

ABSTRACT
The Pseudomonas aeruginosa type III secretion system (T3SS) is a primary virulence factor important for phagocytic avoidance, disruption of host cell signaling, and host cell cytotoxicity. ExsA is the master regulator of T3SS transcription. The expression, synthesis, and activity of ExsA is tightly regulated by both intrinsic and extrinsic factors. Intrinsic regulation consists of the well-characterized ExsECDA partner-switching cascade, while extrinsic factors include global regulators that alter exsA transcription and/or translation. To identify novel extrinsic regulators of ExsA, we conducted a transposon mutagenesis screen in the absence of intrinsic control. Transposon disruptions within gene PA2840, which encodes a homolog of the Escherichia coli RNA-helicase DeaD, significantly reduced T3SS gene expression. Recent studies indicate that E. coli DeaD can promote translation by relieving inhibitory secondary structures within target mRNAs. We report here that PA2840, renamed DeaD, stimulates ExsA synthesis at the posttranscriptional level. Genetic experiments demonstrate that the activity of an exsA translational fusion is reduced in a deaD mutant. In addition, exsA expression in trans fails to restore T3SS gene expression in a deaD mutant. We hypothesized that DeaD relaxes mRNA secondary structure to promote exsA translation and found that altering the mRNA sequence of exsA or the native exsA Shine-Dalgarno sequence relieved the requirement for DeaD in vivo. Finally, we show that purified DeaD promotes ExsA synthesis using in vitro translation assays. Together, these data reveal a novel regulatory mechanism for P. aeruginosa DeaD and add to the complexity of global regulation of T3SS.
IMPORTANCE Although members of the DEAD box family of RNA helicases are appreciated for their roles in mRNA degradation and ribosome biogenesis, an additional role in gene regulation is now emerging in bacteria. By relaxing secondary structures in mRNAs, DEAD box helicases are now thought to promote translation by enhancing ribosomal recruitment. We identify here an RNA helicase that plays a critical role in promoting ExsA synthesis, the central regulator of the Pseudomonas aeruginosa type III secretion system, and provide additional evidence that DEAD box helicases directly stimulate translation of target genes. The finding that DeaD stimulates exsA translation adds to a growing list of transcriptional and posttranscriptional regulatory mechanisms that control type III gene expression.
INTRODUCTION
Pseudomonas aeruginosa is a Gram-negative opportunistic pathogen that expresses a large array of virulence factors to cause both acute and chronic infections (1). P. aeruginosa infections generally initiate as acute infections that can range from self-limiting folliculitis to life-threatening ventilator-associated pneumonia (1). P. aeruginosa utilizes a type III secretion system (T3SS) to promote host cell cytotoxicity and avoid phagocytosis (2, 3). The T3SS is a multiprotein complex that spans the cell envelope and functions by directly injecting effector proteins into the host cell cytoplasm (4). T3SS-deficient strains have reduced cytotoxicity and are attenuated in animal models of acute infection (5–7). Regulation of T3SS gene expression is complex, can occur at various levels (transcriptional, posttranscriptional, and posttranslational), and involves multiple regulatory systems (8). In general, T3SS regulation can be separated into two classes: intrinsic and extrinsic.
Intrinsic control of the T3SS occurs through direct transcriptional regulation by the master regulator ExsA (9). ExsA binds to and activates all T3SS gene promoters (9–14). ExsA activity is controlled by the ExsECD protein-protein interaction cascade (15). Under noninducing conditions, ExsA is sequestered by the anti-activator protein ExsD and is unable to activate T3SS promoters (16). Concurrently, the ExsC chaperone and secreted ExsE protein for a complex (17). In the presence of an inducing signal (host cell contact or growth under low-Ca2+ conditions) ExsE is secreted from the cell via the T3SS apparatus (13, 18, 19). ExsE secretion results in a partner-switching mechanism wherein ExsC preferentially binds ExsD to release ExsA and activate T3SS gene expression (13, 17, 20, 21).
Extrinsic control of T3SS gene expression usually involves regulation of exsA transcription and/or ExsA synthesis by other regulatory systems (8). For example, the cyclic AMP (cAMP)/Vfr signaling (CVS) pathway is an extrinsic regulator of T3SS gene expression (22). Vfr is a cAMP-dependent transcription factor that promotes expression of multiple virulence factors including type IV pili, proteases, and the T3SS (22). Vfr promotes exsA transcription through an unknown mechanism (23). The Gac/Rsm pathway is another extrinsic regulator of T3SS gene expression (8, 24, 25). RsmA belongs to the CsrA family of RNA-binding proteins and functions by directly binding to target mRNAs at conserved 5′-ANGGAAN motifs presented within stem-loop secondary structures (24–27). Binding of CsrA-family proteins to target mRNAs can alter the rate of translation initiation and/or mRNA decay (28). RsmA activity is controlled by two small, noncoding RNAs (RsmY and RsmZ) that sequester RsmA from target mRNAs by presenting multiple competitive binding sites (26, 29–31). RsmA promotes exsA translation through an unknown mechanism (23, 24). A final example of extrinsic control is seen in mucA mutants, which are commonly isolated from cystic fibrosis infections (32–34). Mutants lacking functional MucA have reduced T3SS gene expression (23, 33, 35, 36). The two-component system AlgZR, which has increased activity in mucA mutants, reduces the activity of both the CVS and the Gac/Rsm pathways, ultimately resulting in decreased T3SS gene expression (23, 35).
In the present study, we developed a genetic screen to identify novel extrinsic regulators of T3SS gene expression. By taking advantage of the delayed cytotoxicity phenotype of an exsC mutant, we identified a putative ATP-dependent RNA helicase (PA2840) as a potential regulator of T3SS gene expression. RNA helicases can regulate gene expression at a posttranscriptional level by either associating with the RNA degradosome or by directly unwinding secondary structure in the mRNAs of target genes (37–39). We show that PA2840, renamed DeaD, is a novel extrinsic regulator of ExsA synthesis. Mutants lacking deaD have reduced exsA translation in vivo and purified DeaD promotes ExsA synthesis in vitro. A catalytically inactive variant of DeaD (E168A) is unable to complement a deaD mutant for ExsA or promote ExsA synthesis in vitro. Together, these data identify a novel posttranscriptional regulatory mechanism that promotes P. aeruginosa T3SS gene expression.
MATERIALS AND METHODS
Bacterial strains, culture conditions, and sample preparation.
The bacterial strains used in the present study are listed in Table S1 in the supplemental material. Escherichia coli strains were cultured in Luria-Bertani (LB) medium supplemented with gentamicin (15 μg/ml), ampicillin (100 μg/ml), or tetracycline (15 μg/ml) as necessary. P. aeruginosa strains were cultured on Vogel-Bonner minimal (VBM) medium agar plates or in LB-Miller medium supplemented with gentamicin (100 μg/ml) as necessary. For transcriptional and translational reporter assays, preparation of whole-cell lysates, and total RNA extraction, P. aeruginosa was cultured under inducing conditions for T3SS gene expression in Trypticase soy broth supplemented with 100 mM monosodium glutamate, 1% glycerol, and 1 mM EGTA. In some cases, arabinose was added to induce expression of plasmid-borne genes as indicated in the figure legends. Transcriptional and translational reporter assays, preparation of whole-cell lysates, and immunoblotting were performed as previously described (23).
Plasmid and strain construction.
The plasmids used in the present study, the details of their construction, and the primer sequences are provided in Tables S2 to S4 in the supplemental material. PA103 genomic DNA was used as the template for PCRs. Allelic-exchange vectors and the mini-CTX and Tn7 reporters were integrated into the chromosome of desired strains as previously described (40, 41). Deletion strains and plasmids were verified via sequencing. pEX18GmΔdeaD and pEXG2ΔdeaD were constructed by PCR amplifying upstream (using the primer pair 21372412-21372411) and downstream (primer pair 21372413-21372410) flanking regions of deaD and cloning into the EcoRI-HindIII sites of pEX18Gm and pEXG2, respectively. pEX18GmΔexsA was constructed by PCR amplifying upstream (primer pair 4274568-15945890) and downstream (primer pair 36945052-2715477) flanking regions of exsA and cloning into the EcoRI-HindIII sites of pEX18Gm. The pMini-Tn7-PBAD-exsC vector was constructed by PCR amplifying the PBAD-exsC fragment from pJN-exsC using the primer pair 18918413-18918385 and cloning into the NsiI-KpnI sites in pUC18T-mini-Tn7T-Gm. The pDeaD E168Q and pDeaD E168A expression vectors were constructed by Gibson assembly wherein overlapping upstream (primer pairs 123226612-123226607 and 123226612-123226609) and downstream (primer pairs 123226606-123226613 and 123226608-123226613) portions of deaD containing the desired mutations were cloned into the EcoRI and SacI sites of pJN105. pRBS-ExsA was constructed by PCR amplifying exsA with the Shine-Dalgarno sequence from exsC using the primer pair 124994340-83085368 and cloning into the EcoRI-SacI sites of pJN105. pCEBA-1(p2UY75A) and pCEBA-2(p2UY75B) were constructed by first PCR amplifying the PlacUV5 promoter region from strain UY339 using primer pairs 33041594-33271898 and 20755258-33271898, respectively. These PCR products were used as megaprimers in subsequent PCRs with the primer 83085368, and the resulting products were cloned into the EcoRI-SacI sites of p35B. pET23b-LcrF was constructed by PCR amplifying the 37-nucleotide (nt) exsA leader region and lcrF gene from the hybrid plasmid pLcrF using the primer pair 86360966-130269370. The resulting product was cloned into the XbaI-SacI sites of pET23b+. p3UY51-PlacUV5 – exsCEBAfull′–′lacZ was constructed by PCR amplifying the exsCEBA operon and cloning into the EcoRI-BamHI sites of the previously described p3UY51 vector, fusing exsA codon 277 to the lacZ open reading frame (ORF) (23).
Transposon mutagenesis.
The exsC CTX-PexsD-lacZ:Tn7-PBAD-exsC strain was subjected to Mariner-based transposon mutagenesis via conjugation with E. coli strain SM10 carrying plasmid pBTK30 as previously described (42, 43). Inverse PCR was performed to identify the location of the transposon insertion sites as follows. Genomic DNA isolated from the transposon mutants was digested with HpyCH4IV (New England BioLabs, Beverly, MA) and purified using a QIAquick PCR Purification kit (Qiagen Sciences, Germantown, MD). The digested genomic DNA was ligated en masse and then purified as described above. PCR was performed using the ligated DNA as the template with the transposon-specific primers IPCR-A and IPCR-B (see Table S4 in the supplemental material). Amplified DNA products were gel purified and submitted for DNA sequencing using primer IPCR-A. A BLAST query was performed on the sequences to identify the transposon insertion sites (http://www.pseudomonas.com).
Cytotoxicity assays.
Chinese hamster ovary (CHO) cells (ATCC CCL-61) were cultured in Ham F-12 medium (Invitrogen Corp., Carlsbad, CA) supplemented with 10% fetal calf serum, 50 U of penicillin and streptomycin/ml, 2 mM l-glutamine, 0.12% sodium bicarbonate, and 2.5 mM HEPES at 37°C in 5% CO2. For cytotoxicity assays, CHO cells were seeded at 8 × 104 cells/well into 24-well tissue culture plates (80 to 85% confluence) or 3 × 104 cells/well into 96-well tissue culture plates (95 to 100% confluence) and incubated for 18 to 24 h at 37°C in 5% CO2. P. aeruginosa strains were grown on VBM plates overnight at 37°C, washed with phosphate-buffered saline, diluted in prewarmed Ham F-12 medium, and added to the CHO cells to a multiplicity of infection (MOI) of 10. The plates were centrifuged (500 × g, 5 min, 25°C) and incubated at 37°C in 5% CO2 for the indicated times. After incubation, the plates were centrifuged (500 × g, 5 min, 25°C), and 50 μl of the supernatant was transferred to a 96-well plate and assayed for lactate dehydrogenase (LDH) release using a CytoTox 96 nonradioactive cytotoxicity assay (Promega, Madison, WI). The percent cytotoxicity was calculated by subtracting the optical density at 490 nm of a noninfected control from each sample and using wild-type (wt) PA103 as the positive control normalized to 100%.
Expression and purification of DeaDHis and DeaDHis E168A.
E. coli Tuner (DE3) carrying either the pET16bDeaD or pET16bDeaD E168A expression vectors was inoculated to an initial absorbance at 600 nm (A600) of 0.1 in LB-Miller medium supplemented with ampicillin (100 μg/ml) and incubated 30°C with shaking. When the A600 reached 0.5, 1 mM IPTG (isopropyl-β-d-thiogalactopyranoside) was added to induce protein expression. Cultures were incubated an additional 2 to 4 h at 30°C. Cells were collected by centrifugation (6,000 × g, 10 min, 4°C) and suspended in 20 ml of DeaD buffer (20 mM Tris-HCl [pH 8.0], 500 mM NaCl, 20 mM imidazole) supplemented with 1 protease inhibitor cocktail tablet (Roche Diagnostics, Indianapolis, IN). Cells were lysed using a Microfluidizer (Microfluidics, Newton, MA) and immediately centrifuged (20,000 × g, 20 min, 4°C) to remove cell debris. DeaDHis and DeaDHis E168A were purified from cell extracts using Ni2+-affinity chromatography and dialyzed overnight at 4°C in DeaD buffer lacking imidazole and supplemented with 1 mM dithiothreitol (DTT). The protein concentrations were determined using a DC protein assay (Bio-Rad Laboratories, Hercules, CA).
In vitro transcription/translation of ExsA, LcrF, and Vfr.
A PURExpress in vitro protein synthesis system (New England BioLabs, Ipswich, MA) was used to measure DeaD activity as follows. DNA templates for ExsA, Vfr, and LcrF were PCR amplified from pET23b-exsA1, pET23b-Vfr, and pET23b-LcrF using the primers 127176830 and 124849942. Reaction mixtures (17 μl) contained 1 μl (10.2 μCi) of [35S]methionine (Perkin-Elmer, Waltham, MA), 5 μg of DNA template, 0.5 μl (20 U) of RNAseOUT (Life Technologies, Grand Island, NY), and purified DeaDHis or DeaDHis E168A as indicated. The reaction mixtures were incubated at 37°C for 2 h and then terminated by the addition of 17 μl of 2× SDS-PAGE sample buffer and heating at 95°C. The samples were analyzed by SDS-PAGE, dried, and then visualized using a phosphorimager.
Quantitative reverse transcriptase PCR (qRT-PCR).
P. aeruginosa strains were grown under inducing conditions for T3SS gene expression until the A600 reached 1.0. RNA was stabilized by adding 500 μl of culture to 500 μl of RNAprotect reagent (Qiagen), and the pellets were stored at −80°C. Total mRNA was harvested from cell pellets using the RNeasy minikit (Qiagen) according to the manufacturer's instructions with on-column DNase I digestion (Qiagen). cDNA was generated using primers listed in Table S4 in the supplemental material. Reaction mixtures (12 μl) containing purified RNA (100 ng), 9 μl of RNase-free H2O, 1 μl oligonucleotide mix (2 pmol of each primer/μl), and 1 μl of deoxynucleoside triphosphate mix were heated at 65°C for 5 min. The tubes were then placed on ice for 1 min, and 4 μl of 5× First Strand buffer (Life Technologies), 1 μl of 100 mM DTT, 1 μl (40 U) RNAseOUT, and 1 μl of SuperScript III reverse transcriptase (Life Technologies) were added to the reaction mixtures. The reaction mixtures were then incubated at 50°C for 60 min and terminated by incubation at 70°C for 10 min. cDNA (2 μl) was added to reaction mixtures containing 10 μl of 2× TaqMan universal PCR master mix (Life Technologies), 2 μl of specific PrimeTime qPCR assay (Integrated DNA Technologies, Coralville, IA), and 6 μl of H2O. PCRs were performed by the Iowa Institute of Human Genetics.
To analyze exsA mRNA degradation, strains UY339 and UY339 deaD were cultured as described above. When the culture A600 reached 1.0, 200 μg of rifampin/ml was added, and samples were collected immediately and 5, 10, 15, 20, and 30 min thereafter by adding 500 μl of culture to 500 μl of RNAprotect reagent as described above. Total mRNA from each sample was prepared and subjected to qRT-PCR as described above.
Statistical analyses.
Two-tailed unpaired t tests were used for data analyses when comparing two groups. The one-way analysis of variance test and Tukey's post test were used to determine statistical significance when comparing three or more groups. Statistical analyses were performed using Prism 5.0c (GraphPad Software, Inc., La Jolla, CA).
RESULTS
Identification of novel extrinsic regulators of T3SS gene expression.
Chinese hamster ovary (CHO) cells are rapidly lysed when cocultured with P. aeruginosa strain PA103, a property that is almost entirely attributable to the T3SS (Fig. 1A) (13, 44). Using cytotoxicity as a screen for regulators of T3SS gene expression by transposon mutagenesis, however, is tedious owing to the high rate of insertions that occur within T3SS genes and type IV pilus biosynthetic genes, the latter being required for attachment to host cells (45). To identify novel extrinsic regulators of the T3SS, we utilized a transposon mutagenesis screen that relied on the delayed cytotoxicity of an exsC mutant. In the absence of ExsC, formation of inhibitory ExsA-ExsD complexes prevent high levels of T3SS gene expression (20). For this reason, an exsC mutant is noncytotoxic when cocultured for 2 h with CHO cells as measured by LDH release (Fig. 1A). When cocultured for 4 h, however, the exsC mutant is cytotoxic, owing to low levels of T3SS gene expression. We hypothesized that the cytotoxicity of transposon mutants within T3SS or type IV pilus genes would not be restored by heterologous expression of exsC at 2 h because the underlying defect had not been corrected. Conversely, heterologous expression of exsC should restore cytotoxicity to many of the mutants within extrinsic regulatory genes simply by increasing ExsA activity (Fig. 1B). To test this approach, we integrated an arabinose-inducible exsC expression cassette into an ectopic Tn7 chromosomal integration site in the exsC mutant, resulting in strain exsC-PBAD-exsC. The exsC-PBAD-exsC strain was then cocultured with CHO cells for 2 or 4 h in either the absence or the presence of arabinose. As expected, the cytotoxicity of the exsC-PBAD-exsC strain was not dependent on arabinose-inducible expression of exsC following a 4-h coculture (Fig. 1A). After a 2-h coculture, however, the T3SS-dependent cytotoxicity of the exsC-PBAD-exsC strain was significantly enhanced upon arabinose addition, whereas the exsC mutant lacking the PBAD-exsC cassette resulted in low levels of cytotoxicity (Fig. 1A).
FIG 1.
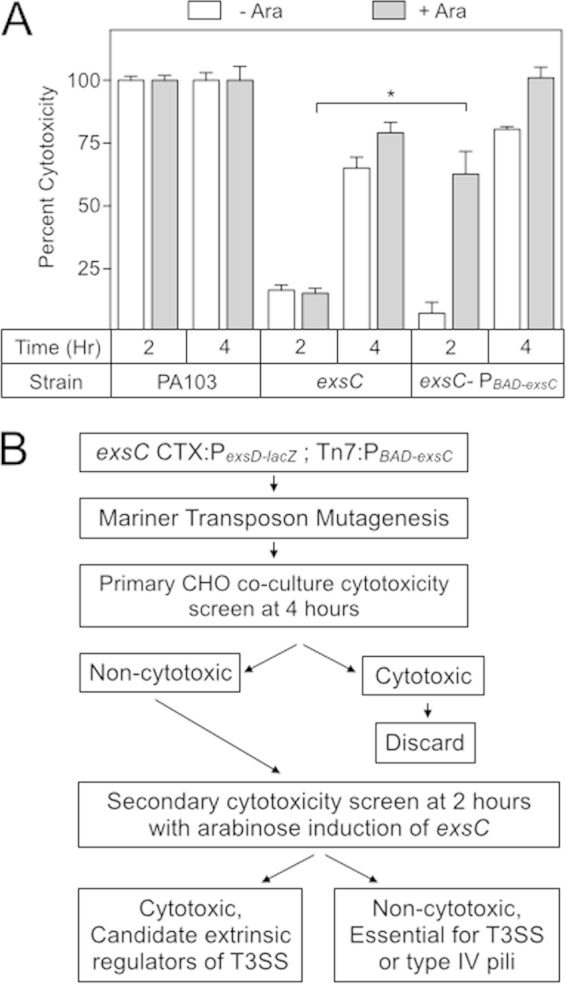
Strategy to identify extrinsic regulators of T3SS gene expression. (A) The indicated strains were cocultured with CHO cells (MOI = 10) for either 2 or 4 h at 37°C. Arabinose (0.1%) was added to induce exsC expression as indicated. The plates were then centrifuged for 5 min at 500 × g, and the coculture supernatant from each well was assayed for LDH activity. The LDH values were normalized to wt PA103 (reported as 100% cytotoxicity) for each condition. *, P < 0.001. (B) Screening strategy to identify extrinsic regulators of T3SS gene expression. PA103 exsC CTX-PexsD-lacZ:Tn7-PBAD-exsC was subjected to Mariner-based transposon mutagenesis. Individual transposon insertion mutants were assayed for cytotoxicity toward CHO cells following a 4-h coculture. Insertion mutants with <50% the activity of wt PA103 were subjected to a secondary cytotoxicity screen consisting of a 2-h coculture with CHO cells in the presence of arabinose and categorized as either cytotoxic (potential extrinsic regulators) and noncytotoxic. The transposon insertion sites were identified by inverse PCR as listed in Tables 1 and 2.
As a primary screen, the exsC-PBAD-exsC strain was subjected to Mariner-based transposon mutagenesis. Approximately 8,000 mutants were selected and screened for cytotoxicity following a 4-h coculture with CHO cells in 96-well tissue culture plates (Fig. 1B). A total of 40 transposon mutants demonstrated a >50% reduction in cytotoxicity at 4 h compared to the parental exsC-PBAD-exsC strain. Mutants were then subjected to a secondary screen by measuring cytotoxicity, following a 2-h coculture in the absence or presence of arabinose. The cytotoxicity defect of 26 transposon mutants was not restored by exsC overexpression (i.e., in the presence of arabinose). With the exception of an insertion mutant in retS, each of remaining 25 transposon insertions mapped to genes required for either the T3SS or type IV pilus biosynthesis (Table 1).
TABLE 1.
Noncytotoxic transposon insertion mutants that were not restored for cytotoxicity by ExsC expression
| PA no. | Gene | Insertion location(s)a | Protein description |
|---|---|---|---|
| PA0395 | pilT | 437474 (3) | T4P pilin biogenesis |
| PA0413 | chpA | 456452* | TCS response regulator, pilin biosynthesis chemotaxis |
| PA0652 | vfr | 706575* | Crp transcriptional regulator |
| PA1690 | pscU | 1840544* | T3SS translocation protein |
| PA1691 | pscT | 1841846 | T3SS translocation protein |
| PA1706 | pcrV | 1852823, 1853092* | T3SS secretion apparatus |
| PA1716 | pscC | 1859782* (2), 1860622, 1860902 | T3SS outer membrane protein |
| PA1723 | pscJ | 1864215*, 1864216 | T3SS export protein |
| PA1698 | popN | 1847813 | T3SS outer membrane protein |
| PA4526 | pilB | 5070795, 5071005 | T4P pilin biogenesis |
| PA4546 | pilS | 5093435* | TCS sensor, pilin biosynthesis |
| PA4554 | pilY1 | 5100979 | T4P tip-associated adhesin |
| PA4556 | pilE | 5104767 | T4P pilin biogenesis |
| PA4856 | retS | 5452585* | TCS sensor, regulator of T3SS and EPS |
| 51530 | exoU | 4581548, 4582345*, 4582694*, 4582943 | T3SS effector, phospholipase |
All open reading frame numbers and insertion locations correspond to the PAO1 genome except for exoU, which corresponds to the PA14 genome (http://www.pseudomonas.com). *, the gentamicin gene of the transposon is transcribed in a forward direction with respect to the annotated PAO1 genome (http://www.pseudomonas.com). Numbers in parentheses indicate that the transposon insertion mutant was isolated either 2 or 3 times.
Arabinose induction in the remaining 14 mutants restored cytotoxicity. As expected, none of these transposon mutants had insertions within T3SS or type IV pilus genes (Table 2). Two transposon insertions mapped to algZ, located immediately upstream of algR. It was previously shown that increased algR expression inhibits T3SS gene expression (23). Overexpression of algR likely accounts for the phenotype of the algZ insertion mutants, since an outwardly facing promoter located within the transposon reads into algR. The identification of algZ served as validation that the screen was capable of identifying extrinsic regulators of T3SS gene expression.
TABLE 2.
Noncytotoxic transposon insertion mutants restored for cytotoxicity by ExsC expression
| PA no. | Gene | Insertion location(s)a | Protein description |
|---|---|---|---|
| PA0754 | 822141 | Hypothetical | |
| PA1056 | shaC | 1146201* | Probable NADH dehydrogenase |
| PA1758 | pabB | 1898365* | para-Aminobenzoate synthase component |
| PA1802 | clpX | 1954857* | ATP-dependent protease |
| PA2840 | deaD | 3195572, 3195569* | Probable ATP-dependent RNA helicase |
| PA3800 | 4259403* | Conserved hypothetical | |
| PA3903 | prfC | 4372629* | Peptide chain release factor |
| PA3980-81 (intergenic) | 4461375 | ||
| PA4284 | recB | 4802593* | Exodeoxyribonuclease V beta chain |
| PA4945 | miaA | 5549122 | tRNA δ2-isopentenylpyrophosphate transferase |
| PA5021 | 5648942* | Probable sodium/hydrogen antiporter | |
| PA5262 | algZ | 5923414*, 5923564* | TCS sensor alginate biosynthesis, T4P |
All open reading frame numbers and insertion locations correspond to the PAO1 genome (http://www.pseudomonas.com). *, the gentamicin gene of the transposon is transcribed in a forward direction with respect to the annotated PAO1 genome (http://www.pseudomonas.com).
DeaD promotes T3SS expression.
The remainder of this study focuses on gene PA2840, identified by two independent transposon insertion events (Table 2) and recently identified in another mutagenesis screen for regulators of T3SS gene expression (46). PA2840, referred to here as deaD, encodes a homolog of Escherichia coli DeaD and is annotated as a probable ATP-dependent RNA helicase (47). To verify the phenotype of the deaD transposon insertion mutants, we constructed an in-frame deaD deletion mutant. The deaD mutant had no discernible growth phenotype when cultured in LB medium (data not shown). Similar to the transposon mutants, the deaD mutant had reduced cytotoxicity compared to wt PA103 and could be complemented by DeaD expressed in trans (Fig. 2A). To verify that the defect in cytotoxicity resulted from reduced T3SS gene expression, we measured the activity of an ExsA-dependent transcriptional reporter (PexsD-lacZ) and ExsA protein levels. Compared to wt PA103, both PexsD-lacZ reporter activity and ExsA steady-state levels were significantly reduced in the deaD mutant and again were restored by deaD expressed in trans (Fig. 2B).
FIG 2.

A PA103 deaD mutant is noncytotoxic and defective for T3SS gene expression. (A) wt PA103 and a deaD mutant carrying either a vector control (pJN105) or a DeaD expression vector (pDeaD) were coincubated with CHO cells for 90 min and then assayed for LDH release. The reported values were normalized to wt PA103 carrying the pJN105 vector control (100% cytotoxicity). (B) A mini-CTX PexsD-lacZ transcriptional reporter was introduced into wt PA103 and the exsD mutant. The resulting strains were cultured under low-Ca2+ conditions (TSB, 1 mM EGTA) in the presence of 25 mM arabinose and assayed for β-galactosidase activity and ExsA quantities by immunoblotting. The reported values were normalized to wt PA103 carrying the vector control (3,553 Miller units). *, P < 0.05; **, P < 0.005.
DeaD is the primary RNA-helicase regulating ExsA synthesis.
DeaD is annotated as a DEAD box RNA helicase, so named for the conserved Asp-Glu-Ala-Asp sequence thought to contribute to either ATPase activity and/or the RNA unwinding function (48, 49). P. aeruginosa DeaD contains a DEAD motif encompassing residues 167 to 170. To determine whether the DEAD motif is required for regulatory activity, we constructed deaD alleles with point mutations resulting in DQAD (DeaD E168Q) and DAAD (E168A) substitutions. Glutamate to glutamine/alanine substitutions within this motif were previously shown to eliminate ATP hydrolysis and helicase activity (50–52). The pDeaD E168Q and pDeaD E168A expression plasmids were introduced into the deaD mutant and assayed for their ability to restore PexsD-lacZ reporter activity. Consistent with the previous mutants in E. coli (50–52), both DeaD E168Q and DeaD E168A mutants were unable to restore PexsD-lacZ activity to levels observed with wt DeaD (Fig. 3A).
FIG 3.
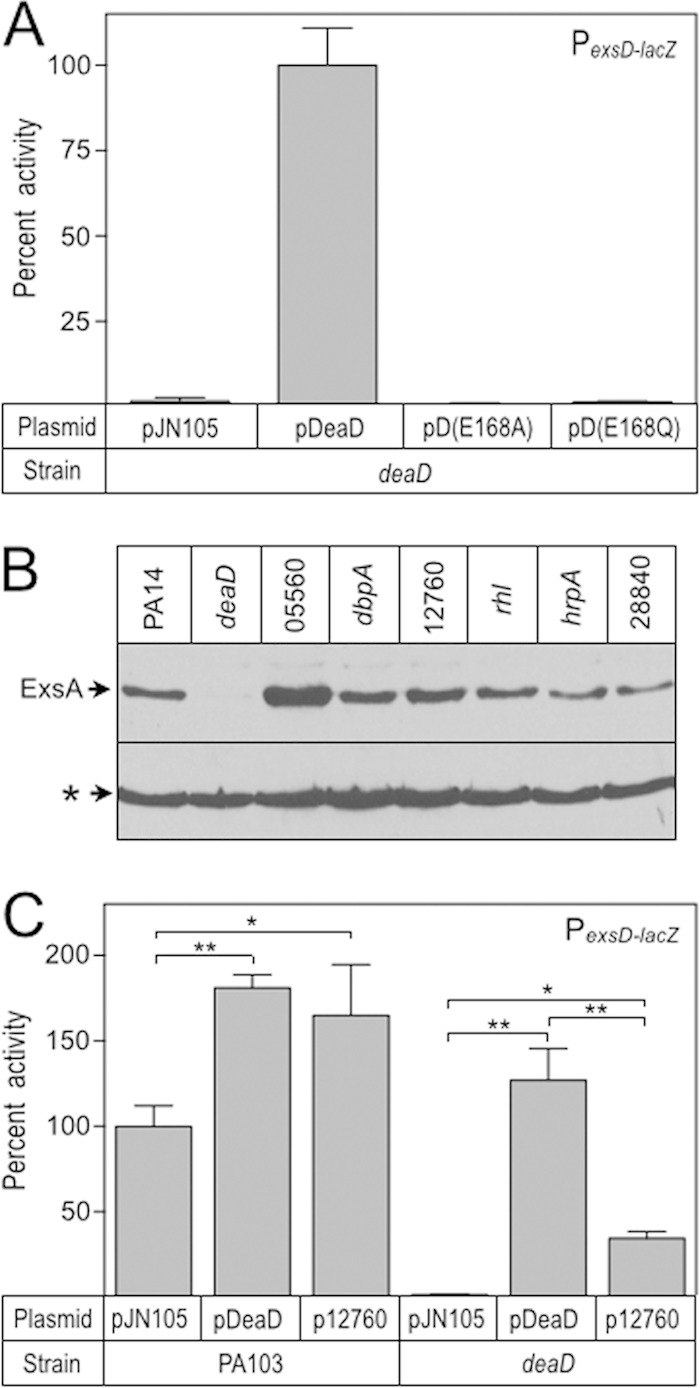
The DEAD motif is critical for T3SS gene expression. (A) The deaD E168Q and deaD E168A alleles were cloned into pJN105 and assayed for their ability to complement a deaD mutant for PexsD-lacZ reporter activity. The reported values were normalized to the deaD mutant carrying pDeaD (4,671 Miller units). (B) PA14 transposon mutants with insertion in annotated RNA-helicases were cultured under inducing conditions for T3SS gene expression and whole-cell lysates were analyzed for ExsA production by immunoblotting. A cross-reactive band (indicated with an asterisk) served as a loading control. (C) An expression plasmid for the PA103 equivalent of the PA14 12760 RNA helicase, which shares 44% identity with DeaD, was introduced into the deaD mutant. The resulting strain was assayed PexsD-lacZ reporter activity. The reported values were normalized to wt PA103 carrying pJN105 (4,133 Miller units). *, P < 0.05; **, P < 0.005.
The P. aeruginosa PA14, PAO1, and PA103 genomes contain seven putative RNA helicases with DEAD box motifs. To determine whether the observed defect in ExsA synthesis was specific to DeaD, we examined ExsA production in each of the RNA helicase transposon mutants available in the PA14 nonredundant mutant library (53). Although ExsA protein levels were elevated in the 05560 mutant and modestly decreased in the hrpA::Tn1 and 28840 mutants, the deaD insertion strain had the most severe decrease in ExsA production (Fig. 3B). To assess whether another RNA helicase could complement the PA103 deaD mutant for T3SS gene expression, we constructed an expression plasmid carrying the 12760 gene, which has the highest similarity (44%) to DeaD. Expression of the 12760 helicase partially activated PexsD-lacZ reporter activity in the deaD mutant, although not as effectively as DeaD, which restored PexsD-lacZ activity to ∼130% that of wt PA103 (Fig. 3C). The latter finding could reflect reduced expression and/or activity of 1270 relative to DeaD. Together, our data suggest that DeaD functions as a DEAD box helicase and is the primary helicase required for activation of T3SS gene expression.
DeaD-based regulation of T3SS gene expression does not involve the Gac/Rsm system.
In E. coli, DeaD posttranscriptionally regulates synthesis of the UvrY response regulator, which in turn controls expression of the small noncoding RNA CsrB (37). CsrB has multiple binding sites for the small RNA-binding protein CsrA and functions by regulating CsrA availability. By analogy, P. aeruginosa GacA activates expression of the small noncoding RNAs RsmY/Z to regulate RsmA availability (29, 54). Because RsmA is also required for T3SS gene expression, we tested the hypothesis that DeaD modulates the activity of the Gac/Rsm system to control the T3SS. We measured PrsmY-lacZ and PrsmZ-lacZ reporter activities in both wt PA103 and the deaD mutant and found no significant change in the activity of either (Fig. 4A). To verify that RsmYZ expression was not affected, we used qRT-PCR to directly measure RsmY and RsmZ RNA levels and again found no significant difference in the expression in the deaD mutant compared to wt PA103 (Fig. 4B). As a final test, we examined the activity of a tssA1′-′lacZ translation reporter, normally repressed by RsmA (24, 27), and again found no difference in activity (Fig. 4C). We conclude that DeaD does not alter RsmYZ expression or disrupt T3SS gene expression through modulation of RsmA activity.
FIG 4.

DeaD does not influence the Gac/Rsm system to control T3SS gene expression. (A) PrsmY-lacZ and PrsmZ-lacZ reporter activity was measured in wt PA103 and a deaD mutant, normalized to wt PA103 (PrsmY-lacZ = 3,620 Miller units; PrsmZ-lacZ = 544 Miller units). (B) RsmY and RsmZ sRNA quantities were measured in wt PA103 and a deaD mutant using qRT-PCR. RNA levels were normalized to the rimM housekeeping gene and reported as a percentage of wt levels. (C) PtssA1′-′lacZ activity was measured in wt PA103 and rsmA and deaD mutants. All activities were normalized to an rsmA mutant (157 CPRG units).
DeaD promotes ExsA synthesis at a posttranscriptional level by activating ExsA translation.
A recent study found that E. coli DeaD can function as a posttranscriptional regulator by relaxing secondary structures in mRNA that presumably interfere with translation initiation (37). Based on our findings that ExsA synthesis is reduced in a deaD mutant and that DeaD likely functions as an RNA helicase, we hypothesized that DeaD regulates ExsA synthesis at a posttranscriptional level through a similar mechanism. ExsA is encoded by the last gene of the exsCEBA operon and autoregulates its own expression through the PexsC promoter. Disruption of ExsA autoregulation, therefore, is necessary to analyze posttranscriptional regulation of ExsA synthesis. To eliminate the complications of positive-feedback control, we took advantage of the previously described UY339 strain in which the native PexsC promoter has been replaced with a constitutive PlacUV5 promoter (23). The deaD mutation was introduced into the UY339 background and the PexsD-lacZ reporter activity was measured in strains carrying an empty vector or the DeaD complementation plasmid. The UY339 deaD mutant had a significant reduction in PexsD-lacZ reporter activity that was complemented by pDeaD (Fig. 5A). To determine whether the defect was specific to ExsA, we measured ExsC and ExsA protein levels via immunoblot. Whereas ExsC synthesis remained consistent across all strain backgrounds, ExsA synthesis was significantly reduced in the UY339 deaD mutant and restored upon complementation with pDeaD (Fig. 5A). Together, these data suggest that DeaD activates ExsA synthesis at a posttranscriptional level.
FIG 5.
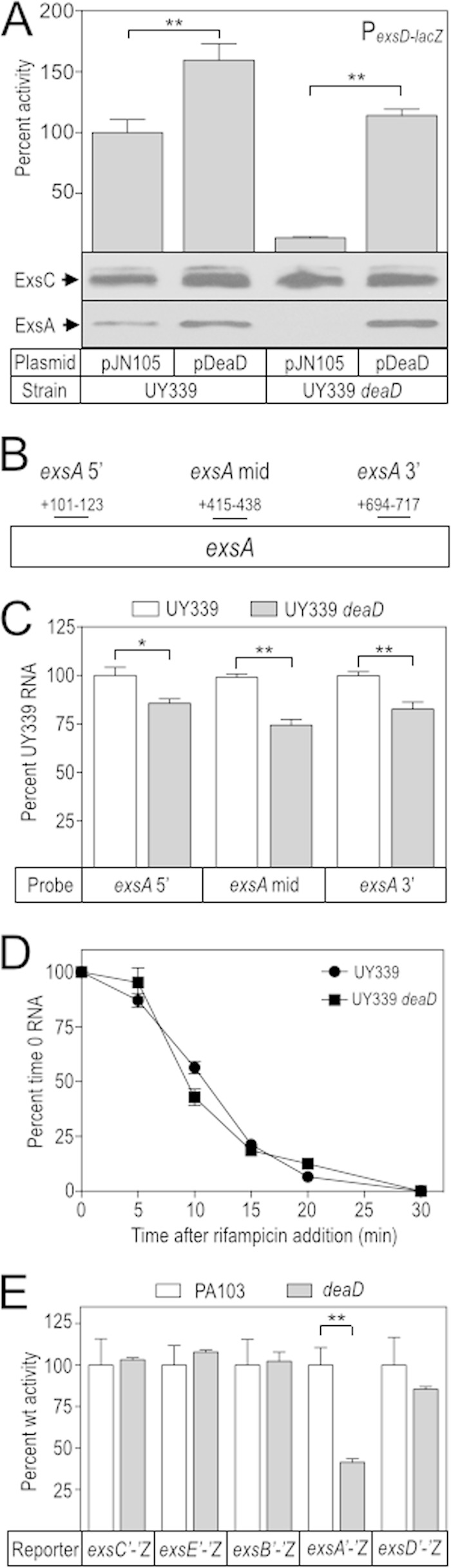
Dead controls ExsA synthesis at a posttranscriptional level. (A) The UY339 and UY339 deaD strains were cultured under inducing conditions for T3SS gene expression in the presence of 25 mM arabinose, and assayed for PexsD-lacZ reporter activity and ExsC and ExsA protein levels. The reported values were normalized to UY339 carrying pJN105 (389 Miller units). (B and C) Total RNA was harvested from the UY339 and UY339 deaD strains and exsA mRNA levels were measured at three independent locations within the ORF as indicated in panel B. We also measured rimM mRNA levels as an internal standard. The values reported in panel C were normalized to UY339 (100%) for each of the exsA probes. (D) The UY339 and UY339 deaD strains were cultured under inducing conditions for T3SS gene expression. When the culture A600 reached 1.0, the cells were treated with 200 μg of rifampin/ml, and RNA samples were then collected every 5 min over a 30-min period. The levels of exsA mRNA were measured using the exsA 5′ probe (B). The reported values were normalized to UY339 at time zero (100%). (E) exsC, exsE, exsB, exsA, and exsD. Expression of the reporters was controlled by a constitutive lacUV5 promoter, and the activity of each was determined in the wt PA103 and deaD backgrounds. The reported values were normalized to the activity of wt PA103 (100%) and reported as CPRG units: exsC′-′lacZ = 648, exsCE′-′lacZ = 100, exsCEB′-′lacZ = 384, exsCEBA′-′lacZ = 287, and exsD′-′lacZ = 306. *, P < 0.05; **, P < 0.005.
DEAD box helicases can function as components of the RNA-degradosome and are important in maintaining RNA homeostasis (38). For this reason, we tested the hypothesis that the stability of the exsA portion of the exsCEBA mRNA is altered in the deaD mutant. As an initial test, we designed quantitative reverse transcriptase PCR (qPCR) primer/probe pairs for three different sites in the exsA mRNA and measured RNA levels in the UY339 and UY339 deaD backgrounds (Fig. 5B and C). Each primer/probe pair revealed a modest decrease in exsA mRNA in the UY339 deaD mutant (Fig. 5C). As a second test of the hypothesis, we collected RNA samples from the UY339 and UY339 deaD strains over a 30 min time course following treatment with rifampin to inhibit further transcription. The exsA mRNA decay rates were similar in both the UY339 and UY339 deaD strains (Fig. 5D). Based on these results, we conclude that reduced stability of the exsA mRNA does not account for DeaD-mediated regulation of ExsA synthesis.
An alternative hypothesis to account for the deaD mutant phenotype is that DeaD helicase activity relaxes an inhibitory mRNA secondary structure to promote exsA translation. To test this hypothesis, we integrated a full-length exsCEBA′-′lacZ translational reporter driven from a constitutive PlacUV5 promoter at the chromosomal CTX phage attachment site. The exsCEBA′-′lacZ translational reporter demonstrated a 60% reduction in the deaD mutant (Fig. 5E). To verify that the translational defect was limited to ExsA, we measured the activity of lacZ translational fusions to exsC, exsE, exsB, and exsD. Each of the control reporters had similar activities in both PA103 and the deaD mutant (Fig. 5E), supporting the conclusion that DeaD specifically promotes exsA translation.
DeaD-dependent activation of ExsA translation is specific to the exsA coding sequence and native Shine-Dalgarno sequence.
Based on our finding that DeaD promotes ExsA translation, we examined the sequence requirements for DeaD-mediated control. ExsA complementation plasmids carrying different portions of the exsA upstream region (Fig. 6A) were introduced into the exsA and deaD, exsA mutants and PexsD-lacZ reporter activity was assayed. Whereas ExsA expressed from plasmids pCEBA-1 (the entire native operon) and pExsA (possessing 37 bp of the upstream untranslated region) complemented the exsA mutant for PexsD-lacZ reporter activity, both plasmids failed to fully restore activity in the deaD exsA double mutant (Fig. 6B). In contrast, pRBS-ExsA, which replaces the native 37-nt untranslated region (UTR)/Shine-Dalgarno from exsA with the 14-bp UTR/Shine-Dalgarno sequence from exsC, fully complemented PexsD-lacZ activity in both exsA and deaD exsA backgrounds (Fig. 6B). Since extrinsic control of T3SS gene expression often influences ExsA synthesis, a common finding is that increasing ExsA activity restores T3SS gene expression in mutants with defects in extrinsic control (55–59). For this reason, the possibility exists that exsA overexpression nonspecifically bypasses the deaD requirement. Nevertheless, we do not feel this is the case because both the pCEBA-1 and pRBS-ExsA expression plasmids generated similar levels of PexsD-lacZ reporter activity (7,496 and 7,925 Miller units, respectively), and yet only expression from the pCEBA-1 plasmid was subject to DeaD-mediated control. We conclude that while most of the sequence upstream of the exsA ORF is dispensable for DeaD-based regulation, the 37-nt leader sequence is required.
FIG 6.
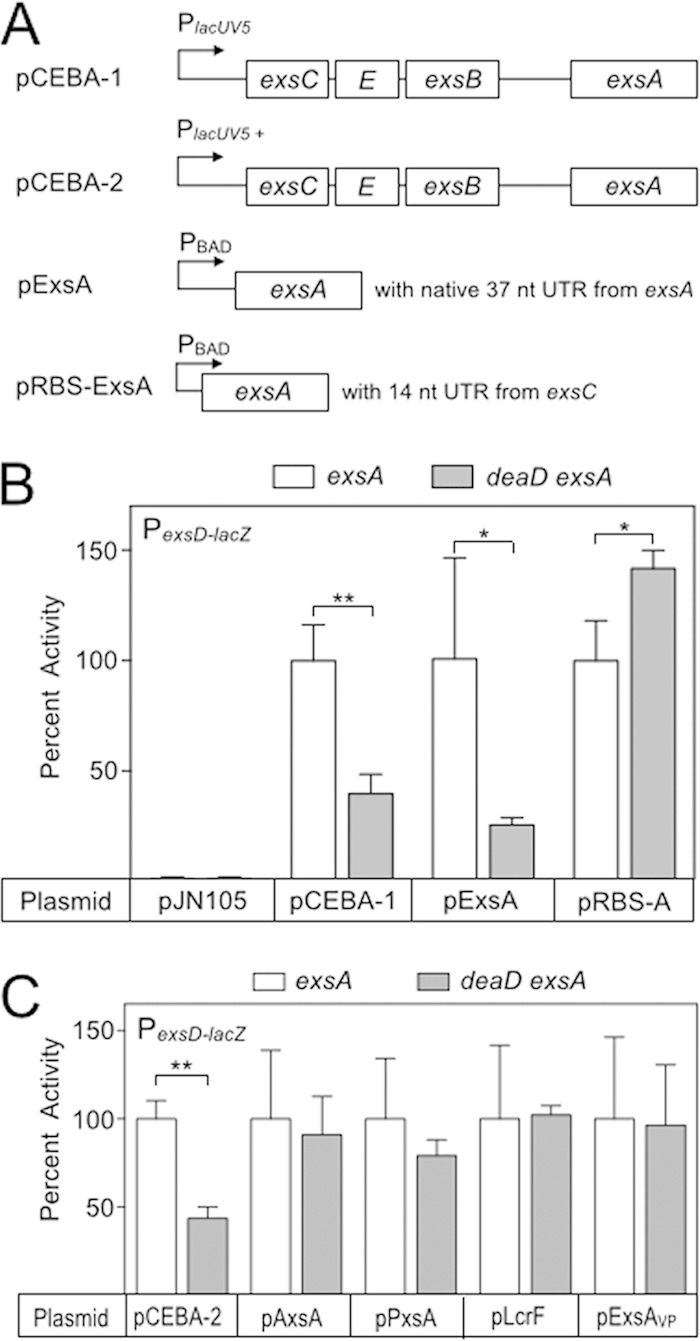
Sequence requirements for DeaD-mediated control of ExsA synthesis. (A) Diagram of the ExsA complementation vectors used for these experiments. pCEBA-1 (p2UY75A) contains the entire exsCEBA operon driven by a constitutive PlacUV5 promoter. pExsA (pEB124) is arabinose inducible and contains 37 nt of the exsA untranslated leader sequence. pRBS-ExsA is arabinose inducible and replaces the native exsA untranslated region with the 14-nt untranslated region from exsC. (B) PA103 exsA and deaD, exsA mutants were transformed with the indicated expression vectors. The resulting strains were cultured under inducing conditions for T3SS gene expression and assayed for PexsD-lacZ reporter activity. The reported values were normalized to the exsA mutant carrying each of the expression plasmids (pJN105 = 34 Miller units, pCEBA-1 = 7,496 Miller units; pExsA = 381 Miller units; pRBS-ExsA = 7,926 Miller units). (C) The exsA and deaD exsA mutants were transformed with plasmids expressing exsA homologs from A. hydrophila (pAxsA), P. luminescens (pPxsA), Y. pestis (pLcrF), and V. parahaemolyticus (pExsAVp). The resulting strains were cultured under inducing conditions for T3SS gene expression and assayed for PexsD-lacZ reporter activity. The reported activities (Miller units) were normalized to the exsA mutant carrying each plasmid (pCEBA-2 = 13,025; pAxsA = 15,447; pPxsA = 4,537; pLcrF = 18,950; pVxsA = 9,903). *, P < 0.05; **, P < 0.005.
Having established that the untranslated region upstream of exsA contributes DeaD-dependent regulation, we next examined whether the exsA coding sequence was required. We took advantage of the fact that ExsA orthologs from Aeromonas hydrophila (AxsA), Photorhabdus luminescens (PxsA), Yersinia pestis (LcrF), and Vibrio parahaemolyticus (ExsAVp) each complement an exsA mutant for PexsD-lacZ reporter activity (60, 61). Although these proteins are highly conserved in their C-terminal DNA-binding domains, their DNA sequences are less conserved ranging from 67 to 75% sequence identity relative to exsA. Each of the genes was placed under the translational control of the 37-nt leader sequence from exsA. As a control for these experiments, we use plasmid pCEBA-2, which contains the entire exsCEBA operon under the transcriptional control of a stronger PlacUV5 promoter, again owing to concerns that exsA overexpression might bypass the DeaD requirement. PexsD-lacZ reporter activity was reduced ∼2-fold in the deaD, exsA double mutant when complemented with pCEBA-2. In contrast, expression of axsA, pxsA, lcrF, and exsAVp in trans restored PexsD-lacZ reporter activity to similar levels in both exsA and deaD exsA mutants (Fig. 6C). These findings suggest that a specific sequence located within the exsA open reading frame contributes to DeaD-dependent control.
DeaD promotes ExsA translation in vitro.
To verify our in vivo findings, we purified DeaDHis and DeaDHis E168A by Ni2+-affinity chromatography (Fig. 7A). To verify that the N-terminal His tag does not interfere with DeaD function, we tested the DeaDHis alleles for their ability to complement a deaD mutant for PexsD-lacZ activity. The DeaDHis protein restored PexsD-lacZ reporter activity in wt PA103, albeit to a lesser degree than untagged DeaD (Fig. 7B). As expected, DeaDHis E168A was unable to restore PexsD-lacZ activity in the deaD mutant, which is consistent with our previous data using the untagged E168A allele (Fig. 7B). Importantly, DeaDHis and DeaDHis E168A were expressed at similar levels when examined by immunoblotting (Fig. 7B).
FIG 7.

Purified DeaDHis promotes ExsA synthesis in vitro. (A) Purified DeaDHis and DeaDHis E168A were analyzed via SDS-PAGE, followed by Coomassie blue staining, and were estimated to be >90% homogeneous. (B) The deaD mutant was transformed with expression vectors encoding untagged DeaD, DeaDHis, or DeaDHis E168A. The resulting strains were cultured under inducing conditions for T3SS gene expression and assayed for PexsD-lacZ reporter activity and DeaDHis production by immunoblotting for the histidine tag. The reported values were normalized to the deaD mutant expressing untagged DeaD (6,230 Miller units). (C) In vitro synthesis reaction mixtures lacking template (lane 1) or containing the exsA template (lanes 2 to 7) were incubated in the absence or presence of the indicated concentrations of DeaDHis E168A (lane 3) or DeaDHis (lanes 4 to 7). Detection was based on incorporation of radiolabeled methionine and phosphorimaging. (D) In vitro synthesis reactions were performed as described above using lcrF or vfr templates. *, P < 0.05; **, P < 0.005.
We next determined whether purified DeaDHis stimulates ExsA synthesis in vitro using a coupled transcription-translation system derived from E. coli. The PCR-generated template consisted of a T7 promoter driving transcription of exsA that was translated from the native Shine-Dalgarno sequence (equivalent to pExsA in Fig. 6A). The expected 31-kDa product for ExsA was absent in reaction mixtures lacking template and present when template was included (Fig. 7C, lanes 1 versus lane 2). Addition of increasing amounts of purified DeaDHis resulted in a dose-dependent increase in ExsA synthesis with maximal stimulation consistently in the 2- to 3-fold range (Fig. 7C, lanes 4 to 7). As a control, we also found that the DeaDHis E168A mutant lacked stimulatory activity (Fig. 7C, lane 3). To rule out the possibility that DeaD nonspecifically stimulates translation from any mRNA, we performed in vitro synthesis reactions with templates for LcrF, which was not subject to DeaD-mediated regulation in vivo (Fig. 6C) and Vfr, a regulator in the CRP family. In both cases, DeaD addition had no effect on synthesis (Fig. 7D). Together, these data demonstrate that DeaD promotes ExsA synthesis at a posttranscriptional level both in vitro and in vivo.
DISCUSSION
While intrinsic control of T3SS gene expression by the ExsECDA cascade is well described, extrinsic regulatory mechanisms are poorly understood. By screening for extrinsic regulators, we identified the DeaD RNA helicase as a novel regulator of T3SS gene expression. Disruption of deaD by either transposon insertion or deletion results in reduced T3SS gene expression and cytotoxicity toward CHO cells (Fig. 2A and B). The underlying defect in the deaD mutant occurs at a posttranscriptional level and specifically reduces exsA translation (Fig. 5E), while little effect on mRNA stability (Fig. 5C and D). Furthermore, alterations of the native exsA Shine-Dalgarno sequence and/or coding sequence relieve the DeaD requirement in vivo (Fig. 6B and C). Finally, purified DeaD activated ExsA synthesis in vitro, whereas DeaD E168A lacked stimulatory activity (Fig. 7C). These data support a model wherein DeaD relieves an inhibitory structure within the exsA mRNA that normally prevents exsA translation. This proposed mechanism is bolstered by the recent findings that E. coli DeaD promotes uvrY translation by relaxing duplex RNA that interferes with ribosomal recruitment (37). The inhibitory duplex RNA results from base pairing between the uvrY untranslated leader region and the proximal coding sequence. Interestingly, RNA folding predictions using the 37-nt exsA untranslated region (required for DeaD-mediated control [Fig. 5]), along with the exsA proximal coding sequence, reveals extensive base pairing with the Shine-Dalgarno sequence (Fig. 8). It seems plausible that the ribosomal access to this region could be enhanced by DeaD. In addition to DeaD, RsmA also stimulates ExsA synthesis at a posttranscriptional level (23, 46). Whether DeaD and RsmA activities function independently or are dependent upon one another will be the subject of future studies.
FIG 8.

Predicted structure of the exsA mRNA region encompassing the 37-nt untranslated region (lowercase) and a portion of the coding sequence (uppercase) as determined using mFOLD. The predicted Shine-Dalgarno sequence is boxed in red, and the AUG start codon is boxed in blue.
Although deaD is essential for T3SS gene expression, expression of the exsCEBA′-′lacZ translational reporter was only reduced ∼2-fold in vivo (Fig. 5E), and the addition of purified DeaD resulted in only a 2- to 3-fold stimulation of ExsA synthesis in vitro (Fig. 7C). Although such effects are seemingly modest, a similar 2-fold decrease in exsCEBA′-′lacZ reporter activity was previously observed in mucA and rsmA mutant backgrounds as well (23). This 2-fold reduction in ExsA translation is adequate to generate T3SS-defective phenotypes in all three mutants (deaD, mucA, and rsmA). The most likely explanations for this is that ExsA autoregulates its own expression through the PexsC promoter and is also subject to negative regulation by its antiactivator ExsD, expression of which is positively controlled by ExsA. Also important to note is that P. aeruginosa T3SS gene expression is bistable, i.e., only a fraction of the cells in a population express the T3SS under inducing conditions (13, 62, 63). These combined observations indicate that intrinsic control by the ExsECDA regulatory cascade is finely balanced and that small changes in the expression and/or activity of any one of the components, including ExsA, can shift the equilibrium resulting in a defect in T3SS gene expression.
In addition to DeaD, our screen identified other potential regulators of cellular toxicity. Based on the design of the screen, the lack of cellular toxicity most likely reflects reduced T3SS gene expression. Most of the genes identified in the screen, however, have no obvious connection to T3SS gene regulation. The miaA insertion mutant is worthy of comment because hfq is located immediately downstream of miaA and in this arrangement may result in hfq overexpression due to the outward-facing promoter in the transposon. Hfq is an RNA-binding protein that facilitates gene expression through either direct interaction with mRNAs or through its sRNA chaperone function (64). Hfq is known to protect RsmY against the degradation (30). This suggests a potential mechanism wherein hfq overexpression could result in increased intracellular concentrations of RsmY, a corresponding decrease in RsmA availability and thus reduced T3SS gene expression.
Many extrinsic regulators of T3SS gene expression, including the CVS and Gac/Rsm systems, also function as global regulators of P. aeruginosa gene expression (22, 27, 65). Although DeaD is required for T3SS gene expression, further studies are required to determine whether this falls under the domain of a simple housekeeping function or a bona fide regulatory function. Only recently has global regulation by E. coli DeaD been investigated by using HITS-CLIP (high-throughput sequencing of RNA isolated by cross-linking immunoprecipitation) (37). This approach identified many targets, one of which was RpoS, itself a global regulator of >100 genes (66). Defining the extent of the P. aeruginosa DeaD regulon will important question for the future.
Supplementary Material
ACKNOWLEDGMENT
Work in the Yahr and Wolfgang laboratories is supported by the National Institutes of Health (AI097264 to M.C.W. and T.L.Y.).
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.00231-15.
REFERENCES
- 1.Bodey GP, Bolivar R, Fainstein V, Jadeja L. 1983. Infections caused by Pseudomonas aeruginosa. Rev Infect Dis 5:279–313. doi: 10.1093/clinids/5.2.279. [DOI] [PubMed] [Google Scholar]
- 2.Hauser AR. 2009. The type III secretion system of Pseudomonas aeruginosa: infection by injection. Nat Rev Microbiol 7:654–665. doi: 10.1038/nrmicro2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Engel J, Balachandran P. 2009. Role of Pseudomonas aeruginosa type III effectors in disease. Curr Opin Microbiol 12:61–66. doi: 10.1016/j.mib.2008.12.007. [DOI] [PubMed] [Google Scholar]
- 4.Chatterjee S, Chaudhury S, McShan AC, Kaur K, De Guzman RN. 2013. Structure and biophysics of type III secretion in bacteria. Biochemistry 52:2508–2517. doi: 10.1021/bi400160a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pukatzki S, Kessin RH, Mekalanos JJ. 2002. The human pathogen Pseudomonas aeruginosa utilizes conserved virulence pathways to infect the social amoeba Dictyostelium discoideum. Proc Natl Acad Sci U S A 99:3159–3164. doi: 10.1073/pnas.052704399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Miyata S, Casey M, Frank DW, Ausubel FM, Drenkard E. 2003. Use of the Galleria mellonella caterpillar as a model host to study the role of the type III secretion system in Pseudomonas aeruginosa pathogenesis. Infect Immun 71:2404–2413. doi: 10.1128/IAI.71.5.2404-2413.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brannon MK, Davis JM, Mathias JR, Hall CJ, Emerson JC, Crosier PS, Huttenlocher A, Ramakrishnan L, Moskowitz SM. 2009. Pseudomonas aeruginosa type III secretion system interacts with phagocytes to modulate systemic infection of zebrafish embryos. Cell Microbiol 11:755–768. doi: 10.1111/j.1462-5822.2009.01288.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Diaz MR, King JM, Yahr TL. 2011. Intrinsic and extrinsic regulation of type III secretion gene expression in Pseudomonas aeruginosa. Front Microbiol 2:89. doi: 10.3389/fmicb.2011.00089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Frank DW. 1997. The exoenzyme S regulon of Pseudomonas aeruginosa. Mol Microbiol 26:621–629. doi: 10.1046/j.1365-2958.1997.6251991.x. [DOI] [PubMed] [Google Scholar]
- 10.Hayes CS, Aoki SK, Low DA. 2010. Bacterial contact-dependent delivery systems. Annu Rev Genet 44:71–90. doi: 10.1146/annurev.genet.42.110807.091449. [DOI] [PubMed] [Google Scholar]
- 11.Vallis AJ, Yahr TL, Barbieri JT, Frank DW. 1999. Regulation of ExoS production and secretion by Pseudomonas aeruginosa in response to tissue culture conditions. Infect Immun 67:914–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim J, Ahn K, Min S, Jia J, Ha U, Wu D, Jin S. 2005. Factors triggering type III secretion in Pseudomonas aeruginosa. Microbiology 151:3575–3587. doi: 10.1099/mic.0.28277-0. [DOI] [PubMed] [Google Scholar]
- 13.Urbanowski ML, Brutinel ED, Yahr TL. 2007. Translocation of ExsE into Chinese hamster ovary cells is required for transcriptional induction of the Pseudomonas aeruginosa type III secretion system. Infect Immun 75:4432–4439. doi: 10.1128/IAI.00664-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McCaw ML, Lykken GL, Singh PK, Yahr TL. 2002. ExsD is a negative regulator of the Pseudomonas aeruginosa type III secretion regulon. Mol Microbiol 46:1123–1133. doi: 10.1046/j.1365-2958.2002.03228.x. [DOI] [PubMed] [Google Scholar]
- 15.Brutinel ED, Yahr TL. 2008. Control of gene expression by type III secretory activity. Curr Opin Microbiol 11:128–133. doi: 10.1016/j.mib.2008.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brutinel ED, Vakulskas CA, Yahr TL. 2010. ExsD inhibits expression of the Pseudomonas aeruginosa type III secretion system by disrupting ExsA self-association and DNA binding activity. J Bacteriol 192:1479–1486. doi: 10.1128/JB.01457-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zheng Z, Chen G, Joshi S, Brutinel ED, Yahr TL, Chen L. 2007. Biochemical characterization of a regulatory cascade controlling transcription of the Pseudomonas aeruginosa type III secretion system. J Biol Chem 282:6136–6142. [DOI] [PubMed] [Google Scholar]
- 18.Rietsch A, Vallet-Gely I, Dove SL, Mekalanos JJ. 2005. ExsE, a secreted regulator of type III secretion genes in Pseudomonas aeruginosa. Proc Natl Acad Sci U S A 102:8006–8011. doi: 10.1073/pnas.0503005102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Urbanowski ML, Lykken GL, Yahr TL. 2005. A secreted regulatory protein couples transcription to the secretory activity of the Pseudomonas aeruginosa type III secretion system. Proc Natl Acad Sci U S A 102:9930–9935. doi: 10.1073/pnas.0504405102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dasgupta N, Lykken GL, Wolfgang MC, Yahr TL. 2004. A novel anti anti-activator mechanism regulates expression of the Pseudomonas aeruginosa type III secretion system. Mol Microbiol 53:297–308. doi: 10.1111/j.1365-2958.2004.04128.x. [DOI] [PubMed] [Google Scholar]
- 21.Lykken GL, Chen G, Brutinel ED, Chen L, Yahr TL. 2006. Characterization of ExsC and ExsD self-association and heterocomplex formation. J Bacteriol 188:6832–6840. doi: 10.1128/JB.00884-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wolfgang MC, Lee VT, Gilmore ME, Lory S. 2003. Coordinate regulation of bacterial virulence genes by a novel adenylate cyclase-dependent signaling pathway. Dev Cell 4:253–263. doi: 10.1016/S1534-5807(03)00019-4. [DOI] [PubMed] [Google Scholar]
- 23.Intile PJ, Diaz MR, Urbanowski ML, Wolfgang MC, Yahr TL. 2014. The AlgZR two-component system recalibrates the RsmAYZ posttranscriptional regulatory system to inhibit expression of the Pseudomonas aeruginosa type III secretion system. J Bacteriol 196:357–366. doi: 10.1128/JB.01199-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Marden JN, Diaz MR, Walton WG, Gode CJ, Betts L, Urbanowski ML, Redinbo MR, Yahr TL, Wolfgang MC. 2013. An unusual CsrA family member operates in series with RsmA to amplify posttranscriptional responses in Pseudomonas aeruginosa. Proc Natl Acad Sci U S A 110:15055–15060. doi: 10.1073/pnas.1307217110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Morris ER, Hall G, Li C, Heeb S, Kulkarni RV, Lovelock L, Silistre H, Messina M, Camara M, Emsley J, Williams P, Searle MS. 2013. Structural rearrangement in an RsmA/CsrA ortholog of Pseudomonas aeruginosa creates a dimeric RNA-binding protein, RsmN. Structure 21:1659–1671. doi: 10.1016/j.str.2013.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pessi G, Williams F, Hindle Z, Heurlier K, Holden MT, Camara M, Haas D, Williams P. 2001. The global posttranscriptional regulator RsmA modulates production of virulence determinants and N-acylhomoserine lactones in Pseudomonas aeruginosa. J Bacteriol 183:6676–6683. doi: 10.1128/JB.183.22.6676-6683.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brencic A, Lory S. 2009. Determination of the regulon and identification of novel mRNA targets of Pseudomonas aeruginosa RsmA. Mol Microbiol 72:612–632. doi: 10.1111/j.1365-2958.2009.06670.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Timmermans J, Van Melderen L. 2010. Post-transcriptional global regulation by CsrA in bacteria. Cell Mol Life Sci 67:2897–2908. doi: 10.1007/s00018-010-0381-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kay E, Humair B, Denervaud V, Riedel K, Spahr S, Eberl L, Valverde C, Haas D. 2006. Two GacA-dependent small RNAs modulate the quorum-sensing response in Pseudomonas aeruginosa. J Bacteriol 188:6026–6033. doi: 10.1128/JB.00409-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sorger-Domenigg T, Sonnleitner E, Kaberdin VR, Blasi U. 2007. Distinct and overlapping binding sites of Pseudomonas aeruginosa Hfq and RsmA proteins on the noncoding RNA RsmY. Biochem Biophys Res Commun 352:769–773. doi: 10.1016/j.bbrc.2006.11.084. [DOI] [PubMed] [Google Scholar]
- 31.Heurlier K, Williams F, Heeb S, Dormond C, Pessi G, Singer D, Camara M, Williams P, Haas D. 2004. Positive control of swarming, rhamnolipid synthesis, and lipase production by the posttranscriptional RsmA/RsmZ system in Pseudomonas aeruginosa PAO1. J Bacteriol 186:2936–2945. doi: 10.1128/JB.186.10.2936-2945.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mathee K, McPherson CJ, Ohman DE. 1997. Posttranslational control of the algT (algU)-encoded σ22 for expression of the alginate regulon in Pseudomonas aeruginosa and localization of its antagonist proteins MucA and MucB (AlgN). J Bacteriol 179:3711–3720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Boucher JC, Yu H, Mudd MH, Deretic V. 1997. Mucoid Pseudomonas aeruginosa in cystic fibrosis: characterization of muc mutations in clinical isolates and analysis of clearance in a mouse model of respiratory infection. Infect Immun 65:3838–3846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ohman DE, Chakrabarty AM. 1981. Genetic mapping of chromosomal determinants for the production of the exopolysaccharide alginate in a Pseudomonas aeruginosa cystic fibrosis isolate. Infect Immun 33:142–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jones AK, Fulcher NB, Balzer GJ, Urbanowski ML, Pritchett CL, Schurr MJ, Yahr TL, Wolfgang MC. 2010. Activation of the Pseudomonas aeruginosa AlgU regulon through mucA mutation inhibits cyclic AMP/Vfr signaling. J Bacteriol 192:5709–5717. doi: 10.1128/JB.00526-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wu W, Badrane H, Arora S, Baker HV, Jin S. 2004. MucA-mediated coordination of type III secretion and alginate synthesis in Pseudomonas aeruginosa. J Bacteriol 186:7575–7585. doi: 10.1128/JB.186.22.7575-7585.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vakulskas CA, Pannuri A, Cortes-Selva D, Zere TR, Ahmer BM, Babitzke P, Romeo T. 2014. Global effects of the DEAD-box RNA helicase DeaD (CsdA) on gene expression over a broad range of temperatures. Mol Microbiol 92:945–958. doi: 10.1111/mmi.12606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kaberdin VR, Blasi U. 2013. Bacterial helicases in posttranscriptional control. Biochim Biophys Acta 1829:878–883. doi: 10.1016/j.bbagrm.2012.12.005. [DOI] [PubMed] [Google Scholar]
- 39.Butland G, Peregrin-Alvarez JM, Li J, Yang W, Yang X, Canadien V, Starostine A, Richards D, Beattie B, Krogan N, Davey M, Parkinson J, Greenblatt J, Emili A. 2005. Interaction network containing conserved and essential protein complexes in Escherichia coli. Nature 433:531–537. doi: 10.1038/nature03239. [DOI] [PubMed] [Google Scholar]
- 40.Hoang TT, Karkhoff-Schweizer RR, Kutchma AJ, Schweizer HP. 1998. A broad-host-range Flp-FRT recombination system for site-specific excision of chromosomally located DNA sequences: application for isolation of unmarked Pseudomonas aeruginosa mutants. Gene 212:77–86. doi: 10.1016/S0378-1119(98)00130-9. [DOI] [PubMed] [Google Scholar]
- 41.Hoang TT, Kutchma AJ, Becher A, Schweizer HP. 2000. Integration-proficient plasmids for Pseudomonas aeruginosa: site-specific integration and use for engineering of reporter and expression strains. Plasmid 43:59–72. doi: 10.1006/plas.1999.1441. [DOI] [PubMed] [Google Scholar]
- 42.Goodman AL, Kulasekara B, Rietsch A, Boyd D, Smith RS, Lory S. 2004. A signaling network reciprocally regulates genes associated with acute infection and chronic persistence in Pseudomonas aeruginosa. Dev Cell 7:745–754. doi: 10.1016/j.devcel.2004.08.020. [DOI] [PubMed] [Google Scholar]
- 43.Wong SM, Mekalanos JJ. 2000. Genetic footprinting with mariner-based transposition in Pseudomonas aeruginosa. Proc Natl Acad Sci U S A 97:10191–10196. doi: 10.1073/pnas.97.18.10191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dasgupta N, Ashare A, Hunninghake GW, Yahr TL. 2006. Transcriptional induction of the Pseudomonas aeruginosa type III secretion system by low Ca2+ and host cell contact proceeds through two distinct signaling pathways. Infect Immun 74:3334–3341. doi: 10.1128/IAI.00090-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sundin C, Wolfgang MC, Lory S, Forsberg A, Frithz-Lindsten E. 2002. Type IV pili are not specifically required for contact-dependent translocation of exoenzymes by Pseudomonas aeruginosa. Microb Pathog 33:265–277. doi: 10.1006/mpat.2002.0534. [DOI] [PubMed] [Google Scholar]
- 46.Li K, Xu C, Jin Y, Sun Z, Liu C, Shi J, Chen G, Chen R, Jin S, Wu W. 2013. SuhB is a regulator of multiple virulence genes and essential for pathogenesis of Pseudomonas aeruginosa. mBio 4:e00419-13. doi: 10.1128/mBio.00419-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Winsor GL, Lam DK, Fleming L, Lo R, Whiteside MD, Yu NY, Hancock RE, Brinkman FS. 2011. Pseudomonas Genome Database: improved comparative analysis and population genomics capability for Pseudomonas genomes. Nucleic Acids Res 39:D596–D600. doi: 10.1093/nar/gkq869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Toone WM, Rudd KE, Friesen JD. 1991. deaD, a new Escherichia coli gene encoding a presumed ATP-dependent RNA helicase, can suppress a mutation in rpsB, the gene encoding ribosomal protein S2. J Bacteriol 173:3291–3302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Py B, Higgins CF, Krisch HM, Carpousis AJ. 1996. A DEAD-box RNA helicase in the Escherichia coli RNA degradosome. Nature 381:169–172. doi: 10.1038/381169a0. [DOI] [PubMed] [Google Scholar]
- 50.Turner AM, Love CF, Alexander RW, Jones PG. 2007. Mutational analysis of the Escherichia coli DEAD box protein CsdA. J Bacteriol 189:2769–2776. doi: 10.1128/JB.01509-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cordin O, Banroques J, Tanner NK, Linder P. 2006. The DEAD-box protein family of RNA helicases. Gene 367:17–37. doi: 10.1016/j.gene.2005.10.019. [DOI] [PubMed] [Google Scholar]
- 52.Tamura M, Kers JA, Cohen SN. 2012. Second-site suppression of RNase E essentiality by mutation of the deaD RNA helicase in Escherichia coli. J Bacteriol 194:1919–1926. doi: 10.1128/JB.06652-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Liberati NT, Urbach JM, Miyata S, Lee DG, Drenkard E, Wu G, Villanueva J, Wei T, Ausubel FM. 2006. An ordered, nonredundant library of Pseudomonas aeruginosa strain PA14 transposon insertion mutants. Proc Natl Acad Sci U S A 103:2833–2838. doi: 10.1073/pnas.0511100103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Brencic A, McFarland KA, McManus HR, Castang S, Mogno I, Dove SL, Lory S. 2009. The GacS/GacA signal transduction system of Pseudomonas aeruginosa acts exclusively through its control over the transcription of the RsmY and RsmZ regulatory small RNAs. Mol Microbiol 73:434–445. doi: 10.1111/j.1365-2958.2009.06782.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dacheux D, Attree I, Toussaint B. 2001. Expression of ExsA in trans confers type III secretion system-dependent cytotoxicity on noncytotoxic Pseudomonas aeruginosa cystic fibrosis isolates. Infect Immun 69:538–542. doi: 10.1128/IAI.69.1.538-542.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dacheux D, Epaulard O, de Groot A, Guery B, Leberre R, Attree I, Polack B, Toussaint B. 2002. Activation of the Pseudomonas aeruginosa type III secretion system requires an intact pyruvate dehydrogenase aceAB operon. Infect Immun 70:3973–3977. doi: 10.1128/IAI.70.7.3973-3977.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Smith RS, Wolfgang MC, Lory S. 2004. An adenylate cyclase-controlled signaling network regulates Pseudomonas aeruginosa virulence in a mouse model of acute pneumonia. Infect Immun 72:1677–1684. doi: 10.1128/IAI.72.3.1677-1684.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Laskowski MA, Osborn E, Kazmierczak BI. 2004. A novel sensor kinase-response regulator hybrid regulates type III secretion and is required for virulence in Pseudomonas aeruginosa. Mol Microbiol 54:1090–1103. doi: 10.1111/j.1365-2958.2004.04331.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Linares JF, Lopez JA, Camafeita E, Albar JP, Rojo F, Martinez JL. 2005. Overexpression of the multidrug efflux pumps MexCD-OprJ and MexEF-OprN is associated with a reduction of type III secretion in Pseudomonas aeruginosa. J Bacteriol 187:1384–1391. doi: 10.1128/JB.187.4.1384-1391.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Brutinel ED, Vakulskas CA, Brady KM, Yahr TL. 2008. Characterization of ExsA and of ExsA-dependent promoters required for expression of the Pseudomonas aeruginosa type III secretion system. Mol Microbiol 68:657–671. doi: 10.1111/j.1365-2958.2008.06179.x. [DOI] [PubMed] [Google Scholar]
- 61.King JM, Schesser Bartra S, Plano G, Yahr TL. 2013. ExsA and LcrF recognize similar consensus binding sites, but differences in their oligomeric state influence interactions with promoter DNA. J Bacteriol 195:5639–5650. doi: 10.1128/JB.00990-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hornef MW, Roggenkamp A, Geiger AM, Hogardt M, Jacobi CA, Heesemann J. 2000. Triggering the ExoS regulon of Pseudomonas aeruginosa: a GFP-reporter analysis of exoenzyme (Exo) S, ExoT, and ExoU synthesis. Microb Pathog 29:329–343. doi: 10.1006/mpat.2000.0398. [DOI] [PubMed] [Google Scholar]
- 63.Rietsch A, Wolfgang MC, Mekalanos JJ. 2004. Effect of metabolic imbalance on expression of type III secretion genes in Pseudomonas aeruginosa. Infect Immun 72:1383–1390. doi: 10.1128/IAI.72.3.1383-1390.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Vogel J, Luisi BF. 2011. Hfq and its constellation of RNA. Nat Rev Microbiol 9:578–589. doi: 10.1038/nrmicro2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Burrowes E, Baysse C, Adams C, O'Gara F. 2006. Influence of the regulatory protein RsmA on cellular functions in Pseudomonas aeruginosa PAO1, as revealed by transcriptome analysis. Microbiology 152:405–418. doi: 10.1099/mic.0.28324-0. [DOI] [PubMed] [Google Scholar]
- 66.Patten CL, Kirchhof MG, Schertzberg MR, Morton RA, Schellhorn HE. 2004. Microarray analysis of RpoS-mediated gene expression in Escherichia coli K-12. Mol Genet Genomics 272:580–591. doi: 10.1007/s00438-004-1089-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.