

Abstract
Nanodiamonds (NDs) have received considerable attention as potential drug delivery vehicles. NDs are small (~5 nm diameter), can be surface modified in a controllable fashion with a variety of functional groups, and have little observed toxicity in vitro and in vivo. However, most biomedical applications of NDs utilize surface adsorption of biomolecules, as opposed to covalent attachment. Covalent modification provides reliable and reproducible ND–biomolecule ratios, and alleviates concerns over biomolecule desorption prior to delivery. The present study has outlined methods for the efficient solid-phase conjugation of ND to peptides and characterization of ND–peptide conjugates. Utilizing collagen-derived peptides, the ND was found to support or even enhance the cell adhesion and viability activities of the conjugated sequence. Thus, NDs can be incorporated into peptides and proteins in a selective manner, where the presence of the ND could potentially enhance the in vivo activities of the biomolecule it is attached to.
Keywords: nanoparticle, collagen, cell adhesion, triple-helical peptide, wound healing, drug delivery
INTRODUCTION
Nanodiamond (ND) is a chemically inert, stable carbon nanoparticle with a rigid diamond core. ND powder is produced in large volumes by detonation, and the raw detonation soot contains 25–30% of ND particles surrounded by graphene shells and amorphous carbon. Following oxidation and acid wash purification, ND particles are ~5 nm in diameter and contain an inert diamond core (sp3 carbon) with numerous surface groups, such as C=O, COOH, OH, CH2, CH3, and patches of sp2 carbon (Figure 1).1,2 The surface layer of functional groups and specific surface area as large as 250–450 m−2 g−1 allows for tailoring ND for numerous applications.1,2
FIGURE 1.

(Left) High resolution transmission electron microscopic photograph of a single detonation ND particle and (right) 5 nm diameter atomistic model illustrating the ND structure where sp3 diamond carbon is shown in grey, sp3 amorphous carbon in green, sp2 carbon in black, oxygen in red, nitrogen in blue, and hydrogen in white. A sector has been cut from the model to show crystalline diamond structure inside the particle.
ND particles have been recognized for their favorable biocompatibility, excellent mechanical properties, flexible surface chemistry, and low production cost.1–6 These properties have sparked considerable interest in the application of ND particles in biomedicine, including as molecular delivery vehicles, polymer matrix components, and fluorescent probes.1,2,7–14 The biodistribution of NDs depends on their quality, functionalization, and size.15 NDs have been recently explored as vectors for delivery of several biomolecules, such as siRNA,16–18 antibodies,19 insulin,20 and small molecules, including paclitaxel21 and doxorubicin.11,22,23 Embedding NDs functionalized with small molecules into films shows promising results for potent sustained drug release.7
Current methods of peptide and protein attachment to NDs primarily rely on adsorption over variable time courses.2 These methods introduce variability on the amount of peptide/protein adsorbed to the NDs, as well as pose concerns on the desorption rate prior to the site of intended delivery.11 Covalent attachment methods are either not discriminatory in terms of where the ND binds the peptide or protein or are limited in the length and complexity of peptide that can be bound.24–27 Furthermore, characterization of the ND surface after biological molecule attachment is very challenging due to the inert nature of the ND particles that makes them refractory for many analytical methods. In contrast to prior studies, where ND has been utilized as the “solid-phase” for direct synthesis of peptides on its surface,24,25 we sought out a different approach that would allow for selective binding of ND within any peptide. More specifically, we considered attachment of NDs following traditional solid-phase peptide synthesis, where the ND would be incorporated in similar fashion as any amino acid. This approach results in covalent ND binding to the peptide, and has not been previously described. For the purposes of future wound-healing applications, here we describe covalent functionalization of NDs with a type I collagen-derived, fluorescently labeled α2β1 integrin-binding peptide (α1(I)fTHP).28 The ND-α1(I)fTHP was characterized herein by MALDI-TOF mass spectrometry, Raman spectroscopy, fluorescence spectroscopy, zeta potential, and particle size distribution. In addition, fibroblast and Chinese hamster ovary cell adhesion and viability was explored for α1(I)fTHP and ND-α1(I)fTHP.
MATERIALS AND METHODS
Solid-Phase Peptide Synthesis
ND bearing free carboxylic acid groups was produced by air oxidation as described29 followed by boiling in aqueous HCl and HNO3 to remove metal impurities.30 Synthesis of α1(I)fTHP [(Gly-Pro-Hyp)4-Gly-Ala-Arg-Gly-Glu-Arg-Gly-Phe-Hyp-Gly-Glu-Arg-(Gly-Pro-Hyp)4-Gly-Pro-Lys(5-Fam)-NH2, where Hyp = 4-hydroxyproline and 5-Fam = 5-carboxyfluorescein] and a control peptide α1(V)THP [(Gly-Pro-Hyp)4-Gly-Pro-Pro-Gly-Val-Val-Gly-Glu-Gln-Gly-Glu-Gln-(Gly-Pro-Hyp)4] followed Nα-(9-fluorenylmethoxycarbonyl) (Fmoc) chemistry procedures well established in our laboratory.31,32 Peptides were assembled using NovaPEG Rink amide resin (substitution level = 0.48 mmol g−1) on the Liberty automated microwave assisted peptide synthesizer (CEM, Matthews, NC) equipped with a Discover microwave module. The side chain protecting group strategy was tBu for Hyp and Glu, triphenylmethyl (Trt) for Gln, and 2,2,4,6,7-pentamethyldihydrobenzofuran-5-sulfonyl (Pbf) for Arg. Fmoc-Lys(Fam) was incorporated manually using 3 equiv of amino acid, 3 equiv of N,N′-diisopropylcarbodiimide, 3 equiv of N-hydroxybenzotriazole (HOBt), and 6 equiv of N,N-diisopropylethylamine (DIPEA). The initial reaction was for 45 min at 40°C, 25 W followed by overnight reaction at room temperature and no microwave power. Unreacted groups were capped with 0.5M acetic anhydride, 0.125M DIPEA, and 0.015M HOBt in DMF. Coupling reactions were performed with 5 equiv of Fmoc-amino acid, 4.9 equiv of 1-H-benzotriazolium-1-[bis(dimethylamino)methylene]-5-chloro-hexafluorophosphate-(1-),3-oxide, and 10 equiv of N-methylmorpholine. For α1(I)fTHP couplings were carried out at 50°C, 25 W for 1200 s, and coupling of Pro11 was repeated. For α1(V)THP couplings were carried out at 50° C, 25 W for 300 s with the exception of Pro14, Pro15, Val18, Gly19, Glu20, and Gln21 where the reaction time was increased to 600 s. Coupling reactions of Pro11, Gly16, and Val17 were repeated overnight at room temperature and no microwave power. For the Fmoc deprotection step, 10% piperazine in DMF was used (30 s, 75° C, 35 W, followed by 180 s, 75°C, 35 W).
Once the peptides were assembled, ND bearing free carboxylic acid groups was acylated to the N-terminus of a portion of the peptide. The amount of ND used for acylation was determined as follows. A 5 nm diameter spherical ND particle has 11,686 carbon atoms, 1560 of which are exposed on the surface. It can be estimated that 20% of the surface atoms are terminated by COOH,29,33 and thus there are 312 COOH groups on each particle. The molecular weight of a 5 nm nonfunction-alized ND particle is 11,686 × 12.0 g mol−1 = 140,232 g mol−1, and thus 1 g of ND has ~2 mmol of COOH. Use of 4 equiv of ND was based on standard, automated peptide synthesis protocols without microwave heating. 37.6 mg (3.5 equiv) of 2-(6-chloro-1H-benzotriazole-1-yl)21,1,3,3-tetramethylaminium hexafluorophosphate, 33.44 μL (8 equiv) of DIPEA, and 48 mg (4 equiv) of ND were dissolved/suspended in 2.5–3 μL tetrahydrofuran (THF). THF was utilized because (a) NDs added to THF results in transparent solutions with low levels of ND aggregation33,34 and (b) THF has appropriate solvent properties for solid-phase synthesis of growing peptide chains.35 This mixture was added to 50 mg (0.024 mmol) of the peptide–resin and the reaction carried out overnight at room temperature. Completion of coupling was evaluated using the ninhydrin test.31 Due to the presence of unreacted amino groups after the overnight reaction the acylation was repeated. Peptides and ND–peptides were released from the resin by treatment with H2O–thioanisole–trifluoroacetic acid (TFA) (1:1:18) for 3.5 h in ambient gas atmosphere. Peptides and ND–peptides were precipitated with cold MTBE, centrifuged, and the resulting pellet dissolved in H2O and lyophilized.
Peptide purity was evaluated using an Agilent 1200 series HPLC (Santa Clara, CA) equipped with a Vydac C18 column (5 μm, 300 Å, 150 mm × 4.6 mm). Solvent A was 0.1% TFA–H2O, solvent B was 0.1% TFA–acetonitrile, the gradient was 2–98% B over 20 min, the flow rate was 1 mL min−1, and detection was at λ = 220 nm. Analytical results were used to determine the optimal preparatory gradient, where 4 mL of H2O-dissolved peptide was injected into a Vydac C18 column (15–20 μm, 300 Å, 250 × 22 mm) on an Agilent 1200 series HPLC. The gradients were 10–35% B over 40 min for α1(I)fTHP and 10–30% B over 40 min for α1(V)THP, the flow rate was 10 mL min−1, and detection was at λ = 220 nm. Peak fractions were analyzed via analytical HPLC and MALDI-TOF mass spectra (Applied Biosystems Voyager DE-PRO Biospectrometry Workstation, Carlsbad, CA). Pure fractions were pooled, frozen, lyophilized, and stored at −20°C in amber vials.
The concentration of α1(I)fTHP and ND-α1(I)fTHP was determined using a Thermo Scientific NanoDrop 1000 Spectrophotometer (Waltham, MA) at λ = 260 nm, εFam = 62,880 M−1 cm−1 (allowing for 3 Fam per triple-helix). The concentration of α1(V)THP and ND-α1(V)THP was determined by weighing. Peptides and ND–peptides were utilized at concentrations so that clear solutions were obtained in aqueous buffers.
MALDI-TOF Analysis
Peptides and undigested or trypsin-digested ND-α1(I)fTHP were subjected to MALDI-TOF mass spectral analysis using a 2,5-dihydroxybenzoic acid matrix. Digestion was carried out by immobilized TPCK–trypsin (Thermo Scientific, Rockford, IL) for 4 h at 37°C in 0.1M NH4HCO3, pH 8.0.
Circular Dichroism (CD) Spectroscopy
Peptides were dissolved in methanol–H2O (1:4) and equilibrated at 4°C (>18 h) to facilitate triple-helix formation. Peptide concentrations were 5–40 μM. Triple-helical structure was evaluated by near-UV CD spectroscopy using a Jasco J-810 Spectropolarimeter (Easton, MD) with a path length of 1 mm. Thermal transition curves were obtained by recording the molar elipticity ([θ]) at λ= 225 nm with temperature increasing by 20° C h−1 from 5 to 80° C. Temperature was controlled by a JASCO PTC-348WI temperature control unit. The peptide melting temperature (Tm) was defined as the inflection point in the transition region (first derivative). The spectra were normalized by designating the highest [θ]225 nm as 100% folded, and the lowest [θ]225 nm as 0% folded.
Fluorescence Spectroscopy
Fluorescence emission scans of α1(I)fTHP and ND-α1(I)fTHP were performed at 8 μM ligand concentration using λexcitation = 490 nm and λemission = 520 nm on a Synergy H4 Hybrid Multi-Mode Microplate Reader (BioTek).
Raman Spectroscopy
Raman spectroscopic measurements were performed with a LabRam HR800-Inv Raman microscope (Horiba Scientific, Edison, NJ). For the drop deposition measurements, 2 mM solutions of ND-α1(I)fTHP, α1(I)fTHP, or 5/6-Fam (AnaSpec, Fremont, CA) were prepared in deionized H2O and 1 μL of each solution was deposited and dried on a #1.5 glass coverslip. The coverslip was placed under the microscope and Raman spectra were recorded for each deposited sample using 2 mW of 647 nm laser light focused by a 100× oil immersion objective. Spectra were measured using 600 g mm−1 grating, 200 mm confocal hole diameter, 125 mm slit width, and 3 min total data collection time. To obtain the ND spectrum, ~0.5 mg of sample was placed on a glass coverslip and mixed with 1 μL of H2O. The spectrum was recorded before the H2O had evaporated, which helped to quench the ND sample during the measurement. For the solution measurement of α1(I)fTHP, 5 mL of 2 μM aqueous solution was placed in a 0.6 mm square inner dimension glass capillary tube (VitroCom, Mountain Lakes, NJ) and sealed at both ends using Critoseal™. The capillary was placed in front of a dedicated 90° macro port of the spectrograph and irradiated with 250 mW of 647 nm laser light for a total of 60 min to record the spectrum.
Zeta Potential and Particle Size Distribution
Zeta potential and particle size distribution of ND and ND-α1(I)fTHP dispersed in deionized H2O followed by 10 s bath sonication were measured at 20°C in back scatter geometry using a Malvern Zetasizer Nano ZS (Malvern Instruments, UK) equipped with a 4 mW He–Ne laser (633 nm).36 Approximately 1 mL of sample was placed in a clear disposable zeta potential and particle size capillary measurement cell (DTS1061, Malvern Instruments), equilibrated at 20°C for 60 s, and then analyzed with scattered light detected at 173° to the direction of the incident light (173° Backscatter NIBS default). Each solution was analyzed at least three times.
Cell Adhesion and Viability
Chinese hamster ovary (CHO)-K1 cells and BJ fibroblasts were obtained from ATCC. CHO-K1 cells were grown in F-12K medium supplemented with 10% fetal bovine serum (FBS) and penicillin–streptomycin, while BJ cells were grown in Eagle’s minimal essential medium (EMEM) supplemented with 10% FBS and penicillin–streptomycin. All cells were grown at 37°C and 5% CO2, 95% relative humidity.
Adhesion assays were carried out as described previously37 with some modifications. Ligands were dissolved in dimethyl sulfoxide–phosphate buffered saline (PBS) (3:1) and further diluted to desired concentrations in PBS. An untreated 96-well Costar High Binding Assay Plate (Corning, Corning, NY) was pretreated with PBS, 10 μM α1(I)fTHP, ND-α1(I)fTHP, or ND-α1(V)THP by shaking at room temperature for 2 h (covered). The plate was washed three times with PBS. Nonspecific adhesion was blocked by adding 2 mg mL−1 bovine serum albumin in PBS and incubating the plate for 2 h at room temperature, then rinsing the plate three times with PBS. Subconfluent cells were rinsed with PBS, and released with Triple cell dissociation reagent (Life Technologies, Carlsbad, CA). Cells were washed and rediluted to 75,000 cells mL−1 (CHO-K1) or 30,000 cells mL−1 (BJ) in the respective growth media. The cells were seeded in 100 μL per well and allowed to adhere for 1 h at 37° C. Cells were washed three times with warm growth medium to remove nonadherent cells, upon which 100 μL PBS was added to each well. Viability was measured with 100 μL of CellTiterGlo added to each well. The plate was incubated for 10 min, upon which luminescence was read by a Synergy H4 Hybrid Multi-Mode Microplate Reader. Each well’s luminescence was expressed in Relative Luminescence Units (RLUs).
RESULTS
Peptide Characterization
MALDI-TOF mass spectral analysis indicated a mass of 4084.7 Da for α1(I)fTHP (theoretical 4083.3 Da) and 3290.2 Da for α1(V)THP (theoretical 3289.6 Da) (data not shown). To examine the triple-helical structure and melting point of α1(I)fTHP and α1(V)THP, the peptides were analyzed by CD spectroscopy. Both α1(I)fTHP and α1(V)THP exhibited CD spectra characteristic of triple-helical structures, with positive molar ellipticities ([θ]) at λ = 225 nm and strongly negative [θ] values at λ = 195 nm (Figure 2, top). Monitoring of [θ]225 nm as a function of temperature resulted in sigmoidal melting curves for α1(I)fTHP and α1(V)THP (Figure 2, bottom), indicative of transitions from triple-helices to monomeric species.38 The melting points (Tm values) for α1(I)fTHP and α1(V)THP were 43.2 and 38.2°C, respectively. A melting temperature above 37°C provides good peptide stability for potential use in physiological-like conditions.
FIGURE 2.

CD properties of α1(I)fTHP and α1(V)THP. (Top) CD spectra of α1(I)fTHP and α1(V)THP. Scans were taken from λ = 180–250 nm. (Bottom) Thermal transition curves of α1(I)fTHP and α1(V)THP. Readings were taken at λ = 225 nm and normalized to fraction folded.
Characterization of ND–Peptides
Several methods have been described to examine ND–peptide/protein conjugates. We initially performed MALDI-TOF mass spectrometric analysis of ND-α1(I)fTHP. There was little ionization of the ND-α1(I)fTHP, unlike the peptide alone (see above). Following trypsin treatment, several species were observed, of molecular mass 1026 Da [corresponding to Gly-Glu-Arg-Gly-Phe-Hyp-Gly-Glu-Arg], 1374 Da [corresponding to (Gly-Pro-Hyp)4-Gly-Ala-Arg], 1731 Da [corresponding to (Gly-Pro-Hyp)4-Gly-Pro-Lys(5-Fam)-NH2], and 4087 Da [corresponding to intact α1(I)fTHP]. Thus, mass spectrometry could be used to characterize peptides or proteins attached to NDs, if a protease is utilized to first liberate and/or fragment the attached peptide or protein.
CD spectra of the ND-α1(I)fTHP could not be obtained, due to light scattering by the ND. The Raman spectra of ND-α1(I)fTHP, α1(I)fTHP, free 5/6-Fam, and free ND were obtained (Figure 3). A comparison of the Fam-labeled peptide spectra with that of 5/6-Fam reveals that the peptide spectra were dominated in the high wavenumber region (1100–1650 cm−1) by bands due to the Fam chromophore, although many of the Fam bands appear to shift on both intensity and position when associated with the peptide. Thus, it appears that Fam interacts strongly with the peptide. Weaker bands that were associated with the peptide include those at 811 cm−1 (νC–C), 1003 cm−1 (Phe ring), and 1031 cm−1 (Phe ring).39,40 Collagen and collagen-like peptides exhibit a characteristic Raman spectroscopic signature that includes a strong band doublet and triplet at ~855/875 cm−1 and ~920/940/970 cm−1, respectively, which are mainly associated with proline ring and skeletal C–C stretches.39,41–46 The same set of bands was evident in the Raman spectra of ND-α1(I)fTHP and α1(I)fTHP at ~870 and 921/940 cm−1 (Figures 3A–3C). ND exhibited a narrow Raman band at 1328 cm−1 (Figure 3E) in accord with previous literature reports.1,29 The appearance of the ND band was obscured in the ND-α1(I)fTHP spectra due to overlap with a strong band at 1335 cm−1 that is likely associated with the Fam chromophore.
FIGURE 3.
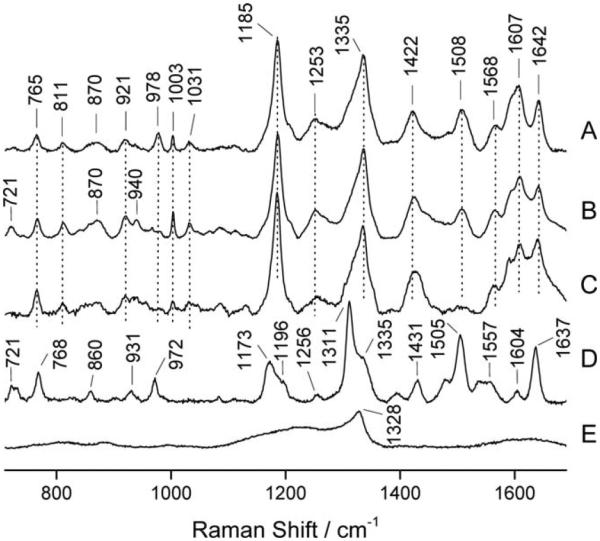
Raman spectra of (A) drop-deposited ND-α1(I)fTHP, (B) drop-deposited α1(I)fTHP, (C) 2 mM aqueous solution of α1(I)fTHP, (D) drop-deposited 5/6-Fam, and (E) aqueous suspension of ND. All spectra were measured with 647 nm laser excitation.
The presence of the 5-Fam group in α1(I)fTHP allowed the use of fluorescence spectroscopy and/or microscopy to evaluate peptide conjugation to ND. Fluorescence emission scans of 8 μM α1(I)fTHP and ND-α1(I)fTHP in PBS revealed a major peak at λ = 530 nm for both species (Figure 4). As expected, ND did not seem to contribute to the overall fluorescence, nor did it emit fluorescence at additional wavelengths scanned.
FIGURE 4.
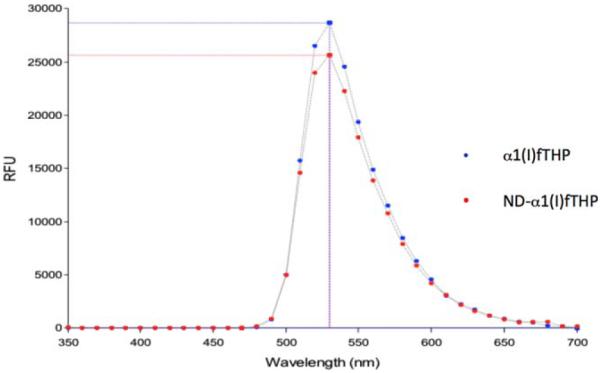
Fluorescence spectra of (blue) α1(I)fTHP and (red) ND-α1(I)fTHP.
The zeta potential and particle size distribution were evaluated for ND and ND-α1(I)fTHP. ND alone had a zeta potential of −32 ± 5 mV (Figure 5, top) while ND-α1(I)fTHP had a zeta potential of 17 6 0.6 mV (Figure 5, bottom). Thus, the zeta potential analysis clearly differentiated between ND alone (which contains a negative total charge resulting from ionization of the functional COOH groups) and ND-α1(I)fTHP (where the peptide adds a positive charge to the ND, based on the presence of 3 Arg and 2 Glu residues in the peptide and amide bond formation). Particle size distribution indicated diameters of 260 ± 141 nm for ND alone (Figure 6, top) and 303 ± 127 nm for ND-α1(I)fTHP (Figure 6, bottom). Both ND and ND-α1(I)fTHP aggregate to form 200–300 nm diameter agglomerates, as primary ND particles are ~5 nm in diameter.36 However, a slightly larger size of agglomerates observed for ND-α1(I)fTHP compared to ND, combined with the shift in the zeta potential, supports the distinction of the peptide-conjugated and unconjugated NDs.
FIGURE 5.

Zeta potentials of (top) ND and (bottom) ND-α1(I)fTHP. The three curves represent three replicates.
FIGURE 6.

Particle size distribution of (top) ND and (bottom) ND-α1(I)fTHP. The five curves represent five replicates.
The scattering intensity also increases between 2000 and 6000 nm for both ND and ND-α1(I)fTHP (Figure 6). However, as the upper detection limit of the instrument is 6000 nm, one cannot determine how high the peak is in this range. As to the origin of this peak, it should be noted that a single large particle can scatter light with intensity several orders of magnitude larger than a smaller particle. Whether the 2000–6000 nm peak originates from impurities or larger agglomerates, their content in the samples is most likely minor compared to the particles giving rise to the main peak at 50–1000 nm.
Cell Adhesion and Viability
α1(I)fTHP is an α2β1 integrin-binding peptide derived from type I collagen that promotes cell adhesion and spreading.28 As a control peptide, we utilized a α1(V)THP, derived from a type V collagen matrix metalloproteinase cleavage site.47 Because α1(I)fTHP promotes cellular activities, we evaluated the effects of ND attachment on peptide biological activity.
Cell adhesion was examined in wells pre-treated with PBS, α1(I)fTHP, ND-α1(I)fTHP, and ND-α1(V)fTHP in a 96-well plate. CHO-K1 cells showed equal adhesion on α1(I)fTHP and ND-α1(I)fTHP, with considerably less adhesion to ND-α1(V)THP and lower adhesion in PBS (Figure 7A). Thus, the ND itself did not promote cell adhesion, but required attachment of a specific cell adhesion sequence. BJ fibroblasts adhered poorly to α1(I)fTHP but very well to ND-α1(I)fTHP, while not at all to ND-α1(V)THP (Figure 7A). This was an intriguing result, as fibroblasts (which are an important component in matrix remodeling) responded only when the cell adhesion sequence was attached to ND.
FIGURE 7.

Adhesion and viability analyses of CHO-K1 cells and BJ fibroblasts. (A) Microscopic images of cell adhesion to peptide and ND–peptide conjugates. Images were taken at 40× magnification under phase contrast microscope. (B) Viability of CHO cells (blue) and BJ fibroblasts (red) following adhesion to PBS, peptide, or ND–peptide conjugates.
In similar fashion to cell adhesion, CHO-K1 cells showed equal viability on α1(I)fTHP and ND-α1(I)fTHP, with little viability on ND-α1(V)THP (Figure 7B). Thus, the ND itself did not promote cell viability, but again required attachment of a specific cell adhesion sequence for cellular activity. The BJ fibroblasts had some viability on α1(I)fTHP but greatly enhanced viability on ND-α1(I)fTHP and almost no viability on ND-α1(V)THP (Figure 7B). As above, fibroblast activity was greatly enhanced when the cell adhesion sequence was attached to ND.
DISCUSSION
The specific attachment of peptides and proteins to ND, and the characterization of the resulting conjugates, are relatively new endeavors and present several challenges. Covalent attachment of peptides and proteins to ND has been achieved by several methods. Hydroxyl groups on the NDs have been reacted with p-benzoquinone, followed by attachment of the activated NDs to free amino groups on proteins.26 All reactions were performed in aqueous buffered solutions, and attachment of protein was characterized by radioactive labeling of the protein.26 Another attachment method utilized carboxyl groups on the ND, whereby the carboxyl groups were activated using N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride and N-hydroxysuccinimide, followed by addition of the cell penetrating peptide TAT.27 All steps were in aqueous buffered solutions, with attachment through the amino groups of the peptide.27 FTIR spectroscopy, zeta potentials, and elemental analyses were used to characterize peptide conjugation to the ND.27 This same strategy was used to attach recombinant fish growth hormone of the yellow grouper (rEaGH) to ND.48 FTIR and Raman spectroscopies were used to characterize rEaGH conjugation to the ND.48 In a related approach that could be applied to peptides and proteins,49 ND carboxyl groups were treated with SOCl2 in the presence of catalytic quantities of DMF, followed by attachment of ethylenediamine.33 The aminated ND was characterized by FTIR spectroscopy, particle size distribution, and zeta potential.33 In all of these cases, attachment to ND was not specific, as any amino or carboxyl group in the peptide or protein could become modified, potentially blocking desired biological activities.
Peptides have been synthesized directly on NDs.24,25 In one case, the ND was modified to possess a (3-aminopropyl)trime-thoxysilane linker, followed by standard Fmoc chemistry (using Fmoc-amino acid chlorides) to incorporate a tetrapeptide.24 The ND peptide was characterized by FTIR spectroscopy and particle size distribution. In a second case, the ND was modified to possess oxyhexanol groups, followed by esterification of Fmoc-Phe using N,N’-dicyclohexylcarbodiimide and 4-dimethylaminopyridine, removal of the Fmoc group, acylation of Fmoc-Val, and removal of the Fmoc group.25 The ND dipeptide was characterized by FTIR spectroscopy. Both the tetrapeptide and dipeptide did not contain any functional side chains, so the approach of direct peptide synthesis on ND may be compromised by the acidic treatment normally used to remove side chain protecting groups. In addition, the length of peptide that could be synthesized directly on the ND may be severely limited, based on observations from solid-phase syntheses on other rigid supports.50 Manipulation of the ND during solid-phase synthesis may also be difficult, due to the small size of the ND, as these prior studies used centrifugation after each step to isolate the modified ND.24,25
We presently examined solid-phase methods to allow for maximum flexibility for attaching ND to peptides and proteins in a specific manner. In this case, the ND was only incorporated at the N-terminus of the peptide, away from any biologically active residues. The attachment protocol was highly efficient. Washing steps that occur after the ND has been conjugated serve to purify the ND and peptide from residual reagents still present following synthesis.
Proper characterization of ND–peptide and protein conjugates requires consideration of multiple analytical techniques. Mass spectrometry is useful for demonstrating peptide/protein attachment to the ND. ND and ND conjugates have poor ionization, and thus mass spectra can be obtained following protease liberation of peptides and proteins from the ND. Raman spectroscopy with 647 nm laser excitation showed independent features of the ND and the peptide. More specifically, we observed a ND band at 1328 cm−1. This indicated that one does not need 325 or 488 nm laser excitation in order to observe purified ND by Raman spectroscopy.29,48 However, an important consideration is that most commercially available NDs have significant nondiamond carbon content, which will obscure the weaker diamond signal during visible light (390–700 nm) excitation. In this situation, resonance Raman conditions are required to selectively enhance the diamond Raman signal, i.e., the excitation wavelength must be close to the material’s band gap. In case of the diamond, the bandgap is 5.5 eV,51 and UV light is required. The observation of a reasonable diamond band in the ND peptide with the 647 nm laser indicated that a well-purified ND was present. Recently, ND Raman spectra have been obtained using a 532 nm laser, where the characteristic peak for the sp3 structural diamond was observed at ~1300 cm−1 and a graphite peak observed at ~1600 cm−1.52 Although not examined here, FTIR spectroscopy can be utilized to characterize ND–peptide/protein conjugates, either instead of or complimentary to Raman spectroscopy.20,24,27,33,53 Fluorescent tags can also be used to monitor peptide and protein attachment to ND. Finally, the attachment of peptides and proteins to ND is readily characterized by zeta potential and particle size distribution unless, in the former case, conjugation of the peptide or protein results in the same overall charge as the ND alone.
Other previously described ND characterization methods were found to be problematic for ND–peptides. Transmission electron microscopy may be useful for association of ND with proteins,20 but likely will not reveal the subtle attachment of peptides.27 While elemental analysis quantifies an increase in nitrogen content upon peptide conjugation,24,27 mass spectrometry provides a precise view of peptide incorporation. Noncontact atomic force microscopy has shown potential for identifying polycyclic aromatic hydrocarbon and fullerene surface modification,54 but has not yet been demonstrated for peptide conjugates. NMR spectroscopy, either solution or solid state,30,55–58 could be used to distinguish between ND and peptides/proteins. At present, NMR spectroscopic analysis of ND conjugates is limited by the concentration of the sample as well as the time needed to process data based on the field strength of the NMR instrumentation. Moreover, the interpretation of ND NMR signal is still controversial.30,58
Attachment of ND to a biologically active peptide via solid-phase methodology demonstrated that biological activity was not compromised by the presence of the ND (Figure 7). In fact, adhesion and viability of fibroblasts on ND-α1(I)fTHP was enhanced compared with α1(I)fTHP. The improved fibroblast activity could stem from the multivalent presentation of the ligand on the ND surface. The ligand-induced clustering of integrins can activate signal transduction pathways that ultimately modulate cellular function.59 Ligand-induced integrin clustering, and subsequent signaling activation, has been observed previously with peptides bound to magnetic microbeads.60 Enhanced cell adhesion could also be due to increasing the surface roughness at the nanoscale level. Addition of modified NDs to biodegradable polymers enhanced in vitro biomineralization, most likely due to increased roughness and the presence of the COOH groups.12
The α1(I)fTHP sequence is only active in triple-helical conformation.28 The peptide alone exhibits stable triple-helical structure, based on CD and Raman spectroscopic analyses (Figures 2 and 3). Although CD spectra of the ND-α1(I)fTHP could not be obtained, Raman spectroscopy supports the presence of a triple-helix in the ND-α1(I)fTHP based on bands at ~870 and 921/940 cm−1 (Figure 3). Thus, the solid-phase incorporation of ND still permitted the active conformation of the peptide to be realized. Taking advantage of orthogonal side chain protecting strategies during solid-phase synthesis would allow for attachment of ND to specific residues within a peptide sequence.
CONCLUSIONS
NDs can be efficiently conjugated to peptides by solid-phase chemistry and the subsequent ND–peptides characterized by a variety of biophysical techniques. Utilizing collagen-derived peptides, the ND was found to support or even enhance the cell adhesion and viability activities of the conjugated sequence. Thus, NDs can be incorporated into peptides and proteins in a selective manner, where the presence of the ND could potentially enhance the in vivo activities of the biomolecule it is attached to. The present study has utilized the ND peptide without purification following solid-phase synthesis. ND–peptides are not amenable to HPLC-based purification methods commonly used for peptides. While this represents a potential limitation for ND attachment to longer peptides, it is also possible that using convergent solid-phase synthesis of protected peptide fragments61 can result in highly pure resin-bound peptides to which NDs can be selectively conjugated. The present work allows for further exploration of ND conjugates assembled by solid-phase approaches for targeted drug delivery.
Acknowledgments
Research reported in this publication was supported by the National Cancer Institute of the National Institutes of Health under Award Number CA098799 (to GBF). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. We thank James K. English and Dan Pepe (Nano Wound Devices, Inc.) for their promotion of these studies. GBF, VM, and YG are Co-Founders of Nano Wound Devices, Inc.
REFERENCES
- 1.Mochalin VN, Shenderova O, Ho D, Gogotsi Y. Nat Nanotech. 2012;7:11–23. doi: 10.1038/nnano.2011.209. [DOI] [PubMed] [Google Scholar]
- 2.Schrand AM, Ciftan Hens SA, Shenderova OA. Crit Rev Solid State Mat Sci. 2009;34:18–74. [Google Scholar]
- 3.Yu S-J, Kang M-W, Chang H-C, Chan K-M, Yu Y-C. J Am Chem Soc. 2005;127:17604–17605. doi: 10.1021/ja0567081. [DOI] [PubMed] [Google Scholar]
- 4.Schrand AM, Huang H, Carlson C, Schlager JJ, Osawa E, Hussain SM, Dai L. J Phys Chem B. 2007;111:2–7. doi: 10.1021/jp066387v. [DOI] [PubMed] [Google Scholar]
- 5.Lien Z-Y, Hsu T-C, Liu K-K, Liao W-S, Hwang K-C, Chao J-I. Biomaterials. 2012;33:6172–6185. doi: 10.1016/j.biomaterials.2012.05.009. [DOI] [PubMed] [Google Scholar]
- 6.Xing Y, Xiong W, Zhu L, Osawa E, Hussin S, Dai L. ACS Nano. 2011;5:2376–2384. doi: 10.1021/nn200279k. [DOI] [PubMed] [Google Scholar]
- 7.Lam R, Chen M, Pierstorff E, Huang H, Osawa E, Ho D. ACS Nano. 2008;2:2095–2102. doi: 10.1021/nn800465x. [DOI] [PubMed] [Google Scholar]
- 8.Krueger A. Chem Eur J. 2008;14:1382–1390. doi: 10.1002/chem.200700987. [DOI] [PubMed] [Google Scholar]
- 9.Schrand AM, Lin JB, Ciftan Hens S, Hussain SM. Nanoscale. 2011;3:435–445. doi: 10.1039/c0nr00408a. [DOI] [PubMed] [Google Scholar]
- 10.Zhang Q, Mochalin VN, Neitzel I, Knoke IY, Han J, Klug CA, Zhou JG, Lelkes PI, Gogotsi Y. Biomaterials. 2011;32:87–94. doi: 10.1016/j.biomaterials.2010.08.090. [DOI] [PubMed] [Google Scholar]
- 11.Mochalin VN, Pentecost A, Li XM, Neitzel I, Nelson M, Wei C, He T, Guo F, Gogotsi Y. Mol Pharm. 2013;10:3728–3735. doi: 10.1021/mp400213z. [DOI] [PubMed] [Google Scholar]
- 12.Zhang Q, Mochalin VN, Neitzel I, Hazeli K, Niu J, Kontsos A, Zhou JG, Lelkes PI, Gogotsi Y. Biomaterials. 2012;33:5067–5075. doi: 10.1016/j.biomaterials.2012.03.063. [DOI] [PubMed] [Google Scholar]
- 13.Mochalin VN, Gogotsi Y. J Am Chem Soc. 2009;131:4594–4595. doi: 10.1021/ja9004514. [DOI] [PubMed] [Google Scholar]
- 14.Neitzel I, Mochalin VN, Niu J, Cuadra J, Kontsos A, Palmese GR, Gogotsi Y. Polymer. 2012;53:5965–5971. [Google Scholar]
- 15.Zhu Y, Li J, Li W, Zhang Y, Yang X, Chen N, Sun Y, Zhao Y, Fan C, Huang Q. Theranostics. 2012;2:302–312. doi: 10.7150/thno.3627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang X-Q, Chen M, Lam R, Xu X, Osawa E, Ho D. ACS Nano. 2009;3:2609–2616. doi: 10.1021/nn900865g. [DOI] [PubMed] [Google Scholar]
- 17.Alhaddad A, Adam MP, Botsoa J, Dantelle G, Perruchas S, Gacoin T, Mansuy C, Lavielle S, Malvy C, Treussart F, Bertrand JR. Small. 2011;7:3087–3095. doi: 10.1002/smll.201101193. [DOI] [PubMed] [Google Scholar]
- 18.Chen M, Zhang X-Q, Man HB, Lam R, Chow EK, Ho D. J Phys Chem Lett. 2010;1:3167–3171. [Google Scholar]
- 19.Smith AH, Robinson EM, Zhang XQ, Chow EK, Lin Y, Osawa E, Xi J, Ho D. Nanoscale. 2011;3:2844–2848. doi: 10.1039/c1nr10278h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shimkunas RA, Robinson E, Lam R, Lu S, Xu X, Zhang X-Q, Huang H, Osawa E, Ho D. Biomaterials. 2009;30:5720–5728. doi: 10.1016/j.biomaterials.2009.07.004. [DOI] [PubMed] [Google Scholar]
- 21.Liu KK, Zheng WW, Wang CC, Chiu YC, Cheng CL, Lo YS, Chen C, Chao JI. Nanotechnology. 2010;21:315106. doi: 10.1088/0957-4484/21/31/315106. [DOI] [PubMed] [Google Scholar]
- 22.Ma XW, Zhao YL, Liang XJ. Acta Pharmacol Sin. 2011;32:543–544. doi: 10.1038/aps.2011.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li Y, Tong Y, Cao R, Tian Z, Yang B, Yang P. Int J Nanomed. 2014;9:1065–1082. doi: 10.2147/IJN.S54864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Krüger A, Liang Y, Jarre G, Stegk J. J Mater Chem. 2006;16:2322–2328. [Google Scholar]
- 25.Zheng W-W, Hsieh Y-H, Chiu Y-C, Cai S-J, Cheng C-L, Chen C. J Mater Chem. 2009;19:8432–8441. [Google Scholar]
- 26.Purtov KV, Petunin AI, Burov AE, Puzyr AP, Bondar VS. Nanoscale Res Lett. 2010;5:631–636. doi: 10.1007/s11671-010-9526-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li C, Shao J, Qin Y, Shao C, Zheng T, Ye L. J Mater Chem. 2011;21:7966–7973. [Google Scholar]
- 28.Knight CG, Morton LF, Onley DJ, Peachey AR, Messent AJ, Smethurst PA, Tuckwell DS, Farndale RW, Barnes MJ. J Biol Chem. 1998;273:33287–33294. doi: 10.1074/jbc.273.50.33287. [DOI] [PubMed] [Google Scholar]
- 29.Osswald S, Yushin G, Mochalin V, Kucheyev SO, Gogotsi Y. J Am Chem Soc. 2006;128:11635–11642. doi: 10.1021/ja063303n. [DOI] [PubMed] [Google Scholar]
- 30.Panich AM, Shames AI, Sergeev NA, Olszewski M, McDonough JK, Mochalin VN, Gogotsi Y. J Phys Condens Matter. 2013;25:245303. doi: 10.1088/0953-8984/25/24/245303. [DOI] [PubMed] [Google Scholar]
- 31.Fields GB, Lauer-Fields JL, Liu R.-q., Barany G. In: In Synthetic Peptides: A User’s Guide. 2nd Grant GA, editor. W.H. Freeman & Co.; New York: 2001. pp. 93–219. [Google Scholar]
- 32.Stawikowski M, Fields GB. In: In Current Protocols in Protein Science. Coligan JE, Dunn B, Ploegh HL, Speicher DW, Wingfield PT, editors. John Wiley & Sons, Inc.; New York: 2012. pp. 18.11.11–18.11.13. [Google Scholar]
- 33.Mochalin VN, Neitzel I, Etzold BJM, Peterson A, Palmese G, Gogotsi Y. ACS Nano. 2011;5:7494–7502. doi: 10.1021/nn2024539. [DOI] [PubMed] [Google Scholar]
- 34.Li C-C, Huang C-L. Colloids Surf. A: Physiochem Eng Aspects. 2010;353:52–56. [Google Scholar]
- 35.Fields GB, Fields CG. J Am Chem Soc. 1991;113:4202–4207. [Google Scholar]
- 36.Pentecost A, Gour S, Mochalin V, Knoke I, Gogotsi Y. ACS Appl Mater Interfaces. 2010;2:3289–3294. doi: 10.1021/am100720n. [DOI] [PubMed] [Google Scholar]
- 37.Lauer-Fields JL, Malkar NB, Richet G, Drauz K, Fields GB. J Biol Chem. 2003;278:14321–14330. doi: 10.1074/jbc.M212246200. [DOI] [PubMed] [Google Scholar]
- 38.Fields GB, Prockop DJ. Biopolymers (Pept Sci) 1996;40:345–357. doi: 10.1002/(SICI)1097-0282(1996)40:4%3C345::AID-BIP1%3E3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 39.Merlino A, Sica F, Mazzarella L, Zagari A, Vergara A. Biophys Chem. 2008;137:24–27. doi: 10.1016/j.bpc.2008.06.008. [DOI] [PubMed] [Google Scholar]
- 40.Ye J, Fox SA, Cudic M, Rezler EM, Lauer JL, Fields GB, Terentis AC. J Am Chem Soc. 2010;132:980–988. doi: 10.1021/ja9043196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bonifacio A, Sergo V. Vib Spectrosc. 2010;53:314–317. [Google Scholar]
- 42.Carcamo JJ, Aliaga AE, Clavijo RE, Branes MR, Campos-Vallette MM. Spectrochim Acta Part A Mol Biomol Spectrosc. 2012;86:360–365. doi: 10.1016/j.saa.2011.10.049. [DOI] [PubMed] [Google Scholar]
- 43.Diem M, Bhatnagar RS, Druyan ME, Renugopalakrishnan V. Biopolymers. 1984;23:2955–2961. doi: 10.1002/bip.360231218. [DOI] [PubMed] [Google Scholar]
- 44.Goheen SC, Lis LJ, Kauffman JW. Biochim Biophys Acta. 1978;536:197–204. doi: 10.1016/0005-2795(78)90065-x. [DOI] [PubMed] [Google Scholar]
- 45.Terentis AC, Fox SA, Friedman SJ, Spencer ES. J Raman Spectrosc. 2013;44:1205–1216. [Google Scholar]
- 46.Wang YN, Galiotis C, Bader DL. J Biomechanics. 2000;33:483–486. doi: 10.1016/s0021-9290(99)00194-3. [DOI] [PubMed] [Google Scholar]
- 47.Lauer-Fields JL, Sritharan T, Stack MS, Nagase H, Fields GB. J Biol Chem. 2003;278:18140–18145. doi: 10.1074/jbc.M211330200. [DOI] [PubMed] [Google Scholar]
- 48.Cheng CY, Perevedentseva E, Tu JS, Chung PH, Cheng CL, Liu KK, Chao JI, Chen PH, Chang CC. Appl Phys Lett. 2007;90:163903. [Google Scholar]
- 49.Prato M, Bianco A, Maggini M, Scorrano G, Toniolo C, Wudl F. J Org Chem. 1993;58:5578–5580. [Google Scholar]
- 50.Meldal M. Meth Enzymol. 1997;289:83–104. doi: 10.1016/s0076-6879(97)89045-3. [DOI] [PubMed] [Google Scholar]
- 51.Kittel C. Introduction to Solid State Physics. 7th Wiley; New York: 1995. [Google Scholar]
- 52.Nigamatullin RR, Baleanu D, Povarova D, Salah N, Habib SS, Memic A. Math Prob Eng. 2013;2013:847076. [Google Scholar]
- 53.Huang L-C, Chang H-C. Langmuir. 2004;20:5879–5884. [PubMed] [Google Scholar]
- 54.Gross LA, Mohn F, Moll N, Schuler B, Criado A, Guitián E, Pena D, Gourdon A, Meyer G. Science. 2012;337:1326–1329. doi: 10.1126/science.1225621. [DOI] [PubMed] [Google Scholar]
- 55.Komatsu N, Kadota N, Kimura T, Osawa E. Chem Lett. 2007;36:398–399. [Google Scholar]
- 56.Fang X, Mao J, Levin EM, Schmidt-Rohr K. J Am Chem Soc. 2009;131:1426–1435. doi: 10.1021/ja8054063. [DOI] [PubMed] [Google Scholar]
- 57.Dubois M, Guérin K, Batisse N, Petit E, Hamwi A, Komatsu N, Kharbache H, Pirotte P, Masin F. Solid State Nucl Magn Reson. 2011;40:144–154. doi: 10.1016/j.ssnmr.2011.10.003. [DOI] [PubMed] [Google Scholar]
- 58.Cui J-F, Fang X-W, Schmidt-Rohr K. J Phys Chem C. 2014;118:9621–9627. [Google Scholar]
- 59.Gimond C, Sonnenberg A. In: In Integrin-Integrin Interaction. Eble JA, Kühn K, editors. R.G. Landes Company; Austin, TX: 1997. pp. 219–240. [Google Scholar]
- 60.Plopper GE, McNamee HP, Dike LE, Bojanowski K, Ingber DE. Mol Biol Cell. 1995;6:1349–1365. doi: 10.1091/mbc.6.10.1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Borgia JA, Fields GB. Trends Biotech. 2000;18:243–251. doi: 10.1016/s0167-7799(00)01445-1. [DOI] [PubMed] [Google Scholar]