,1-dimethylheptyl)resorcinol (0.86 g, 3.44 mmol) in dry dichloromethane (125 mL) and under argon atmosphere, was added slowly a solution of 4-hydroxy-myrtenyl pivalate (0.88 g, 3.49 mmol) in dry dichloromethane (30 mL). The mixture was stirred at room temperature for 1.5 h. A saturated NaHCO3 solution was added, and the organic layer was then washed twice with water, dried, and evaporated. The residue was purified by column chromatography on silica gel in ether/petroleum ether (5:95) to give 0.71 g (43%) of 2-(3-myrtenyl pivalate)-5-(1,1-dimethylheptyl)resorcinol, a colorless oil.
Methylation.
Methyl iodide (0.75 mL, 12 mmol) was added to a solution of 2-(3-myrtenyl pivalate)-5-(1,1-dimethylheptyl)resorcinol (0.71 g, 1.51 mmol) and K2CO3 (1.6 g, 12 mmol) in dry dimethylformamide (DMF) (5 mL). After stirring at room temperature for 24 h, the mixture was diluted with water (40 mL) and extracted with ether. The organic layer was washed with water, dried, and evaporated. Purification by column chromatography on silica gel with ether/ petroleum ether (5:95) gave 0.51 g (67%) of 2-(3-myrtenyl pivalate)dimethoxy-5-(1,1dimethylheptyl)resorcinol (HU-433; pivalate) a colorless oil.
Preparation of HU-433.
HU-433 pivalate (0.35 g, 0.7 mmol) in dry ether (20 mL) was added slowly, under argon to a suspension of LiAlH4 (64 mg, 1.68 mmol) an dry ether (5 mL). The mixture was refluxed for 2 h. The excess LiAlH4 was destroyed with ethyl acetate followed by a saturated solution of MgSO4 until a clear ether solution was obtained. The ether layer was decanted, dried, and evaporated. The product precipitated out from pentane to give HU-433, m.p. 44–45 °C, [α]D= −120°.
HU-433 Spectral Data.
1H-NMR (CDCl3): 3.741 (s, 6H CH3O-), 3.998 (t, J = 2.1Hz, 1H allylic), 4.069 (d, J = 5.7 Hz, 2H, -CH2OH), 5.704(t, J = 1.2, 1H, olefinic) 6.483 (s, 2H, aromatic). MS calc/found: m/z 414.37/437.27 (m+Na).
Acknowledgments
Research in the R.M. laboratory was supported by the Kessler Family Foundation and by National Institute on Drug Abuse Grant DA-9789.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
3Deceased October 18, 2014.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1503395112/-/DCSupplemental.
References
- 1.Pacher P, Mechoulam R. Is lipid signaling through cannabinoid 2 receptors part of a protective system? Prog Lipid Res. 2011;50(2):193–211. doi: 10.1016/j.plipres.2011.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ofek O, et al. Peripheral cannabinoid receptor, CB2, regulates bone mass. Proc Natl Acad Sci USA. 2006;103(3):696–701. doi: 10.1073/pnas.0504187103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Basu S, Ray A, Dittel BN. Cannabinoid receptor 2 is critical for the homing and retention of marginal zone B lineage cells and for efficient T-independent immune responses. J Immunol. 2011;187(11):5720–5732. doi: 10.4049/jimmunol.1102195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Galiègue S, et al. Expression of central and peripheral cannabinoid receptors in human immune tissues and leukocyte subpopulations. Eur J Biochem. 1995;232(1):54–61. doi: 10.1111/j.1432-1033.1995.tb20780.x. [DOI] [PubMed] [Google Scholar]
- 5.Pertwee RG. Cannabinoid pharmacology: the first 66 years. Br J Pharmacol. 2006;147(Suppl 1):S163–S171. doi: 10.1038/sj.bjp.0706406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cabral GA, Raborn ES, Griffin L, Dennis J, Marciano-Cabral F. CB2 receptors in the brain: Role in central immune function. Br J Pharmacol. 2008;153(2):240–251. doi: 10.1038/sj.bjp.0707584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Elphick MR, Egertová M. The neurobiology and evolution of cannabinoid signalling. Philos Trans R Soc Lond B Biol Sci. 2001;356(1407):381–408. doi: 10.1098/rstb.2000.0787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rajesh M, et al. Cannabinoid-2 receptor agonist HU-308 protects against hepatic ischemia/reperfusion injury by attenuating oxidative stress, inflammatory response, and apoptosis. J Leukoc Biol. 2007;82(6):1382–1389. doi: 10.1189/jlb.0307180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Toguri JT, et al. Anti-inflammatory effects of cannabinoid CB(2) receptor activation in endotoxin-induced uveitis. Br J Pharmacol. 2014;171(6):1448–1461. doi: 10.1111/bph.12545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hanus L, et al. HU-308: A specific agonist for CB(2), a peripheral cannabinoid receptor. Proc Natl Acad Sci USA. 1999;96(25):14228–14233. doi: 10.1073/pnas.96.25.14228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huffman JW, et al. 3-(1′,1′-Dimethylbutyl)-1-deoxy-delta8-THC and related compounds: Synthesis of selective ligands for the CB2 receptor. Bioorg Med Chem. 1999;7(12):2905–2914. doi: 10.1016/s0968-0896(99)00219-9. [DOI] [PubMed] [Google Scholar]
- 12.Horváth B, et al. A new cannabinoid CB2 receptor agonist HU-910 attenuates oxidative stress, inflammation and cell death associated with hepatic ischaemia/reperfusion injury. Br J Pharmacol. 2012;165(8):2462–2478. doi: 10.1111/j.1476-5381.2011.01381.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Han S, Thatte J, Buzard DJ, Jones RM. Therapeutic utility of cannabinoid receptor type 2 (CB(2)) selective agonists. J Med Chem. 2013;56(21):8224–8256. doi: 10.1021/jm4005626. [DOI] [PubMed] [Google Scholar]
- 14.Ofek O, et al. CB2 cannabinoid receptor targets mitogenic Gi protein-cyclin D1 axis in osteoblasts. J Bone Miner Res. 2011;26(2):308–316. doi: 10.1002/jbmr.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ariëns EJ. Stereochemistry, a basis for sophisticated nonsense in pharmacokinetics and clinical pharmacology. Eur J Clin Pharmacol. 1984;26(6):663–668. doi: 10.1007/BF00541922. [DOI] [PubMed] [Google Scholar]
- 16.Tamura T, et al. New resorption assay with mouse osteoclast-like multinucleated cells formed in vitro. J Bone Miner Res. 1993;8(8):953–960. doi: 10.1002/jbmr.5650080808. [DOI] [PubMed] [Google Scholar]
- 17.Zaidi M, Moonga BS, Huang CL. Calcium sensing and cell signaling processes in the local regulation of osteoclastic bone resorption. Biol Rev Camb Philos Soc. 2004;79(1):79–100. doi: 10.1017/s1464793103006262. [DOI] [PubMed] [Google Scholar]
- 18.Sulcova E, Mechoulam R, Fride E. Biphasic effects of anandamide. Pharmacol Biochem Behav. 1998;59(2):347–352. doi: 10.1016/s0091-3057(97)00422-x. [DOI] [PubMed] [Google Scholar]
- 19.Bab I, Ofek O, Tam J, Rehnelt J, Zimmer A. Endocannabinoids and the regulation of bone metabolism. J Neuroendocrinol. 2008;20(Suppl 1):69–74. doi: 10.1111/j.1365-2826.2008.01675.x. [DOI] [PubMed] [Google Scholar]
- 20.Klauke AL, et al. The cannabinoid CB₂ receptor-selective phytocannabinoid beta-caryophyllene exerts analgesic effects in mouse models of inflammatory and neuropathic pain. Eur Neuropsychopharmacol. 2014;24(4):608–620. doi: 10.1016/j.euroneuro.2013.10.008. [DOI] [PubMed] [Google Scholar]
- 21.Mimura T, Ueda Y, Watanabe Y, Sugiura T. The cannabinoid receptor-2 is involved in allergic inflammation. Life Sci. 2012;90(21-22):862–866. doi: 10.1016/j.lfs.2012.04.005. [DOI] [PubMed] [Google Scholar]
- 22.Kou J, et al. Anti-inflammatory activities of aqueous extract from Radix Ophiopogon japonicus and its two constituents. Biol Pharm Bull. 2005;28(7):1234–1238. doi: 10.1248/bpb.28.1234. [DOI] [PubMed] [Google Scholar]
- 23.Włodarski KH. Properties and origin of osteoblasts. Clin Orthop Relat Res. 1990;(252):276–293. [PubMed] [Google Scholar]
- 24.Kurihara N, et al. Generation of osteoclasts from isolated hematopoietic progenitor cells. Blood. 1989;74(4):1295–1302. [PubMed] [Google Scholar]
- 25.Prussin C, Metcalfe DD. 4. IgE, mast cells, basophils, and eosinophils. J Allergy Clin Immunol. 2003;111(2) Suppl:S486–S494. doi: 10.1067/mai.2003.120. [DOI] [PubMed] [Google Scholar]
- 26.Palczewski K, et al. Crystal structure of rhodopsin: A G protein-coupled receptor. Science. 2000;289(5480):739–745. doi: 10.1126/science.289.5480.739. [DOI] [PubMed] [Google Scholar]
- 27.Xie X-Q, Chen J-Z, Billings EM. 3D structural model of the G-protein-coupled cannabinoid CB2 receptor. Proteins. 2003;53(2):307–319. doi: 10.1002/prot.10511. [DOI] [PubMed] [Google Scholar]
- 28.Hurst DP, et al. A lipid pathway for ligand binding is necessary for a cannabinoid G protein-coupled receptor. J Biol Chem. 2010;285(23):17954–17964. doi: 10.1074/jbc.M109.041590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang R, Xie X. Tools for GPCR drug discovery. Acta Pharmacol Sin. 2012;33(3):372–384. doi: 10.1038/aps.2011.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pacher P, Kunos G. Modulating the endocannabinoid system in human health and disease—successes and failures. FEBS J. 2013;280(9):1918–1943. doi: 10.1111/febs.12260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ross RA, et al. Agonist-inverse agonist characterization at CB1 and CB2 cannabinoid receptors of L759633, L759656, and AM630. Br J Pharmacol. 1999;126(3):665–672. doi: 10.1038/sj.bjp.0702351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Thomas A, et al. Evidence that the plant cannabinoid Δ9-tetrahydrocannabivarin is a cannabinoid CB1 and CB2 receptor antagonist. Br J Pharmacol. 2005;146(7):917–926. doi: 10.1038/sj.bjp.0706414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cheng Y, Prusoff WH. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem Pharmacol. 1973;22(23):3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- 34.Bolognini D, Cascio MG, Parolaro D, Pertwee RG. AM630 behaves as a protean ligand at the human cannabinoid CB2 receptor. Br J Pharmacol. 2012;165(8):2561–2574. doi: 10.1111/j.1476-5381.2011.01503.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kato M, et al. Cbfa1-independent decrease in osteoblast proliferation, osteopenia, and persistent embryonic eye vascularization in mice deficient in Lrp5, a Wnt coreceptor. J Cell Biol. 2002;157(2):303–314. doi: 10.1083/jcb.200201089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Asagiri M, Takayanagi H. The molecular understanding of osteoclast differentiation. Bone. 2007;40(2):251–264. doi: 10.1016/j.bone.2006.09.023. [DOI] [PubMed] [Google Scholar]
- 37.Bajayo A, et al. Central IL-1 receptor signaling regulates bone growth and mass. Proc Natl Acad Sci USA. 2005;102(36):12956–12961. doi: 10.1073/pnas.0502562102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yirmiya R, et al. Depression induces bone loss through stimulation of the sympathetic nervous system. Proc Natl Acad Sci USA. 2006;103(45):16876–16881. doi: 10.1073/pnas.0604234103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Parfitt AM, et al. Report of the ASBMR Histomorphometry Nomenclature Committee Bone histomorphometry: Standardization of nomenclature, symbols, and units. J Bone Miner Res. 1987;2(6):595–610. doi: 10.1002/jbmr.5650020617. [DOI] [PubMed] [Google Scholar]
- 40.Magrane M, Consortium U. UniProt Knowledgebase: A hub of integrated protein data. Database (Oxford) 2011;2011:bar009. doi: 10.1093/database/bar009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol. 1993;234(3):779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 42.Choi G, Landin J, Xie XQ. The cytoplasmic helix of cannabinoid receptor CB2, a conformational study by circular dichroism and (1)H NMR spectroscopy in aqueous and membrane-like environments. J Pept Res. 2002;60(3):169–177. doi: 10.1034/j.1399-3011.2002.21012.x. [DOI] [PubMed] [Google Scholar]
- 43.Frisch MJ, et al. Gaussian 09, Revision D.01. Gaussian; Wallingford, CT: 2009. [Google Scholar]
- 44.Morris GM, et al. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J Comput Chem. 2009;30(16):2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gasteiger J, Marsili M. Iterative partial equalization of orbital electronegativity - a rapid access to atomic charges. Tetrahedron. 1980;36(22):3219–3228. [Google Scholar]
- 46.Wang J, Kollman PA, Kuntz ID. Flexible ligand docking: A multistep strategy approach. Proteins-Structure Function and Genetics. 1999;36(1):1–19. [PubMed] [Google Scholar]
- 47.Dundas J, et al. CASTp: Computed atlas of surface topography of proteins with structural and topographical mapping of functionally annotated residues. Nucleic Acids Res. 2006;34(Web Server Issue):W116-8. doi: 10.1093/nar/gkl282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang Y, et al. Mutagenesis and computer modeling studies of a GPCR conserved residue W5.43(194) in ligand recognition and signal transduction for CB2 receptor. Int Immunopharmacol. 2011;11(9):1303–1310. doi: 10.1016/j.intimp.2011.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Poso A, Huffman JW. Targeting the cannabinoid CB2 receptor: Modelling and structural determinants of CB2 selective ligands. Br J Pharmacol. 2008;153(2):335–346. doi: 10.1038/sj.bjp.0707567. [DOI] [PMC free article] [PubMed] [Google Scholar]