

Abstract
Excess or insufficient lipid storage in white adipose tissue lipid droplets is associated with dyslipidemia, insulin resistance and increased risk for diabetes type 2. Thus, maintenance of adipose lipid droplet growth and function is critical to preserve whole body insulin sensitivity and energy homeostasis. Progress in understanding biology of lipid droplets has underscored the role of proteins that interact with lipid droplets. Here, we review the current knowledge of adipose specific lipid droplet proteins, which share unique functions controlling adipocyte lipid storage, limiting lipid spill-over and lipotoxic effects thought to contribute to disease. This article is part of a Special Issue entitled: Modulation of Adipose Tissue in Health and Disease.
Keywords: Adipose tissue, Lipid droplet associated protein, Lipid droplet growth, Obesity, Insulin resistance
1. Introduction
Lipid droplets (LDs) are the lipid storage organelles of all organisms. Adipose tissue is the body’s largest energy reservoir in mammalians and birds. Energy is stored in fat cell LDs as triacylglycerols (TGs). In the past fifty years, drastic life style and environmental changes have contributed to a worldwide pandemic of obesity and co-morbidities that demands a better understanding of adipose LDs, their role in maintaining energy homeostasis and impact on development of metabolic diseases. In recent years, our general knowledge of the biology of LDs has increased, reviewed extensively elsewhere [1–6]. This review is focused on specific aspects of adipose LD biology as it relates to metabolic diseases.
2. Critical role of adipose LDs in mammalian physiology and diseases
2.1. White adipose energy storing LDs
LDs in mammalian adipocytes in white adipose tissue (WAT) serve as the main long-term energy store and play a crucial role maintaining energy homeostasis [7,8]. The remarkable lipid storage capacity of white adipocyte LDs can be readily visualized by microscopic observation [5]. Most mature adipocytes have a single LD, whose size can range from 25 to 150 μm diameter, occupying most of the cell volume and thereby determining the cell size. Adipose depots grow by either increasing fat cell/LD size (hypertrophy) or increasing the number of fat cells (hyperplasia) (Fig. 1). Importantly, both mechanisms require coordinated and extensive cellular structural changes that accommodate the emergence and growth of the LD. In the fed state, adipose LDs store excess energy as TG. During fasting, when glucose becomes limiting, TGs in adipose LDs are rapidly hydrolyzed into non-esterified fatty acids (NEFAs) and glycerol. NEFA and glycerol leave the adipose and are transported via the bloodstream to other tissues (for glycerol mainly to the liver and for NEFA mainly to skeletal muscle and heart). During fasting plasma NEFA is almost entirely from hydrolysis of TG stored in the adipose LDs (Fig. 1) [9].
Fig. 1.
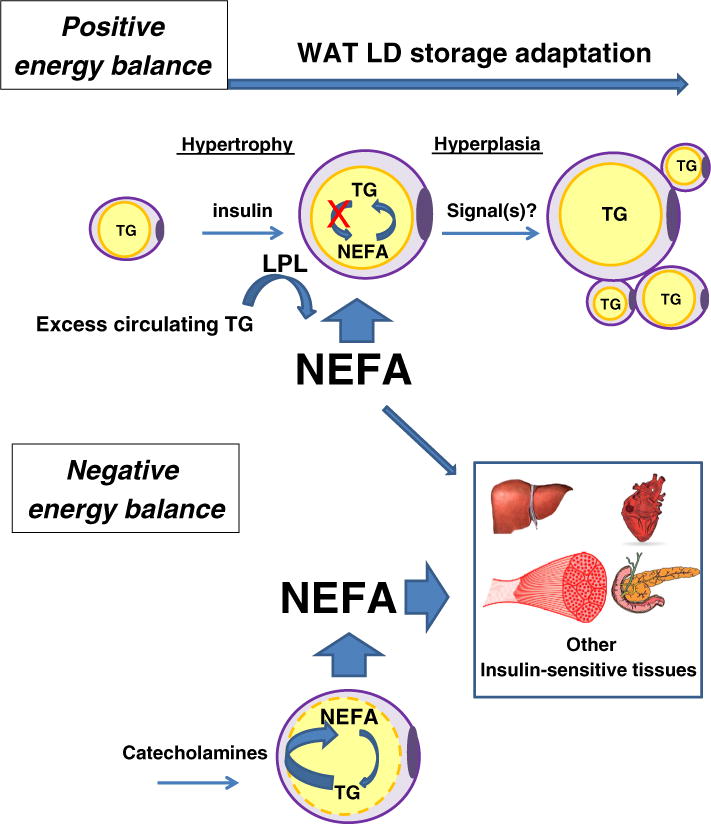
Healthy adipocyte lipid storage expansion and lipid release. In the fed state with positive energy balance, excess circulating lipids are hydrolyzed by lipoprotein lipase (LPL) synthesized in adipose tissue parenchymal cells and spread along the vascular mesh. LPL-released non-esterified fatty acids (NEFAs) are then re-esterified into triglycerides (TG) and stored in adipocyte LDs. It is accepted that once LD growth has reached its capacity (hypertrophic growth), new preadipocytes are either recruited or undergo maturation (hyperplasia). This requires effective tissue remodeling including, adequate implementation of the adipogenic program, angiogenesis and extracellular matrix remodeling supported by endocrine paracrine and neuronal factors. Signaling from adipocytes filled to their LD storage capacity helping recruit preadipocytes are possible, but have yet to be identified.
When energy and macronutrient levels are saturated by chronic overfeeding, surplus energy is stored in adipose LDs and leads to obesity, generally defined as excessive accumulation of TG in WAT. Concomitant with increased adipocyte LD size, pathological overgrowth of adipose tissue is associated with a cluster of changes including hypoxia, inadequate angiogenesis, increase in adipocyte cell death, macrophage infiltration, fibrosis and adipose tissue insulin resistance (Fig. 2). The adipocyte’s micro-environment is severely impacted and limits adipose tissue expandability by inhibiting recruitment of new lipid storage units (preadipocytes) and/or preventing their maturation (Fig. 2) [10,11]. This scenario is supported by recent murine and human studies. Overexpressing adiponectin in murine adipose tissue, an adipokine with known anti-inflammatory and anti-insulin resistance properties [12], or down-regulating collagen VI, a highly enriched extracellularmatrix component of adipose tissue [13], leads to expansion of adipocytes but paradoxically is associated with substantial improvements in whole-body energy homeostasis, both with high-fat diet exposure and in leptin deficient background [12,13]. The main difference found between fat cell/LD size from insulin-sensitive and insulin-resistant obese patient adipocytes is a propensity for a small fat cell/LD size in obese insulin resistant patients, suggesting a defect in LD growth and maturation [14]. Alternatively, treatment with pharmacological agents that promotes adipose tissue expandability such as thiazolidinediones (TZDs) reverses it [15].
Fig. 2.
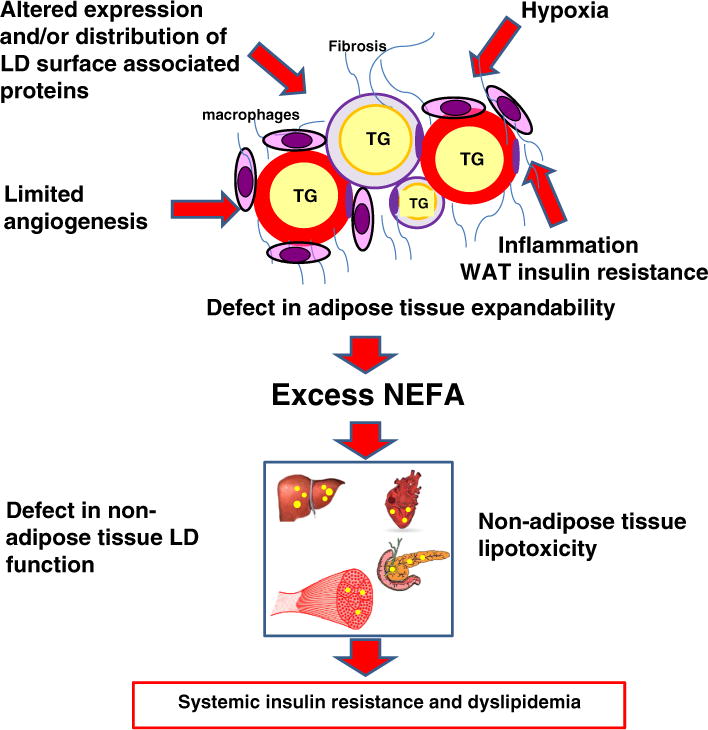
Unhealthy adipocyte lipid storage expansion associated with obesity. White adipose tissue (WAT) maladaptation to chronic excess lipids includes altered expression and/or distribution of LD surface associated proteins, inadequate angiogenesis, hypoxia, increased adipocyte cell death, increased macrophage infiltration, increased inflammation, increased fibrosis and adipose tissue insulin resistance that leads to reduced adipose tissue capability to store lipids. Excess spill over lipids are then directed to non-adipose tissues with a limited capability for LD storage. Defects in non-adipose tissue LD lead to tissue lipotoxicity, whole body imbalance of glucose/lipid energy homeostasis and development of pathological states such as T2D.
More importantly, obesity is thought to be the most common cause of systemic insulin resistance and it is a key factor in the etiology of a number of diseases, including type 2 diabetes (T2D) [16]. Insulin resistance is defined as an inadequate response to insulin in target tissues, such as skeletal muscle, liver, and adipose tissue, reducing physiologic effects of circulating insulin. The hallmark of impaired insulin sensitivity in WAT is a reduced ability of insulin to inhibit LD lipolysis, resulting in elevated circulating NEFAs. An evidence based consensus is that high NEFA release from WAT causes insulin resistance in skeletal muscle, liver and other tissues [17,18]. It is inferred that these tissues are unable to store or oxidize the lipid influx. The lipid then floods cellular pathways and compartments, causing dysfunction labeled lipotoxicity (Fig. 2) [19]. It is purposed that the link between obesity and systemic insulin resistance is the result of ineffective lipid partitioning to adipocyte LDs and these lipids disrupt adipokine and cytokine secretion [20].
The proposed basis for the relationship between obesity and systemic insulin resistance relies on WAT LDs acting as a lipid sink for excess nutritional lipids, storing them in the form of neutral lipids. This LD centric view argues that as long as nutrient excess can be efficiently sequestered in insulin sensitive white adipose LDs, non-adipose tissues are protected from lipotoxicity. This concept was further shaped from observations that animals and humans with lipodystrophy, in which adipose tissue fails to develop properly or is ill-distributed, also have ectopic LD deposition, contributing to insulin resistance and eventually to decreased insulin secretion [21,22]. Supporting the importance of WAT LD function is the fact that monogenic mutations responsible for 95% of lipodystrophies were found to effect either adipogenesis or LD growth and function. These adipogenesis gene mutations include peroxisome proliferator-activated receptor-γ (PPARγ), Akt2, or lamin A/C. LD growth gene mutations found include perilipin 1 (PLIN1), cell death-inducing DFF45-like effector C (CIDEC), cav-1 (CAV-1), Polymerase 1 and Transcript Release Factor (PTRF/cavin-1), seipin (BSCL2), and 1-acylglycerol-3 phosphate-oacyltransferase 2 (AGPAT-2) [21].
In this context, understanding mechanisms that control expandability of adipose LD storage may be essential to determining risk factors for development of diabetes with obesity and better treatments for lipodystrophies.
2.2. Brown and beige/brite adipose energy consuming LDs
Whereas white adipocyte LDs store fat, brown adipocyte LDs are adapted to dissipate stored energy as heat for thermogenesis. Since 2007, several independent research teams have shown conclusively that adult humans have functional brown adipose tissue (BAT) [23–27]. This discovery of functional BAT in adult humans re-energized the obesity research field. BAT is primarily localized in discrete locations such as the supraclavicular and axillary regions but can also be found interspersed within WAT and skeletal muscle tissues in rodents [28,29]. Brown fat cells that emerge in white fat depots under certain conditions have been described as “beige” or “brite”. The current thinking is that beige cells and brown fat cells come from separate cell lineages [30]. But some reports suggest that WAT could trans-differentiate to BAT and vice versa [31,32]. The thermogenic capacity of even small amounts of BAT makes it an attractive therapeutic target for weight loss, and for anti-obesity or anti-T2D therapies increasing energy expenditure.
The origin, differentiation, function and physiological importance of BAT have been reviewed [33,34], but while LDs in WAT have been extensively studied, much less is known about their biology and function in BAT. The understanding of BAT LDs and their regulation relies heavily on knowledge of WAT LDs, but advances have been limited to date since the brown or beige/brite LD proteomes have yet to be fully characterized. Intriguingly, over-expression and loss of function of some WAT LD associated proteins in transgenic murine models have resulted in an increased presence of brown adipose cells and improved systemic insulin sensitivity. Here we will review in this context some of the studies.
3. LD surface-associated proteins as a critical interface for LD function
The importance of the normal function of WAT LDs to preserve whole body insulin sensitivity and energy homeostasis has spurred interest in extensive characterization of white adipocyte LDs, the most adapted fat-storing organelle. Seminal work by Dr. Londos and colleagues led to identification of the first LD surface-associated protein, Plin1, in white adipocytes [35]. Their discovery revoked the classic view of LDs as passive storage structures of lipids and gave momentum to new lines of investigation aiming to better understand the biology of this organelle in physiological and pathological conditions [1–8]. LDs are now known to be dynamic organelles that have a key regulatory role in the cellular turnover of lipids, and they exist in most cell types and organisms [36–39]. All cytosolic LDs have a spherical shape, giving the least surface area. They comprise a core of neutral lipids, and in adipocytes the most abundant species is TG, with a phospholipid monolayer surface containing free cholesterol [40] and proteins that regulate the LD function. White adipocyte LD proteome must be well adapted to efficiently control storage and release of lipids. This remarkable lipid storage capacity is characterized by presence of a unilocular droplet, occupying most of the cell volume, imposing a lack of droplet motility and a unique physical close proximity with the plasma membrane.
Results from four proteomic studies using LDs isolated from primary adipocytes and adipogenic 3T3-L1 cells identified over two hundred LD proteins [41–44]. Overall, the classification profile of this unexpected large panel of proteins in the adipocyte LD proteome is quite similar to other reported mammalian proteomic profiles obtained from other cell types or from Drosophila fat bodies, confirming the status of the LD as a defined and conserved organelle. It includes the predominant representation proteins that belong to the perilipin protein family, lipid metabolism related enzymes and modifiers, intracellular trafficking including many Rab proteins, chaperone proteins, cytoskeleton elements, ER and mitochondria proteins. These observations underscore the dynamic surface of LDs and the importance of communication with other intracellular organelles though specific interactions. It remains a possibility that the use of cell fractionation techniques, the highly hydrophobic nature of the lipid droplet, the relative abundance of adipocyte proteins and their close structural association with ER and mitochondria might confound the proteomic analyses. Secondary analysis using imaging and functional studies is necessary to confirm a subset of these proteins as bona fide LD proteins, and to localize them on the LD surface. The most surprising finding of these studies is that despite a remarkable morphological difference between adipose and non-adipose LDs, a relatively small number of specifically enriched adipose LD associated proteins have been identified so far, but includes Plin1, Fsp27/Cidec, caveolins and cavins. Intriguingly these proteins all have direct functional links with development of systemic insulin resistance.
4. Adipocyte-specific LD proteins with direct links to insulin resistance
4.1. LD proteins controlling adipose LD size and hydrolysis
4.1.1. Perilipin 1
Perilipin 1 (Plin1) was the first member recognized of the perilipin protein family. The family is defined by sequence similarity across species and currently has five members [45]. A comprehensive overview of the perilipin family has been published elsewhere [46–48]. The perilipins constitute a proteome “signature” for LDs that consistently includes at least one of the five members. A perilipin is always present and quantitatively represents the most abundant protein, suggesting at least an important structural role for this class of proteins in LD machinery [45–47]. Perilipin distribution is also tissue and FA utilization dependent. Plin1 and perilipin 4 (Plin4, previously S3–12) are highest in adipose tissue. Perilipin 2 (Plin2, previously adipophilin, ADRP) and perilipin 3 (Plin3, previously Tip47) are ubiquitous, although Plin2 is highly abundant in the liver. Perilipin 5 (Plin5, previously MLDP, OXPAT, LSDP5) is found primarily in oxidative tissues, including BAT or subcutaneous WAT treated with peroxisome proliferator-activated receptor gamma (PPAR-γ) agonists [48]. In mice and humans, a single Plin1 gene gives rise to at least three isoforms, Plin1A, B and C, with a common N-terminal region but differing in C-terminal length [45]. Plin1A and 1B are highly expressed in adipose tissues while Plin1C is preferentially found in steroidogenic tissues. Applying fluorescence activated cell sorting (FACS) to separate fluorescently labeled LDs, it was recently demonstrated that isoforms of Plin1 differentially coat either TG (Plin1A and B) or cholesterol ester (CE) specific LDs (Plin1C), emphasizing diversity of function for the different Plin1 isoforms [49]. So far, there is little understanding of the physiological importance of expression and regulation of these isoforms. Plin1A, often referred to as Plin1, is the most abundant form and constitutive of the LDs as well as the major PKA substrate in adipocytes. Its transcription has been found regulated by estrogen receptor-related receptor alpha (ERR-α), peroxisome proliferator-activated receptor gamma (PPAR-γ) and more recently by liver X receptor alpha (LXR-α) [50–53]. During the past ten years, using cell culture studies and transgenic mice models, several laboratories demonstrated an important role of Plin1 orchestrating both TG and diacylglycerol hydrolysis in adipocytes in response to phosphorylation by protein kinase A (PKA) [54–58]. Plin1 regulates substrate access of adipose triglyceride lipase (ATGL) and hormone sensitive lipase (HSL), two key adipose LD hydrolytic enzymes with triacylglycerol lipase and diacylglycerol lipase activity, respectively [59]. Plin1 serves as a scaffolding protein at the LD surface mediating protein/protein interactions with key players in LD hydrolysis. The present accepted model is that when lipolysis is suppressed by insulin (basal conditions), comparative gene identification-58 (CGI-58), a co-activator of ATGL, preferentially binds to un-phosphorylated perilpin-1 at the surface of the LD [47,59,60]. Under these conditions, ATGL is located in both the cytosol and on LDs, whereas HSL is only cytosolic. Upon β-adrenergic stimulation, HSL and Plin1 are both phosphorylated by PKA, resulting in a reorganization so that HSL binds Plin1 at the LD surface via at an N-terminal region [58,61] and dissociation of CGI-58 from Plin1, which promotes CGI-58 interaction with ATGL [62]. Natural occurring genetic errors in humans and in vitro truncation studies have identified a protein–protein surface in teraction that involves Plin1 C-terminal with the cap region (pro180 to Leu280) of CGI-58 [63].
4.1.2. Links between Plin1 and insulin sensitivity and energy homeostasis
Transgenic mice with Plin1 loss of expression helped establish a link between Plin1 and systemic insulin sensitivity [64,65]. Plin1 null mice, generated by two separate groups of investigators, show a lean phenotype and systemic insulin resistance with aging [64,65]. Isolated adipocytes from Plin1 null mice showed elevated constitutive (un-stimulated) lipolysis attributed to loss of a TG protective function of Plin1. They also showed dramatically attenuated stimulation of lipolytic activity. Intriguingly, the usual adipose parameters implicated in development of insulin resistance, NEFA, leptin and adiponectin plasma levels, were not substantially different between Plin1 null and wild type mice. Indeed, despite demonstrated increased rates of constitutive un-stimulated lipolysis in Plin1 null adipocytes, circulating NEFAs were either decreased [65] or unchanged [64]. A drastic reduction of adipose LD size and a compensatory increase of adipose FA β-oxidation likely contribute to the observed lack of expected increase in circulating NEFA in this mice model [66]. Thus, the exact mechanisms underlying a functional relationship between Plin1 and systemic insulin resistance in the Plin1 null mice are yet to be fully understood. Interactions of Plin1 with other modifier genes that effect both weight and insulin sensitivity with age are possible, but remain to be identified. A further complication in understanding Plin1 role in insulin resistance is an unexpected effect found with over-expression of Plin1 in adipose tissue resulting in a lean phenotype, resistance to diet induced obesity and improvement of whole body insulin sensitivity. Further analyses indicated compensatory mechanisms including increased β-oxidation and decreased levels of Fsp27, a LD associated protein involved in LD growth. Although the translational aspect of these studies is difficult to interpret, they do reveal LD surface limitation for protein binding and murine adipocyte adaptation to shunt excess NEFA via mitochondria β-oxidation.
Two separate heterozygous missense mutations in PLIN1, which alters amino acids in the C-terminus of the protein, were recently identified in six patients and associated with a novel form of familial partial lipodystrophy, hepatic steatosis, dyslipidemia, insulin resistance, and severe T2D [67]. Histological analysis of WAT showed increased presence of macrophage infiltration and fibrosis. Follow-up with in vitro mechanistic studies showed that both mutations fail to inhibit un-stimulated lipolysis due to an inability to bind and stabilize CGI-58 [63,67].
Overall, these rare PLIN1 human mutations present a stronger systemic phenotype than observed in the Plin1 null mice. The differences may be due to species differences in adipose tissue metabolism. Studies comparing transgenic murine models with rare human polymorphisms are extremely valuable to establish in vivo gene function. So far, only a limited number of human studies have investigated links between adipocyte PLIN1expressionand obesity phenotypes, with inconsistent results. An early study showed adipose PLIN1 levels to be greater in non-diabetic obese individuals than in lean individuals and positively correlated with percent body fat, but unrelated to insulin resistance or inflammatory markers [69]. In contrast, two studies comparing morbidly obese vs non-obese patients or obese vs non-obese women showed lower PLIN1 levels in the obese populations [70,71]. In the latter study, a greater rate of lipolysis was associated to a specific PLIN1 polymorphism [71]. Finally, several studies found a number of PLIN1 polymorphisms that potentially influence body weight and risk of metabolic disease, although this was not fully reproduced in other studies. The impact of PLIN1 polymorphisms on health has been recently reviewed [72].
4.1.3. Cidea and Fsp27/Cidec
The CIDE family currently consists of three proteins, Cidea, Cideb and Fsp27 (murine)/Cidec (human) based on primary sequence homology and has been reviewed elsewhere [73,74]. CIDE family expression is tissue-dependent with Cidea primarily in brown adipocytes, Fsp27/Cidec mostly in white adipocytes and Cideb in the liver. In humans, higher expression of Cidea occurs in white adipocytes along with Cidec. All three CIDE proteins can be found at the ER and at the LD surface [75,76]. Work from several laboratories has clearly demonstrated importance of Fsp27/Cidec and Cidea in the regulation of adipocyte LD size and adipocyte TG storage.
An earlier report showed that ectopic expression of Fsp27 in adipogenic 3T3-L1 cells resulted in larger LD size while knockdown of Fsp27 in fully differentiated adipocytes resulted in smaller LD particle size and increased un-stimulated lipolysis, compared to wild type cells [76]. Recent progress has been made in understanding molecular events underlying Fsp27/Cidec function. Using confocal imaging and fluorescent recovery after photobleaching, both Fsp27 and Cidea were shown to facilitate clustering of droplets [77,78] and to bundle at discrete LD–LD contact sites, mediating transfer of bulk lipids from smaller LDs to larger LDs, thus promoting LD growth [77]. This process differs from vesicle fusion by a dependency on LD size and extended time scale. Fsp27-mediated LD growth does not seem to be dependent on the presence of adipose specific proteins, especially Plin1, as ectopic Fsp27 is able to promote LD growth in non-adipose cultured cells.
Further studies are necessary to identify if other protein(s) are involved and to understand the specifics of Fsp27-mediated reorganization of lipids and proteins at the LD surface. This will require obtaining native purified Fsp27 protein to perform in vitro biochemical reconstitution of Fsp27-mediated lipid transfer. A recent publication of a preliminary partial analysis of the crystal structure of the CIDE-N domain of Fsp27 promises to help elucidate structure–function of this protein [79]. The molecular mechanisms by which Fsp27 controls lipolysis remain unclear but Fsp27 was recently shown to constitutively limit LD association of ATGL [80]. Further analyses are required to distinguish if Fsp27/Cidec competes with ATGL for lipid binding sites on the LD surface or if re-organization of other proteins/lipid at LD surface prevents ATGL binding.
New exciting findings indicate that Cidea and Fsp27 may exert cellular dual functions as both proteins were reported to localize in nuclei [81,82]. However, a nuclear localization signal (NLS) sequence has yet to be identified and tested in these proteins, leaving the underlying mechanisms for their nuclear translocation to be investigated. Human Cidea sequence analysis reveals two potential motifs for nuclear receptor binding [81]. Cidea is shown to bind to LXR in in vitro and ex vivo systems and to repress LXR-regulated reported constructs in 3T3-L1 [81]. Interestingly, LXRs have been implicated in lipolytic regulation in several studies [53]. The increased in basal lipolysis caused by LXR was dependent on the decreased expression of Plin1. Cidea nuclear activity may also be important to regulate human adipose LD hydrolysis.
In addition, Cidea acts as a transcriptional co-activator regulating mouse mammary gland secretion of milk lipids by directly interacting with CCAAT/enhancer-binding protein (C/EBP-β) in mammary epithelial cells and in BAT [82]. Fsp27 was also found to interact with C/EBP-β in differentiated 3T3-L1 cells and BAT [82]. Intriguingly, recent studies suggested an unexpected and additional function for Fsp27, regulating nuclear factor of activated-T cells 5 (NFAT5) intracellular localization and downstream signaling [83]. NFAT5 is a member of the Rel family of transcription factors localized in the cytosol, activated in response to osmotic stress and translocating to the nuclei where it regulates, osmoprotective and inflammatory genes such as MCP1 and TNF-α, two major proteins known to be involved in chronic inflammation associated with adipose insulin resistance [20]. Protein–protein interaction between Fsp27 and NFAT5 was identified in a yeast two-hybrid screen. NFAT5 has a highly conserved NLS sequence motif, playing an important role in the nuclear translocation. Over-expression of Fsp27 decreases the nuclear trafficking of NFAT5 after induction of cellular hypertonic stress and inhibited NFAT5 transcriptional activation of MCP1. Future investigations are needed to show how these functions of Fsp27, LD growth and regulation of NFAT5, integrate in physiological conditions or in obesity and T2D.
Overall, these novel findings support an interesting emerging concept of LD proteins playing a role in nuclear transcription.
4.1.4. Links between Cidea and Fsp27/Cidec with insulin sensitivity and energy homeostasis
Fsp27 null mice were generated independently by two laboratories [84,85]. Phenotypic consequences of Fsp27 loss of function are similar to those observed in Plin1-null mice, including reduced fat mass, increased lipolysis and increased adipocyte β-oxidation [84]. Isolated adipocytes from Fsp27 null mice showed elevated constitutive lipolysis, attenuated β-adrenergic-stimulation and elevated lipid oxidation with up-regulated mitochondrial activity [84]. Fsp27 null mice have increased energy expenditure and are protected from genetic or diet-induced obesity [84,85]. A noticeable difference at the cellular level between Plin1 and Fsp27 null mice was the observation that Fsp27 null white adipocytes contained multilocular droplets while they remain unilocular in Plin1 null. Plin1 null mice develop insulin resistance by 6 months, but Fsp27 null mice remained insulin sensitive through a 4-month study duration. It remains un-tested if absence of Fsp27 impacts glucose homeostasis at a later age. However, insulin sensitivity of the leptin deficient ob/ob mice was improved with Fsp27 deficiency. Thus, while both Plin1 and Fsp27 deficiency in mice affects adipose LD size, LD hydrolysis and β-oxidation, it appears to result in different outcomes for systemic insulin sensitivity, perhaps because of differences in degree of compensatory increase of adipose mitochondria β-oxidation or in levels of adipokine and cytokine secretion.
Cidea null mice exhibit increased lipolysis in BAT and are resistant to high fat diet-induced obesity and diabetes, an indication that Cidea may share some of the function of Fsp27 in this tissue [86].
A rare homozygous nonsense mutation in the human Cidec gene resulting in a truncated protein unable to bind to LDs was recently identified in one patient. Unlike mice but similar to human PLIN mutations, this human CIDEC mutation was found to be associated with partial lipodystrophy, T2D, hypertriglyceridemia, and hepatic steatosis [87]. Furthermore, adipose expression levels of human Cidea and Cidec were found positively correlated with insulin sensitivity in obese patients matched for BMI [71]. These human studies appear to be in contradiction with transgenic murine models and future studies are needed to confirm if this discrepancy between mice and man indicates the existence of species-specific differences in the compensatory metabolic response to defects in LD biology.
4.3. Scaffolding proteins enriched in adipose LD proteome and role controlling LD size
4.3.1. Caveolins and cavins, potential key players in adipose LD size
Caveolins (cav) and cavins (cavin) are primarily known as the two major protein components of caveolae, invaginations in the plasma membranes (PM) of most mammalian cell types. They are particularly abundant in white adipocytes where they are thought to play an important role in lipid metabolism [88–90]. It was proposed that some subtypes of caveolae are discrete sites of TG synthesis in adipose tissue plasma membranes, a possible tissue specific adaptation to adipose high TG volume storage [90]. In addition, while cav-1 and -2 and cavin-1 are highly expressed in adipose tissues and localized at the PM, a direct association of caveolins with the LD surface was reported by several groups [92–94]. Furthermore, evidence now support interactions of cav-1 and cavin-1 with LD proteins involved in the lipolytic pathway. Co-immunoprecipitation experiments revealed close association of cav-1 with Plin1 but not Plin2, underscoring a unique and specialized structural lipid/protein organization of the adipose LD surface [95,96,93]. Finally, cavin-1, present in the adipose proteome, was reported to interact with HSL in primary human adipocytes and suggested to act in concert with HSL to regulate lipolysis [97].
Overall these studies support a role for cav-1 and cavin-1 in adipocyte “ins and outs” of NEFA and thus as potential key regulators of LD size. This was confirmed by observations that LD cav-1 protein expression is positively correlated with adipocyte LD size and that absence of cav-1 or cavin-1 in mice and humans results in a marked reduction of adipocyte LD size [96,98–102]. The reasons why adipocytes remain atrophic in the total absence of cav-1 are not linked to overt alterations in food intake, nutrient absorption or energy expenditure, and this triggered an interest to understand the basis for relationship existing between caveolins, cavins and adipose LD size.
Caveolae have a requirement for cholesterol for their role in PM, and cav-1 is known to interact with cholesterol in membranes [103]. Changes in free cholesterol LD surface content could affect lipid surface composition and interactions with LD surface proteins, thus potentially altering LD growth. This was tested by comparing LD proteomes from cav-1 deficient and wild-type adipocytes [43]. Indeed, relative abundance of surface phospholipid species, phosphatidylserine and lysophospholipids, is reduced in cav-1 deficient cells. But analysis of the cav-1 null adipocyte LD proteome indicated that undetected proteins were only those normally found in association with cav-1 in the caveolae protein complex, including EH-domain containing 2 (EHD2) and cavin-1 as well as some cytoskeleton proteins [43]. These results confirmed integrity of caveolae structure at the LD surface. However, the qualitative proteomic approach cannot entirely rule out subtle differences in LD protein composition that may still affect LD growth or maintenance.
Cav-1 and cavin-1 may control adipose LD size via lipolysis. Caveolae structures and expression of caveolar proteins in white adipocytes are increased with fasting, and their interactions with key players in the lipolytic pathway were established [95–97]. But primary isolated cav-1 deficient adipocytes also lacking cavin-1 have an impaired β-adrenergic agonist-stimulated lipolysis and a specific impairment in PKA-dependent Plin1 and HSL phosphorylation [95]. In contrast, cav-1 deficient MEF cells retained cavin-1 expression, have enhanced β-adrenergic stimulated lipolysis without alteration in PKA-Plin1 or HSL phosphorylation [104]. These results point to differential functions for cav-1 and cavin-1, and a potential role of cavin-1 in regulation of lipolysis, but fail to explain the lower LD size observed in the primary cav-1 deficient adipocytes. Finally, it was reported that cav-1 deficient adipocytes have increased autophagy but it remains to be experimentally demonstrated whether this mechanism contributes to decreased LD size by channeling LD lipids to lysosomal degradation [105].
Discrepancies between in vivo and in vitro experiments raise concerns that perhaps the decreased adipose LD size observed in the cav-1 null mice may only be secondary to adipose lipotoxicity, induced by absence of caveolae to protect the adipocyte against fluctuations of NEFA traffic in and out [98]. Adipose lipotoxicity could be responsible for a detrimental sequence of events observed in cav-1 null mice, including release of cytokines, macrophage infiltration and development of fibrosis and lipoatrophy. However, expression of endothelium-derived caveolin in the caveolin KO mice curtailed macrophage infiltration and local levels of cytokines (except adipose secreted PAI-1) without rescuing the lipoatrophy phenotype, an indication that cav-1 and or cavin-1 may control LD size in absence of severe local inflammation [106].
Overall, the specific mechanism(s) enabling cav-1 and cavin-1 to control LD size remains difficult to tease apart as these proteins are assembled in the caveolae structure and absence of one of these proteins effects expression of the others and hence the caveolae structure itself. Future studies are needed to elucidate how cav-1 and cavin-1 regulate adipose LD size, their intracellular dynamics and the details of their interactions with other players of the LD hydrolysis.
4.3.2. Links between caveolins, cavins and insulin sensitivity and energy homeostasis
Mice lacking cav-1 are lean, have progressive lipoatrophy, and are hyperlipidemic, insulin-resistant and resistant to diet-induced obesity [95]. Mice lacking cavin-1 have a similar phenotype, albeit they develop hyperinsulinemia between 8 and 12 weeks of age [98]. The aggravated systemic phenotype of the cavin-1 null may be a consequence of the absence of skeletal muscle caveolae in this transgenic model [98].
In agreement with mice, a human homozygous null mutation in the CAV-1 gene was identified in one patient associated with near absence of adipose tissues, severe insulin resistance, T2D and dyslipidemia [99]. Additional heterozygous mutations were later identified with partial lipodystrophy [100]. Cavin-1 homozygous mutations were identified in 21 patients to cause congenital generalized lipodystrophy, associated in some of these patients with additional skeletal and cardiac muscle pathologies [101,102]. Interestingly, when examined, signs for adipose tissue macrophage infiltration and fibrosis were observed in human adipose tissue histological samples from CAV-1 and cavin-1 null mutation carriers. The identification of these mutations provides strong human genetic evidence for a critical role of these proteins in adipocyte lipid storage and systemic glucose/lipid homeostasis.
5. Conclusions
The white adipocyte has been termed the “professional” lipid storage cell [107], a reference to its long accepted function in whole organisms. Recent advances in LD biology have underscored the importance of LD associated proteins in tissue-specific lipid storage and utilization. Comparatively few adipocyte-specific proteins, associated with LDs within fat cells, have been identified to control lipid stores so far. Although the exact roles of these proteins, especially Cidec, Cidea, cav-1 and cavin-1 remain to be clarified all these proteins facilitate sequestration of circulating NEFAs in the form of esterified lipids, control NEFA release when needed, and prevent metabolic complications due to lipotoxicity. The cellular distribution of these proteins ranging from largely only PM and LD for cav-1 and cavin-1, only ER and LD for Cidea and Cidec and finally exclusively LD for Plin1, revealed structural organization specific to the adipocyte presumed to maximize the efficiency and coordination of unesterified lipid flux in and out of cells (Table 1). Genetic ablation of these proteins, whether by design in mice or by nature in humans, results in various degrees of lipoatrophy and lipodystrophy, insulin resistance, dyslipidemia and increased adipose tissue inflammation. Expression and distribution of these proteins are found altered in insulin resistance associated with obesity, but dynamics of these proteins and their respective protein interactions need to be identified (Table 3).
Table 1.
Dynamics of known LD surface-associated proteins and key interaction partners in adipocyte found associated with positive/negative energy balance or with obesity/insulin resistance state.
| PM | Cytosol | ER | LD surface | |
|---|---|---|---|---|
| Positive energy balance | Cav-1 | HSL | Cidec | ATGL |
| Cavin-1 | ATGL | Cidec | ||
| Cavin-1 | Plin1 | |||
| Cav-1 | ||||
| CGI-58 | ||||
| Protein interactions | Cavin-1 ↔ HSL | Cav-1 ↔ Plin1 | ||
| CGI-58 ↔ Plin1 | ||||
| Negative energy balance | Cav-1 | CGI-58 (?) | Cidec | HSL-P |
| Cavin-1 | Cidec | |||
| ATGL | ||||
| Plin1-P | ||||
| CGI-58 | ||||
| Protein interactions | Plin1-P ↔ HSL-P | |||
| CGI-58 ↔ ATGL (?) | ||||
| Obesity/insulin resistance | Cav-1 (?) | HSL | Cidec (?) | Cav-1 |
| Cavin-1 (?) | ATGL (?) | Cavin-1 | ||
| Cavin-1 (?) | Plin1 | |||
| CGI-58 (?) | Cidec | |||
| ATGL (?) | ||||
| CGI-58(?) | ||||
| Protein interactions | Cavin-1 ↔ HSL (?) | CGI-58 ↔ ATGL (?) |
LD proteomic studies have identified a handful of murine white adipose specific lipid droplet proteins, perilipin 1 (Plin1), Cidec, Caveolin-1 (Cav-1) and Cavin-1, all sharing activities in LD growth and lipolysis. Cellular distribution of these proteins indicates importance of close spatial relationships between plasma membranes (PM), endoplasmic reticulum (ER) and lipid droplet (LD) surface to support the unique “professional” role of adipose LD to store a high volume of circulating unesterified lipids while preventing local cellular lipotoxicity. Font size represents relative amount of protein. P indicates if protein is phosphorylated. Protein expression, distribution or interaction not yet experimentally demon strated is noted with a question mark.
Abnormal adipose LD growth may stunt the ability to recruit and mature new adipocytes by either initiating a pro-inflammatory microenvironment or preventing release of yet uncharacterized sensing mechanisms whereby once adipocytes reach a critical volume they secrete factors essential to promote the recruitment of new adipocytes. While studying LD associated proteins has highlighted their importance to maintain adipose LD growth and integrity, they are also valuable to open the door to a better understanding to adipose tissue lipid storage expandability in health and metabolic diseases.
Acknowledgments
We honor the memory of Dr. Dean Londos who inspired so many of us to focus our research interest on lipid droplet biology. We also sincerely thank Dr. Rosalind Coleman and Dr. Dawn Brasaemle for having launched the FASEB lipid droplet meetings, their intelligent initiative gave the perfect venue to catalyze this field of investigation. Finally, we thank Dr. Martin Woodle for careful review and helpful editing of the manuscript.
This work was supported by a career development award 1-05-CD-17 from the American Diabetes Association (to C.S.), a grant from NIH 1RO1 DK 075017 (to C.S.), a grant in aid 11GRNT7600027 from the American Heart Association (to C.S.), the Geriatric Research, Education and Clinical Center, Baltimore Veterans Affairs Health Care Center, the Clinical Nutrition Research Unit of Maryland (DK072488), and the Intramural Research Programs of NIDDK.
Footnotes
References
- 1.Walther TC, Farese RV., Jr Lipid droplets and cellular lipid metabolism. Annu Rev Biochem. 2012;81:687–714. doi: 10.1146/annurev-biochem-061009-102430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brasaemle DL, Wolins NE. Packaging of fat: an evolving model of lipid droplet assembly and expansion. J Biol Chem. 2012;287(4):2273–2279. doi: 10.1074/jbc.R111.309088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mak HY. Lipid droplets as fat storage organelles in Caenorhabditis elegans: thematic review series: lipid droplet synthesis and metabolism: from yeast to man. J Lipid Res. 2012;53:28–33. doi: 10.1194/jlr.R021006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Murphy DJ. The dynamic roles of intracellular lipid droplets: from archaea to mammals. Protoplasma. 2012;249:541–585. doi: 10.1007/s00709-011-0329-7. [DOI] [PubMed] [Google Scholar]
- 5.Suzuki M, Shinohara Y, Ohsaki Y, Fujimoto T. Lipid droplets: size matters. J Electron Microsc (Tokyo) 2011;60(Suppl. 1):S101–S116. doi: 10.1093/jmicro/dfr016. [DOI] [PubMed] [Google Scholar]
- 6.Fujimoto T, Parton RG. Not just fat: the structure and function of the lipid droplet. Cold Spring Harb Perspect Biol. 2011;3 doi: 10.1101/cshperspect.a004838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Greenberg AS, Coleman RA, Kraemer FB, McManaman JL, Obin MS, Puri V, Yan QW, Miyoshi H, Mashek DG. The role of lipid droplets in metabolic disease in rodents and humans. J Clin Invest. 2011;121:2102–2110. doi: 10.1172/JCI46069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Le Lay S, Dugail I. Connecting lipid droplet biology and the metabolic syndrome. Prog Lipid Res. 2009;48:191–195. doi: 10.1016/j.plipres.2009.03.001. [DOI] [PubMed] [Google Scholar]
- 9.Karpe F, Dickmann JR, Frayn KN. Fatty acids, obesity, and insulin resistance: time for a reevaluation. Diabetes. 2011;60:2441–2449. doi: 10.2337/db11-0425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sun K, Kusminski CM, Scherer PE. Adipose tissue remodeling and obesity. J Clin Invest. 2011;121:2094–2101. doi: 10.1172/JCI45887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee MJ, Wu Y, Fried SK. Adipose tissue remodeling in pathophysiology of obesity. Curr Opin Clin Nutr Metab Care. 2010;13:371–376. doi: 10.1097/MCO.0b013e32833aabef. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim JY, van de Wall E, Laplante M, Azzara A, Trujillo ME, Hofmann SM, Schraw T, Durand JL, Li H, Li G, Jelicks LA, Mehler MF, Hui DY, Deshaies Y, Shulman GI, Schwartz GJ, Scherer PE. Obesity-associated improvements in metabolic profile through expansion of adipose tissue. J Clin Invest. 2007;117:2621–2637. doi: 10.1172/JCI31021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Khan T, Muise ES, Iyengar P, Wang ZV, Chandalia M, Abate N, Zhang BB, Bonaldo P, Chua S, Scherer PE. Metabolic dysregulation and adipose tissue fibrosis: role of collagen VI. Mol Cell Biol. 2009;6:1575–1591. doi: 10.1128/MCB.01300-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McLaughlin T, Sherman A, Tsao P, Gonzalez O, Lamendola C, Reaven GM, Cushman SW. Enhanced proportion of small adipose cells in insulin-resistant vs. insulin-sensitive obese individuals implicates impaired adipogenesis. Diabetologia. 2007;50:1707–1715. doi: 10.1007/s00125-007-0708-y. [DOI] [PubMed] [Google Scholar]
- 15.Rosen ED, Sarraf P, Troy E, Bradwin G, Moore K, Milstone DS, Speigelman BM, Mortensen RM. PPARgamma is required for the differentiation of adipose tissue in vivo and invitro. Mol Cell. 1999;4:611–617. doi: 10.1016/s1097-2765(00)80211-7. [DOI] [PubMed] [Google Scholar]
- 16.Reaven GM. Insulin resistance: the link between obesity and cardiovascular disease. Med Clin North Am. 2011;95:875–892. doi: 10.1016/j.mcna.2011.06.002. [DOI] [PubMed] [Google Scholar]
- 17.Opie LH, Walfish PG. Plasma free fatty acid concentrations in obesity. N Engl J Med. 1963;26:757–760. doi: 10.1056/NEJM196304042681404. [DOI] [PubMed] [Google Scholar]
- 18.Boden G, Chen X, Ruiz J, White JV, Rossetti L. Mechanisms of fatty acid-induced inhibition of glucose uptake. J Clin Invest. 1994;93:2438–2446. doi: 10.1172/JCI117252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Unger RH, Clark GO, Scherer PE, Orci L. Lipid homeostasis, lipotoxicity and the metabolic syndrome. Biochim Biophys Acta. 2010;1801:209–214. doi: 10.1016/j.bbalip.2009.10.006. [DOI] [PubMed] [Google Scholar]
- 20.Romeo GR, Lee J, Shoelson SE. Metabolic syndrome, insulin resistance, and roles of inflammation—mechanisms and therapeutic targets. Arterioscler Thromb Vasc Biol. 2012;32:1771–1776. doi: 10.1161/ATVBAHA.111.241869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vigouroux C, Caron-Debarle M, Le Dour C, Magré J, Capeau J. Molecular mechanisms of human lipodystrophies: from adipocyte lipid droplet to oxidative stress and lipotoxicity. Int J Biochem Cell Biol. 2011;43:862–876. doi: 10.1016/j.biocel.2011.03.002. [DOI] [PubMed] [Google Scholar]
- 22.Garg A. Clinical review#: lipodystrophies: genetic and acquired body fat disorders. J Clin Endocrinol Metab. 2011;96:3313–3325. doi: 10.1210/jc.2011-1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cypess AM, Lehman S, Williams G, Tal I, Rodman D, Goldfine AB, Kuo FC, Palmer EL, Tseng YH, Doria A, Kolodny GM, Kahn CR. Identification and importance of brown adipose tissue in adult humans. N Engl J Med. 2009;360:1500–1508. doi: 10.1056/NEJMoa0810780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Marken Lichtenbelt WD, Vanhommerig JW, Smulders NM, Drossaerts JM, Kemerink GJ, Bouvy ND, Schrauwen P, Teule GJ. Cold-activated brown adipose tissue in healthy men. N Engl J Med. 2009;360:1509–1517. doi: 10.1056/NEJMoa0808718. [DOI] [PubMed] [Google Scholar]
- 25.Virtanen KA, Lidell ME, Orava J, Heglind M, Westergren R, Niemi T, Taittonene M, Laine J, Savisto NJ, Enerbach S, Nuutila P. Functional brown adipose tissue in healthy adults. N Engl J Med. 2009;360:1518–1525. doi: 10.1056/NEJMoa0808949. [DOI] [PubMed] [Google Scholar]
- 26.Zingaretti MC, Crosta F, Vitali A, Guerrieri M, Frontini A, Cannon B, Nedergaard J, Cinti S. The presence of UCP1 demonstrates that metabolically active adipose tissue in the neck of adult humans truly represents brown adipose tissue. FASEB J. 2009;23:3113–3120. doi: 10.1096/fj.09-133546. [DOI] [PubMed] [Google Scholar]
- 27.Saito M, Okamatsu-Ogura Y, Matsushita M, Watanabe K, Yoneshiro T, Kobayashi J, Iwanaga T, Myagawa M, Kameya T, Nakada K, Kawai Y, Tsujisaki M. High incidence of metabolically active brown adipose tissue in healthy adult humans: effects of cold exposure and adiposity. Diabetes. 2009;58:1526–1531. doi: 10.2337/db09-0530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Smorlesi A, Frontini A, Giordano A, Cinti S. The adipose organ: white–brown adipocyte plasticity and metabolic inflammation. Obes Rev. 2012;13:83–96. doi: 10.1111/j.1467-789X.2012.01039.x. [DOI] [PubMed] [Google Scholar]
- 29.Almind K, Manieri M, Sivitz WI, Cinti S, Kahn CR. Ectopic brown adipose tissue in muscle provides a mechanism for differences in risk of metabolic syndrome in mice. Proc Natl Acad Sci USA. 2007;104:2366–2371. doi: 10.1073/pnas.0610416104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wu J, Cohen P, Spiegelman BM. Adaptive thermogenesis in adipocytes: is beige the new brown? Genes Dev. 2013;27:234–250. doi: 10.1101/gad.211649.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Barbatelli G, Murano I, Madsen L, Hao Q, Jimenez M, Kristiansen K, Giacobino JP, De Matteis R, Cinti S. The emergence of cold-induced brown adipocytes in mouse white fat depots is determined predominantly by white to brown adipocyte transdifferentiation. Am J Physiol Endocrinol Metab. 2010;298:E1244–E1253. doi: 10.1152/ajpendo.00600.2009. [DOI] [PubMed] [Google Scholar]
- 32.Petrovic N, Walden TB, Shabalina IG, Timmons JA, Cannon B, Nedergaard J. Chronic peroxisome proliferator-activated receptor gamma (PPARgamma) activation of epididymally derived white adipocyte cultures reveals a population of thermogenically competent, UCP1-containing adipocytes molecularly distinct from classic brown adipocytes. J Biol Chem. 2010;285:7153–7164. doi: 10.1074/jbc.M109.053942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cannon, Nedergaard J. Brown adipose tissue, function and physiological significance. Physiol Rev. 2004;84:277–359. doi: 10.1152/physrev.00015.2003. [DOI] [PubMed] [Google Scholar]
- 34.Wolf G. Brown adipose tissue: the molecular mechanism of its formation. Nutr Rev. 2009;67:167–171. doi: 10.1111/j.1753-4887.2009.00184.x. [DOI] [PubMed] [Google Scholar]
- 35.Greenberg AS, Egan JJ, Wek SA, Garty NB, Blanchette-Mackie EJ, Londos C. Perilipin, a major hormonally regulated adipocyte-specific phosphoprotein associated with the periphery of lipid storage droplets. J Biol Chem. 1991;266:11341–11346. [PubMed] [Google Scholar]
- 36.Beller M, Thiel K, Thul PJ, Jäckle H. Lipid droplets: a dynamic organelle moves into focus. FEBS Lett. 2010;584:2176–2182. doi: 10.1016/j.febslet.2010.03.022. [DOI] [PubMed] [Google Scholar]
- 37.Beller M, Sztalryd C, Southall N, Bell M, Jäckle H, Auld DS, Oliver B. COPI complex is a regulator of lipid homeostasis. PLoS Biol. 2008;6:e292. doi: 10.1371/journal.pbio.0060292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zehmer JK, Huang Y, Peng G, Pu J, Anderson RG, Liu P. A role for lipid droplets in inter-membrane lipid traffic. Proteomics. 2009;9:914–921. doi: 10.1002/pmic.200800584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Guo Y, Walther TC, Rao M, Stuurman N, Goshima G, Terayama K, Wong JS, Vale RD, Walter P, Farese RV. Functional genomic screen reveals genes involved in lipid-droplet formation and utilization. Nature. 2008;453:657–661. doi: 10.1038/nature06928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Prattes S, Horl G, Hammer A, Blaschitz A, Graier WF, Sattler W, Zechner R, Steyrer E. Intracellular distribution and mobilization of unesterified cholesterol in adipocytes: triglyceride droplets are surrounded by cholesterol-rich ER-like surface layer structures. J Cell Sci. 2000;113:2977–2989. doi: 10.1242/jcs.113.17.2977. [DOI] [PubMed] [Google Scholar]
- 41.Brasaemle DL, Dolios G, Shapiro L, Wang R. Proteomic analysis of proteins associated with lipid droplets of basal and lipolytically stimulated 3T3-L1 adipocytes. J Biol Chem. 2004;279:46835–46837. doi: 10.1074/jbc.M409340200. [DOI] [PubMed] [Google Scholar]
- 42.Ding Y, Wu Y, Zeng R, Liao K. Proteomic profiling of lipid droplet-associated proteins in primary adipocytes of normal and obese mouse. Acta Biochim Biophys. 2012;44:394–406. doi: 10.1093/abbs/gms008. [DOI] [PubMed] [Google Scholar]
- 43.Blouin CM, Le Lay S, Eberl A, Köfeler HC, Guerrera IC, Klein C, Le Liepvre X, Lasnier F, Bourron O, Gautier JF, Ferré P, Hajduch E, Dugail I. Lipid droplet analysis in caveolin-deficient adipocytes: alterations in surface phospholipid composition and maturation defects. J Lipid Res. 2010;51:945–956. doi: 10.1194/jlr.M001016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cho SY, Shin ES, Park PJ, Shin DW, Chang HK, Kim D, Lee HH, Lee JH, Kim SH, Song MJ, Chang IS, Lee OS, Lee TR. Identification of mouse Prp19p as a lipid droplet-associated protein and its possible involvement in the biogenesis of lipid droplets. J Biol Chem. 2007;282:2456–2465. doi: 10.1074/jbc.M608042200. [DOI] [PubMed] [Google Scholar]
- 45.Bickel PE, Tansey JT, Welte MA. PAT proteins, an ancient family of lipid droplet proteins that regulate cellular lipid stores. Biochim Biophys Acta. 2009;1791:419–440. doi: 10.1016/j.bbalip.2009.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Londos D, Sztalryd C, Tansey JT, Kimmel AR. Role of PAT proteins in lipid metabolism. Biochimie. 2005;87:45–49. doi: 10.1016/j.biochi.2004.12.010. [DOI] [PubMed] [Google Scholar]
- 47.Brasaemle DL. Thematic review series: adipocyte biology. The perilipin family of structural lipid droplet proteins: stabilization of lipid droplets and control of lipolysis. J Lipid Res. 2007;48:2547–2559. doi: 10.1194/jlr.R700014-JLR200. [DOI] [PubMed] [Google Scholar]
- 48.Wang H, Sztalryd C. Oxidative tissue: perilipin 5 links storage with the furnace. Trends Endocrinol Metab. 2011;22:197–203. doi: 10.1016/j.tem.2011.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hsieh K, Lee YK, Londos C, Raaka BM, Dalen KT, Kimmel AR. Perilipin family members preferentially sequester to either triacylglycerol-specific or cholesterylester-specific intracellular lipid storage droplets. J Cell Sci. 2012;125:4067–4076. doi: 10.1242/jcs.104943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Akter MH, Yamaguchi T, Hirose F, Osumi T. Perilipin, a critical regulator of fat storage and breakdown, is a target gene of estrogen receptor-related receptor alpha. Biochem Biophys Res Com. 2008;368:563–568. doi: 10.1016/j.bbrc.2008.01.102. [DOI] [PubMed] [Google Scholar]
- 51.Arimura N, Horiba T, Imagawa M, Shimizu M, Sato R. The peroxisome proliferator-activated receptor gamma regulates expression of the perilipin gene in adipocytes. J Biol Chem. 2004;279:10070–10076. doi: 10.1074/jbc.M308522200. [DOI] [PubMed] [Google Scholar]
- 52.Dalen KT, Schoonjans K, Ulven SM, Weedon-Fekjaer MS, Bentzen TG, Koutnikova H, Auwerx J, Nebb HI. Adipose tissue expression of the lipid droplet-associating proteins S3-12 and perilipin is controlled by peroxisome proliferator-activated receptor-gamma. Diabetes. 2004;53:1243–1252. doi: 10.2337/diabetes.53.5.1243. [DOI] [PubMed] [Google Scholar]
- 53.Stenson BM, Rydén M, Venteclef N, Dahlman I, Pettersson AM, Mairal A, Aström G, Blomqvist L, Wang V, Jocken JW, Clément K, Langin D, Arner P, Laurencikiene J. Liver X receptor (LXR) regulates human adipocyte lipolysis. J Biol Chem. 2011;286:370–379. doi: 10.1074/jbc.M110.179499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Brasaemle DL, Rubin B, Harten IA, Gruia-Gray J, Kimmel AR, Londos C. Perilipin A increases triacylglycerol storage by decreasing the rate of triacylglycerol hydrolysis. J Biol Chem. 2000;275:38486–38493. doi: 10.1074/jbc.M007322200. [DOI] [PubMed] [Google Scholar]
- 55.Miyoshi H, Souza SC, Zhang HH, Strissel KJ, Christoffolete MA, Kovsan J, Rudich A, Kraemer FB, Bianco AC, Obin MS, Greenberg AS. Perilipin promotes hormone-sensitive lipase-mediated adipocyte lipolysis via phosphorylation-dependent and -independent mechanisms. J Biol Chem. 2006;281:15837–15844. doi: 10.1074/jbc.M601097200. [DOI] [PubMed] [Google Scholar]
- 56.Granneman JG, Moore HP, Krishnamoorthy R, Rathod M. Perilipin controls lipolysis by regulating the interactions of AB-hydrolase containing 5 (Abhd5) and adipose triglyceride lipase (Atgl) J Biol Chem. 2009;284:34538–34544. doi: 10.1074/jbc.M109.068478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tansey JT, Huml AM, Vogt R, Davis KE, Jones JM, Fraser KA, Brasaemle DL, Kimmel AR, Londos C. Functional studies on native and mutated forms of perilipins. A role in protein kinase A-mediated lipolysis of triacylglycerols. J Biol Chem. 2003;278:8401–8406. doi: 10.1074/jbc.M211005200. [DOI] [PubMed] [Google Scholar]
- 58.Wang H, Hu L, Dalen K, Dorward H, Marcinkiewicz A, Russell D, Gong D, Londos C, Yamaguchi T, Holm C, Rizzo MA, Brasaemle D, Sztalryd C. Activation of hormone-sensitive lipase requires two steps, protein phosphorylation and binding to the PAT-1 domain of lipid droplet coat proteins. J Biol Chem. 2009;284:32116–32125. doi: 10.1074/jbc.M109.006726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lass A, Zimmermann R, Oberer M, Zechner R. Lipolysis—a highly regulated multi-enzyme complex mediates the catabolism of cellular fat stores. Prog Lipid Res. 2011;50:14–27. doi: 10.1016/j.plipres.2010.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Granneman JG, Moore HP. Location, location: protein trafficking and lipolysis in adipocytes. Trends Endocrinol Metab. 2008;19:3–9. doi: 10.1016/j.tem.2007.10.006. [DOI] [PubMed] [Google Scholar]
- 61.Shen WJ, Patel S, Mioshi H, Greenberg AS, Kraemer FB. Functional interaction of hormone sensitive lipase and perilipin in lipolysis. J Lipid Res. 2009;50 doi: 10.1194/jlr.M900176-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Granneman JG, More HP, Krishnamoorhy R, Rahtod M. Perilipin control lipolysis by regulating the interactions of AB-hydrolase containing5 (Abhd5) and adipose triglyceride lipase (Atgl) J Biol Chem. 2009;284:34538–34544. doi: 10.1074/jbc.M109.068478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gandotra S, Lim K, Girousse A, Saudek V, O’Rahilly S, Savage DB. Human frame shift mutations affecting the carboxyl terminus of perilipin increase lipolysis by failing to sequester the adipose triglyceride lipase (ATGL) coactivator AB-hydrolase-containing 5 (ABHD5) J Biol Chem. 2011;286:34998–35006. doi: 10.1074/jbc.M111.278853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Martinez-Botas J, Anderson JB, Tessier D, Lapillonne A, Chang BH, Quast MJ, Gorenstein D, Chen KH, Chan L. Absence of perilipin results in leanness and reverses obesity in Lepr(db/db) mice. Nat Genet. 2000;26:474–479. doi: 10.1038/82630. [DOI] [PubMed] [Google Scholar]
- 65.Tansey JT, Sztalryd C, Gruia-Gray J, Roush DL, Zee JV, Gavrilova O, Reitman ML, Deng CX, Li C, Kimmel AR, Londos C. Perilipin ablation results in a lean mouse with aberrant adipocyte lipolysis, enhanced leptin production, and resistance to diet-induced obesity. Proc Natl Acad Sci USA. 2001;98:6494–6499. doi: 10.1073/pnas.101042998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Saha PK, Kojima H, Martinez-Botas J, Sunehag AL, Chan L. Metabolic adaptations in the absence of perilipin: increased beta-oxidation and decreased hepatic glucose production associated with peripheral insulin resistance but normal glucose tolerance in perilipin-null mice. J Biol Chem. 2004;279:35150–35158. doi: 10.1074/jbc.M405499200. [DOI] [PubMed] [Google Scholar]
- 67.Gandotra S, Le Dour C, Bottomley W, Cervera P, Giral P, Reznik Y, Charpentier G, Auclair M, Delépine M, Barroso I, Semple RK, Lathrop M, Lascols O, Capeau J, O’Rahilly S, Magré J, Savage DB, Vigouroux C. Perilipin deficiency and autosomal dominant partial lipodystrophy. N Engl J Med. 2011;364:740–748. doi: 10.1056/NEJMoa1007487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Straub BK, Stoeffel P, Heid H, Zimbelmann R, Schirmacher P. Differential pattern of lipid droplet-associated proteins and de novo perilipin expression in hepatocyte steatogenesis. Hepatology. 2008;47:1936–1946. doi: 10.1002/hep.22268. [DOI] [PubMed] [Google Scholar]
- 69.Kern PA, Di Gregorio G, Lu T, Rassouli N, Ranganathan G. Perilipin expression in human adipose tissue is elevated with obesity. J Clin Endocrinol Metab. 2004;89:1352–1358. doi: 10.1210/jc.2003-031388. [DOI] [PubMed] [Google Scholar]
- 70.Mottagui-Tabar S, Ryden M, Lofgren P, Faulds G, Hoffstedt J, Brookes AJ, Andersson I, Arner P. Evidence for an important role of perilipin in the regulation of human adipocyte lipolysis. Diabetologia. 2003;46:789–797. doi: 10.1007/s00125-003-1112-x. [DOI] [PubMed] [Google Scholar]
- 71.Puri V, Ranjit S, Konda S, Nicoloro SM, Staubhaar J, Chawla A, Chouinard M, Lin C, Burkart A, Corvera S, Perugini RA, Czech MP. Cidea is associated with lipid droplets and insulin sensitivity in humans. Proc Natl Acad Sci USA. 2008;105:7833–7838. doi: 10.1073/pnas.0802063105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Smith CE, Ordovás JM. Update on perilipin polymorphisms and obesity. Nutr Rev. 2012;70:611–621. doi: 10.1111/j.1753-4887.2012.00515.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gong J, Sun Z, Li P. CIDE proteins and metabolic disorders. Curr Opin Lipidol. 2009;20:121–126. doi: 10.1097/MOL.0b013e328328d0bb. [DOI] [PubMed] [Google Scholar]
- 74.Xu L, Zhou L, Li P. CIDE proteins and lipid metabolism. Arterioscler Vasc Biol. 2012;32:1094–1098. doi: 10.1161/ATVBAHA.111.241489. [DOI] [PubMed] [Google Scholar]
- 75.Qi J, Gong J, Zhao T, Zhao J, Lam P, Ye J, Li JZ, Wu J, Zhou HM, Li P. Downregulation of AMP-activated protein kinase by Cidea-mediated ubiquitination and degradation in brown adipose tissue. EMBO J. 2008;27:1537–1548. doi: 10.1038/emboj.2008.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Puri V, Konda S, Ranjit S, Aouadi M, Chawla A, Chakladar M, Czech MP. Fat-specific protein 27, a novel lipid droplet protein that enhances triglyceride storage. J Biol Chem. 2007;282:34213–34218. doi: 10.1074/jbc.M707404200. [DOI] [PubMed] [Google Scholar]
- 77.Gong J, Sun Z, Wu L, Xu W, Schieber N, Xu D, Shui G, Yang H, Parton RG, Li P. Fsp27 promotes lipid droplet growth by lipid exchange and transfer at lipid droplet contact sites. J Cell Biol. 2011;195:953–963. doi: 10.1083/jcb.201104142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Jambunathan S, Yin J, Khan W, Tamori Y, Puri V. FSP27 promotes lipid droplet clustering and then fusion to regulate triglyceride accumulation. PLoS One. 2011;6 doi: 10.1371/journal.pone.0028614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wang X, Zhang B, Xu D, Gao J, Wang L, Wang Z, Shan Y, Yu X. Purification, Crystallization and preliminary X-ray crystallographic analysis of the. CIDE-N domain of Fsp27. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2012;68:1529–1533. doi: 10.1107/S1744309112043989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Yang X, Heckmann BL, Zhang X, Smas CM, Liu J. Distinct mechanisms regulate ATGL-mediated adipocyte lipolysis by lipid droplet coat proteins. Mol Endocrinol. 2013;27:116–126. doi: 10.1210/me.2012-1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kulyte A, Pettersson AT, Antoson P, Stenson BM, Langib D, Gustafsson JA, Staels B, Ryden M, Arner P, Laurencikiene J. CIDEA interacts with liver X receptors in white fat cells. FEBBS Lett. 2011;585:744–748. doi: 10.1016/j.febslet.2011.02.004. [DOI] [PubMed] [Google Scholar]
- 82.Wang W, Lv N, Zhang S, Shui G, Qian H, Zhang J, Chen Y, Ye J, Xie Y, Shen Y, Wenk MR, Li P. Cidea is an essential transcriptional coactivator regulating mammary gland secretion of milk lipids. Nat Med. 2012;18:235–243. doi: 10.1038/nm.2614. [DOI] [PubMed] [Google Scholar]
- 83.Ueno M, Shen WJ, Patel S, Greenberg AS, Azhar S, Kraemer FB. Fat specific protein 27 modulates nuclear factor of activated-T cells 5 and the cellular response to stress. J Lipid Res. 2012 doi: 10.1194/jlr.M033365. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Nishino N, Tamori Y, Tateya S, Kawaguchi T, Shibakusa T, Mizunoya W, Inoue K, Kitazawa R, Kitazawa S, Matsuki Y, Hiramatsu R, Masubuchi S, Omachi A, Kimura K, Saito M, Amo T, Ohta S, Yamaguchi T, Osumi T, Cheng J, Fujimoto T, Akao NH, Nakao K, Aiba A, Okamura H, Fushiki T, Kasuga M. Fsp27 contributes to efficient energy storage in murine white adipocytes by promoting the formation of unilocular lipid droplets. J Clin Invest. 2008;118:2808–2821. doi: 10.1172/JCI34090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Toh SY, Gong J, Du G, Li JZ, Yang S, Ye J, Yao H, Zhang Y, Xue B, Li Q, Yang H, Wen Z, Li P. Up-regulation of mitochondrial activity and acquirement of brown adipose tissue-like property in the white adipose tissue of fsp27 deficient mice. PLoS One. 2008;3:e2890. doi: 10.1371/journal.pone.0002890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zhou Z, Toh S Yon, Chen Z, Guo K, Ng CP, Ponniah S, Lin SC, Hong W, Li P. Cidea-deficient mice have lean phenotype and are resistant to obesity. Nat Genet. 2003;35:49–56. doi: 10.1038/ng1225. [DOI] [PubMed] [Google Scholar]
- 87.Rubio-Cabezas O, Puri V, Murano I, Saudek V, Semple RK, Dash S, Hyden CS, Bottomley W, Vigouroux C, Magré J, Raymond-Barker P, Murgatroyd PR, Chawla A, Skepper JN, Chatterjee VK, Suliman S, Patch AM, Agarwal AK, Garg A, Barroso I, Cinti S, Czech MP, Argente J, O’Rahilly S, Savage DB. LD Screening Consortium, Partial lipodystrophy and insulin resistant diabetes in a patient with a homozygous nonsense mutation in CIDEC. EMBO Mol Med. 2009;1:280–287. doi: 10.1002/emmm.200900037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Pilch P, Liu L. Fat caves: caveolae, lipid trafficking and lipid metabolism in adipocytes. Trends Endocrinol Metab. 2011;22:318–324. doi: 10.1016/j.tem.2011.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Le Lay S, Blouin ICM, Hajduch E, Dugail I. Filling up adipocytes with lipids. Lesson from caveolin-1 deficiency. 2009;1791:514–518. doi: 10.1016/j.bbalip.2008.10.008. [DOI] [PubMed] [Google Scholar]
- 90.Briand N, Dugail I, Le Lay S. Cavin proteins: new players in the caveolae field. Biochimie. 2011;93:71–77. doi: 10.1016/j.biochi.2010.03.022. [DOI] [PubMed] [Google Scholar]
- 91.Ost A, Ortegren U, Gustavsson J, Nystrom FH, Strålfors P. Triacylglycerol is synthesized in a specific subclass of caveolae in primary adipocytes. J Biol Chem. 2005;280:5–8. doi: 10.1074/jbc.C400429200. [DOI] [PubMed] [Google Scholar]
- 92.Fujimoto H, Kogo K, Ishiguro K, Tauchi R, Nomura K. Caveolin-2 is targeted to lipid droplets, a new “membrane domain” in the cell. J Cell Biol. 2001;152:1079–1085. doi: 10.1083/jcb.152.5.1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ostermeyer AG, Paci JM, Zeng Y, Lublin DM, Munro S, Brown DA. Accumulation of caveolin in the endoplasmic reticulum redirects the protein to lipid storage droplets. J Cell Biol. 2001;152:1071–1078. doi: 10.1083/jcb.152.5.1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Pol A, Luetterforst R, Lindsay M, Heino S, Ikonen E, Parton RG. A caveolin dominant negative mutant associates with lipid bodies and induces intracellular cholesterol imbalance. J Cell Biol. 2001;152:1057–1070. doi: 10.1083/jcb.152.5.1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Cohen AW, Razani B, Schubert W, Williams TM, Wang XB, Iyengar P, Brasaemle DL, Scherer PE, Lisanti MP. Role of caveolin-1 in the modulation of lipolysis and lipid droplet formation. Diabetes. 2004;53:1261–1270. doi: 10.2337/diabetes.53.5.1261. [DOI] [PubMed] [Google Scholar]
- 96.Storey SM, McIntosh AL, Senthivinayagam S, Moon KC, Atshaves BP. The phospholipid monolayer associated with perilipin-enriched lipid droplets is a highly organized rigid membrane structure. Am J Physiol Endocrinol Metab. 2011;301:E991–E1003. doi: 10.1152/ajpendo.00109.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Aboulaich N, Ortegren U, Vener AV, Strålfors P. Association and insulin regulated translocation of hormone-sensitive lipase with PTRF. Biochem Biophys Res Commun. 2006;350:657–661. doi: 10.1016/j.bbrc.2006.09.094. [DOI] [PubMed] [Google Scholar]
- 98.Liu L, Brown D, McKee M, Lebrasseur NK, Yang D, Albrecht KH, Ravid K, Pilch PF. Deletion of Cavin/PTRF causes global loss of caveolae, dyslipidemia, and glucose intolerance. Cell Metab. 2008;8:310–317. doi: 10.1016/j.cmet.2008.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kim CA, Delepine M, Boutet E, Mourabit EL, Le Lay S, Meier M, Nemani M, Bridel E, Leite CC, Bertola DR, Semple RK, O’Rahilly S, Dugail I, Capeau J, Lathrop M, Magre J. Association of a homozygous non sense caveolin-1 mutation with beradinelli-seipin congenital lipodystrophy. J Clin Endocrinol Metab. 2008;93:1129–1134. doi: 10.1210/jc.2007-1328. [DOI] [PubMed] [Google Scholar]
- 100.Cao H, Alston L, Ruschman J, Hegele RA. Heterozygous CAV1 frameshift mutations (MIM 601047) in patients with atypical partial lipodystrophy and hypertriglyceridemia. Lipids Health Dis. 2008;31:7,3. doi: 10.1186/1476-511X-7-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Hayashi YK, Matsuda C, Gawa OM, Goto K, Tominaga K, Mitsuhashi S, Park YE, Nonaka I, Hino-Fukuyo N, Haginoya K, Sugano H, Nishino I. Human PTRF mutations cause secondary deficiency of caveolins resulting in muscular dystrophy with generalized lipodystrophy. J Clin Invest. 2009;119:2623–2633. doi: 10.1172/JCI38660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Shastry S, Delgado MR, Dirik E, Turkmen M, Agarwal AK, Garg A. Congenital generalized lipodystrophy, type 4 (CGL4) associated with myopathy due to novel PTRF mutations. Am J Med Genet A 152A. 2010:2245–2253. doi: 10.1002/ajmg.a.33578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Hayer A, Stoeber M, Bissig C, Helenius A. Biogenesis of caveolae: stepwise assembly of large caveolin and cavin complexes. Traffic. 2010;3:361–382. doi: 10.1111/j.1600-0854.2009.01023.x. [DOI] [PubMed] [Google Scholar]
- 104.Meshulam T, Breen MR, Liu L, Parton RG, Pilch PF. Caveolins/caveolae protect adipocytes from fatty acid-mediated lipotoxicity. 2011;52:1526–1532. doi: 10.1194/jlr.M015628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Le Lay S, Briand N, Blouin CM, Chateau D, Prado C, Lasnier F, Le Liepvre X, Hajduch E, Dugail I. The lipoatrophic caveolin-1 deficient mouse model reveals autophagy in mature adipocytes. Autophagy. 2010;6:754–763. doi: 10.4161/auto.6.6.12574. [DOI] [PubMed] [Google Scholar]
- 106.Briand N, Le Lay S, Sessa WC, Ferré P, Dugail I. Distinct roles of endothelial and adipocyte caveolin-1 in macrophage infiltration and adipose tissue metabolic activity. Diabetes. 2011;60:448–453. doi: 10.2337/db10-0856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Reue K. A thematic review series: lipid droplet storage and metabolism: from yeast to man. J Lipid Res. 2011;52:1865–1868. doi: 10.1194/jlr.E020602. [DOI] [PMC free article] [PubMed] [Google Scholar]