

Summary
Dual and triple combination therapies with RAF inhibitors plus other targeted agents have demonstrated promising clinical utility in BRAF V600-mutant solid tumors. However, despite vertical inhibition at multiple nodes on the MAPK signaling pathway, resistant tumors emerge. Ahronian and colleagues show that in BRAF mutant colorectal cancer, resistance involves re-activation of RAS/RAF/MEK/ERK signaling and may be overcome by newly emerging ERK inhibitors.
Mutations in the serine-threonine kinase, BRAF, at amino acid position V600, occur in 8–15% of colorectal cancers (CRC). BRAF V600 mutations also occur in other tumor types, such as melanoma, non-small cell lung cancer (NSCLC), and hairy cell leukemia (HCL), suggesting that mutant BRAF could be broadly targeted regardless of histology. Unfortunately, response rates to mutant-specific BRAF inhibitors, such as vemurafenib and dabrafenib, vary by tumor type. While BRAF inhibitors have achieved a mere 5% response rate in BRAF V600-mutant CRC, response rates in melanoma, NSCLC, and HCL are ~52%, 32%, and 96%, respectively (1–3). Thus, at least in solid tumors, the presence of a BRAF mutation is not a ‘slam dunk’ for single-agent BRAF inhibition.
Mechanisms of resistance to single-agent BRAF inhibition also differ by histological subtype. In BRAF V600-mutant CRC cells treated with vemurafenib, MAP kinase (MAPK) signaling is reactivated by feedback activation of EGFR (Fig. 1A) (3). Amplification of mutant BRAF has also been implicated in preclinical resistance to BRAF inhibitors in this context (4). Consistent with these findings, combined BRAF/MEK and BRAF/EGFR inhibition more effectively inhibits growth of BRAF V600-mutant CRC cells than BRAF inhibition alone (3). Other studies have shown that PIK3CA mutations and PTEN loss may also contribute to vemurafenib resistance in BRAF V600-mutant CRC cell lines (5). Additional molecular alterations across different tumor types (mostly melanoma) that modulate response to BRAF inhibition in BRAF V600-mutant tumors include RAS mutations, loss of RAS negative regulator NF1, BRAF splice variants, increased expression of CRAF and COT, MEK1 mutations, RB1 inactivation, and activation of MET, IGF-1R, and HER3 receptor tyrosine kinases, among others (3). Clearly, reactivation of MAPK signaling is a recurring theme (3, 5).
Figure 1. Schematic representation of mechanisms of resistance to BRAF/EGFR or BRAF/MEK inhibitor combinations in BRAF V600-mutant colorectal cancer (CRC).
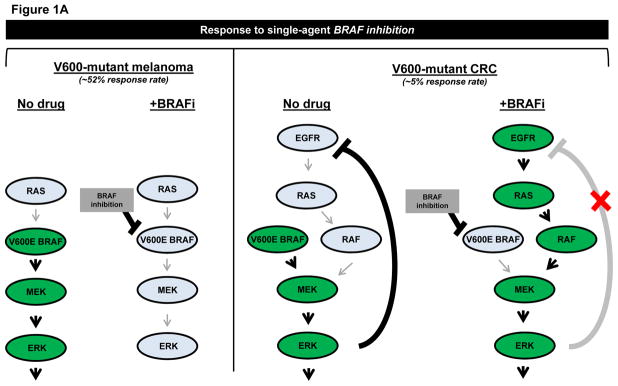
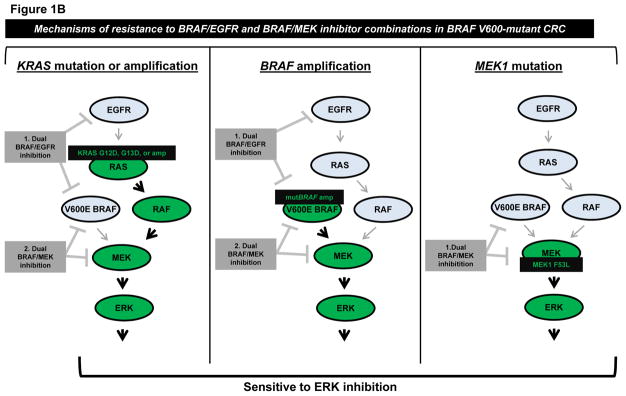
A, Comparison of response of V600-mutant melanoma versus CRC to single-agent BRAF inhibition. Left, BRAF inhibitors effectively inhibit MAPK signaling and induce tumor regression in over half of BRAF V600-mutant tumors, when used as monotherapy. Mechanisms of resistance in non-responsive tumors are not depicted. Right, BRAF inhibitors result in reactivation of EGFR-mediated MAPK signaling in BRAF V600-mutant CRC, contributing to the low overall response rate to single-agent BRAF inhibition. B, Mechanisms of resistance to various combinations of BRAF/EGFR or BRAF/MEK inhibitors were identified by Ahronian and colleagues via in vitro modeling of acquired resistance or analysis of tumor samples after progression on combination therapy. KRAS G12D/KRAS G13D mutations and amplification of wild-type KRAS or mutant BRAF were found to confer resistance to dual inhibition of BRAF/EGFR and BRAF/MEK. A MEK1 F53L substitution mutation was found to confer resistance to dual BRAF/MEK inhibition. All models of resistance to combination BRAF/EGFR and BRAF/MEK inhibition retained sensitivity to ERK inhibition.
Based on the identification that EGFR bypass signaling can mediate resistance to vemurafenib in BRAF V600-mutant CRC, clinical trials testing combinations of BRAF/MEK and BRAF/EGFR inhibitors in these patients are underway. Such combinations have demonstrated somewhat improved outcomes relative to BRAF inhibition alone, but acquired resistance still emerges (6, 7). In this issue of Cancer Discovery, Ahronian and colleagues provide the first description of mechanisms of acquired resistance to combined BRAF/MEK and combined BRAF/EGFR inhibition in BRAF V600-mutant CRC (4). By utilizing both in vitro cell line models and clinical samples, they describe multiple distinct mechanisms of resistance to these BRAF inhibitor combinations, all of which reactivate the MAPK signaling pathway.
In vitro modeling of acquired resistance to combined BRAF/EGFR inhibition (vemurafenib/cetuximab) and combined BRAF/MEK inhibition (vemurafenib/selumetinib) resulted in acquired KRAS G12D and G13D substitution mutations, respectively (Fig. 1B). The authors further showed that these activating KRAS mutations increased phosphorylation of CRAF, MEK, ERK, and RSK, suggesting that mutant-KRAS-mediated constitutive activation of the MAPK signaling pathway is responsible for resistance in this setting. Importantly, these cell line models of KRAS-mediated acquired resistance to combined BRAF/EGFR inhibition displayed cross-resistance to combined BRAF/MEK inhibition, and vice versa.
Ahronian and colleagues also performed whole-exome and/or RNA sequencing of paired pre-treatment and post-resistance biopsies from three separate patients with BRAF V600-mutant CRC who progressed on either BRAF/EGFR or BRAF/MEK combination therapy. In the two described cases of acquired resistance to BRAF/EGFR combination therapy, the authors identified amplification of BRAF (dabrafenib/panitumumab resistance) and KRAS (encorafenib/cetuximab resistance) unique to the resistant tumor (Fig. 1B). Of note, the KRAS amplification was identified in a lesion that had progressed through BRAF/MEK (dabrafenib/trametinib) combination therapy and continued to progress after the patient was switched to the BRAF/EGFR (encorafenib/cetuximab) regimen. This finding further validates the authors’ in vitro result that KRAS-mediated activation of MAPK signaling can confer cross-resistance to both BRAF/MEK and BRAF/EGFR inhibition.
Additionally, the authors identified co-occurring acquired missense mutations in MEK1 and ARAF in a BRAF V600-mutant CRC sample with acquired resistance to BRAF/MEK inhibition (dabrafenib/trametinib). Follow-up in vitro analysis revealed that the MEK1 F53L mutation, but not the ARAF Q489L mutation, was sufficient to confer resistance to dabrafenib/trametinib in vitro, although the two mutations were not tested for cooperativity within the same cell (Fig. 1B). As the authors point out, this result highlights a separate important principle regarding the significance of findings revealed by comprehensive mutational profiling, i.e. that thorough and efficient biological validation of novel mutations is critical to distinguish passenger versus driver mutations, especially when two obvious signaling molecules in the same pathway harbor mutations.
Taken together, the translational data from Ahronian and colleagues have multifaceted implications for our ongoing understanding of the underlying biology and clinical treatment of BRAF V600-mutant tumors. Context-dependent differences in sensitivity to single-agent RAF inhibition reveal varying levels of “addiction to mutant BRAF signaling” across histological subtypes. These differences appear to extend to the setting of combination therapies as well. Although combined BRAF/EGFR and combined BRAF/MEK inhibition thus far demonstrate improved clinical outcomes in BRAF-mutant CRC compared to BRAF inhibition alone, response rates to combination therapy still range from only 12–29%. By comparison, in BRAF-mutant melanoma, combined BRAF/MEK inhibition has demonstrated a response rate of 64% (8). Biological mechanisms underlying these phenomena are not yet fully understood but could be explained by differences in factors such as cell lineage, epigenetics, and microenvironment of the tumor, etc. The 12–29% response rate indicates that it will take more than dual combination therapy to inhibit BRAF V600-mutant CRC to the same extent as BRAF V600-mutant melanoma; however, it is also evident that even in BRAF V600-mutant melanoma, dual BRAF/MEK inhibition is not curative.
Finally, the authors of this study leave us with the hope that newly developed ERK inhibitors may successfully overcome MAPK-driven resistance to combined BRAF/EGFR or BRAF/MEK inhibition in BRAF V600-mutant CRC. Recent preclinical studies have also demonstrated that ERK inhibitors may be effective at overcoming acquired resistance to BRAF/MEK inhibition in BRAF V600-mutant melanoma (9). It remains to be seen whether ERK inhibitors will be effective in overcoming all mechanisms of resistance to BRAF/MEK/EGFR targeted therapies, as well as whether ERK inhibitors will be useful as first-line therapies across different histological groups of BRAF-mutant tumors. Factors mediating sensitivity to ERK inhibition will probably vary according to cellular context, as we have seen with BRAF inhibitors. Biomarkers of sensitivity to ERK inhibition will be important as clinicians are faced with questions about how best to administer these therapies to provide maximal benefit to patients.
These studies spark a broader discussion regarding the utility of monotherapy versus combination therapy in personalized cancer medicine. Maximizing combination therapy in the first-line setting makes intuitive sense, with the goal being to limit any residual resistant disease that will ultimately result in tumor progression or recurrence. However, the current reality in metastatic solid tumors is that resistance almost invariably develops for the targeted agents in clinical use. It remains to be seen whether the addition of new ‘downstream’ ERK inhibitors will result in longer time to resistance. It is also unknown whether resistance to such newer therapies will result in more aggressive tumors that are unresponsive to other targeted agents (10).
In the case of BRAF V600-mutant CRC, for example, addition of an ERK inhibitor to the BRAF/MEK or BRAF/EGFR combination could significantly extend progression-free survival. However, if acquired resistance after addition of an ERK inhibitor occurs within a similar time frame as seen with BRAF/MEK and BRAF/EGFR inhibitor combinations, a patient could potentially benefit from first receiving the dual inhibitor combination first, followed by an ERK inhibitor once the tumor progressed. The question of which strategy will provide the best outcome for patients can only be answer clinically. In the meantime, studies such as those performed by Ahronian and colleagues are critical to pave the road ahead.
Acknowledgments
Financial support:
The authors acknowledge support from the NIH grants R01-CA12120 and P01-CA129243.
Footnotes
Conflicts of interest:
WP: Consulting: AstraZeneca, Bristol-Myers Squibb, Exelixis; Research funding: AstraZeneca, Symphogen, Bristol-Myers Squibb; Other: WP is currently an employee of Roche.
References
- 1.Hyman DM, Blay J-Y, Chau I, Raje NS, Fernandez MEE, Wolf J, et al. VE-BASKET, a first-in-kind, phase II, histology-independent “basket” study of vemurafenib (VEM) in nonmelanoma solid tumors harboring BRAF V600 mutations (V600m) J Clin Oncol 2014. 2014;32(suppl):5s, Abstract nr 2533. [Google Scholar]
- 2.Jain P, Polliack A, Ravandi F. Novel therapeutic options for relapsed hairy cell leukemia. Leuk Lymphoma. 2015:1–9. doi: 10.3109/10428194.2014.1001988. [DOI] [PubMed] [Google Scholar]
- 3.Lito P, Rosen N, Solit DB. Tumor adaptation and resistance to RAF inhibitors. Nat Med. 2013;19:1401–9. doi: 10.1038/nm.3392. [DOI] [PubMed] [Google Scholar]
- 4.Ahronian LG, Sennott EM, Van Allen EM, Wagle N, Kwak EL, Faris JE, et al. Clinical acquired resistance to RAF inhibitor combinations in BRAF-mutant colorectal cancer through MAPK pathway alterations. Cancer Discov. 2015 doi: 10.1158/2159-8290.CD-14-1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boussemart L, Malka-Mahieu H, Girault I, Allard D, Hemmingsson O, Tomasic G, et al. eIF4F is a nexus of resistance to anti-BRAF and anti-MEK cancer therapies. Nature. 2014;513:105–9. doi: 10.1038/nature13572. [DOI] [PubMed] [Google Scholar]
- 6.Corcoran RB, Atreya CE, Falchook GS, Infante JR, Hamid O, Messersmith WA, et al. Phase 1–2 trial of the BRAF inhibitor dabrafenib (D) plus MEK inhibitor trametinib (T) in BRAF V600 mutant colorectal cancer (CRC): Updated efficacy and biomarker analysis. ASCO Meeting Abstracts. 2014;32:3515. [Google Scholar]
- 7.Tabernero J, Chan E, Baselga J, Blay J-Y, Chau I, Hyman DM, et al. VE-BASKET, a Simon 2-stage adaptive design, phase II, histology-independent study in nonmelanoma solid tumors harboring BRAF V600 mutations (V600m): Activity of vemurafenib (VEM) with or without cetuximab (CTX) in colorectal cancer (CRC) ASCO Meeting Abstracts. 2014;32:3518. [Google Scholar]
- 8.Robert C, Karaszewska B, Schachter J, Rutkowski P, Mackiewicz A, Stroiakovski D, et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N Engl J Med. 2015;372:30–9. doi: 10.1056/NEJMoa1412690. [DOI] [PubMed] [Google Scholar]
- 9.Morris EJ, Jha S, Restaino CR, Dayananth P, Zhu H, Cooper A, et al. Discovery of a novel ERK inhibitor with activity in models of acquired resistance to BRAF and MEK inhibitors. Cancer Discov. 2013;3:742–50. doi: 10.1158/2159-8290.CD-13-0070. [DOI] [PubMed] [Google Scholar]
- 10.Meador CB, Jin H, de Stanchina E, Nebhan CA, Pirazzoli V, Wang L, et al. Optimizing the Sequence of Anti-EGFR-Targeted Therapy in EGFR-Mutant Lung Cancer. Mol Cancer Ther. 2015;14:542–52. doi: 10.1158/1535-7163.MCT-14-0723. [DOI] [PMC free article] [PubMed] [Google Scholar]