

Abstract
Firefly luciferase is homologous to fatty acyl-CoA synthetases. We hypothesized that the firefly luciferase substrate d-luciferin and its analogs are fatty acid mimics that are ideally suited to probe the chemistry of enzymes that release fatty acid products. Here, we synthesized luciferin amides and found that these molecules are hydrolyzed to substrates for firefly luciferase by the enzyme fatty acid amide hydrolase (FAAH). In the presence of luciferase, these molecules enable highly sensitive and selective bioluminescent detection of FAAH activity in vitro, in live cells, and in vivo. The potency and tissue distribution of FAAH inhibitors can be imaged in live mice, and luciferin amides serve as exemplary reagents for greatly improved bioluminescence imaging in FAAH-expressing tissues such as the brain.
Firefly luciferase is best known for its light emission chemistry with d-luciferin, but it is also a long-chain fatty acyl-CoA synthetase (ACSL) that can bind fatty acid substrates such as arachidonic acid (Figure 1).1 Conversely, we have recently shown that an ACSL from the fruit fly Drosophila melanogaster is a latent luciferase that can emit light with a synthetic luciferin.2 In both cases, adenylation of a carboxylic acid is the first step in catalysis. Furthermore, both enzymes can bind fatty acids ranging from octanoic acid to arachidonic acid, suggesting that d-luciferin and aminoluciferin analogs3−5 are acting as fatty acid mimics. Based in part on this observation, we hypothesized that luciferins are ideally suited to probe the chemistry of enzymes that release fatty acid products.
Figure 1.

(A) Firefly luciferase catalyzes light emission from d-luciferin. (B) Firefly luciferase is also a fatty acyl-CoA synthetase. (C) FAAH cleaves anandamide to arachidonic acid. (D) Luciferin amides could allow bioluminescence imaging of FAAH activity.
Fatty acid amide hydrolase (FAAH) is a serine hydrolase that limits the lifetime and sphere of action of fatty acid amide second messengers by hydrolysis to their corresponding fatty acids (Figure 1).6,7 Most notably, arachidonoyl ethanolamine (anandamide) is a locally generated agonist for the cannabinoid receptor CB1. Inhibition of FAAH prolongs the action of anandamide and is therefore an attractive drug target for the treatment of pain, anxiety, and cannabinoid dependence.6 Many FAAH inhibitors are being developed as potential therapeutics, and there is great interest in detecting FAAH activity in vivo.8 Current techniques to assay FAAH inhibitors in mice primarily require sacrificing the mouse, homogenizing the tissues, adding radioactive lipid substrates, and HPLC analysis of the products.9 This places large demands on time and quantities of mice required to evaluate inhibitors and furthermore cannot give longitudinal data from the same animal. Some inroads have been made with PET imaging probes for FAAH, but these are specialized and expensive tools with low throughput and signal-to-noise that lack the specificity for whole-body imaging.8
FAAH readily accepts a wide range of saturated and unsaturated fatty acid amides in addition to anandamide7,10 and has been shown to hydrolyze ethanolamides, primary amides, and methyl amides.7,10,11 We therefore hypothesized that FAAH could hydrolyze luciferin amides to their respective carboxylates, resulting in the formation of a luminogenic luciferase substrate (Figure 1). Here we show that luciferin amides allow exquisitely selective and sensitive imaging of endogenous FAAH activity in live cells and in live mice. FAAH is both necessary and sufficient for bioluminescence to occur and is the only enzyme activating these probes. The performance of FAAH inhibitors can be imaged in live mice, and inhibitors that cross the blood-brain barrier can be readily distinguished from those that cannot. Moreover, the amount of luciferin amide probe needed to perform this imaging is >1000-fold lower than typical d-luciferin imaging conditions but nonetheless improves overall signal from the brain. Thus, luciferin amides also excel at delivering luciferins into FAAH-expressing cells and tissues.
To test our initial hypothesis, we synthesized four luciferin amides (Figure 1) by the condensation of electrophilic nitriles3,5 with a d-cysteine amide (see Supporting Information). Without a free carboxylate, these luciferin analogs are not light-emitting substrates for purified firefly luciferase (Figures 2 and S1). Pretreatment of the luciferin amides with recombinant rat FAAH12 restores luminescent activity and could be specifically blocked by incubation with FAAH inhibitors such as PF3845 (Figures 2 and S1).13 The presence of FAAH or FAAH inhibitors has no effect on light emitted from the parent luciferins (Figures 2 and S1). Thus, luciferin amides can be used to detect FAAH activity and inhibition in vitro.
Figure 2.
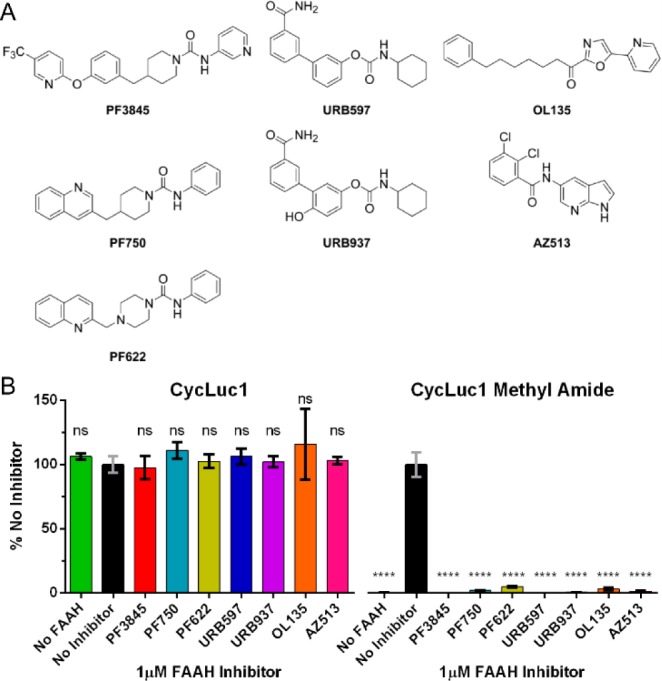
(A) FAAH inhibitor structures. (B) Photon flux from the indicated luciferin analog (10 μM) normalized to emission in the presence of FAAH with no FAAH inhibitor. The assay was performed in triplicate and is represented as the mean ± SEM.
We next sought to determine whether luciferin amides were specific to FAAH and sensitive enough to enable the detection of FAAH activity in live cells. Chinese hamster ovary (CHO) cells are known to express FAAH,14 an integral membrane protein,6 but at levels insufficient to detect with fluorescence-based assays.15 In contrast, treatment of luciferase-expressing CHO cells with luciferin amides resulted in robust bioluminescence (Figures 3A and S2). Potentially, in the complex environment of the cell, luciferin amides could be cleaved by proteases or other serine hydrolases. However, treatment with PF3845, which specifically inhibits FAAH but no other serine hydrolases,13 blocked emission from luciferin amides but had no effect on luciferase activity in the presence of the parent luciferin (Figure 3A). Furthermore, inhibitors of other serine hydrolases had no effect, and we evaluated the potency of a wide range of FAAH inhibitors in the natural context of live cell membranes (Figures S3 and S4). Lacking an ionized carboxylate, the luciferin amides also served as excellent luciferin delivery vehicles in these FAAH-expressing cells, yielding higher bioluminescence signals than their parent luciferins at concentrations <100 μM (Figure S2). CycLuc1-methyl amide achieved higher maximal photon flux than CycLuc1-amide, presumably because uncleaved CycLuc1-amide can ultimately inhibit luciferase, while CycLuc1-methyl amide cannot (Figures S2 and S5).
Figure 3.
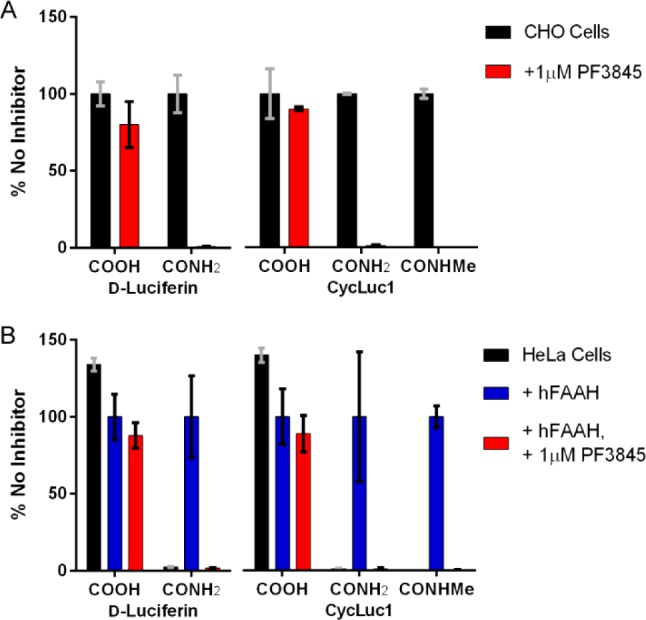
(A) Relative photon flux from live luciferase-expressing CHO cells treated with the indicated luciferins and luciferin amides (125 μM) in the absence (black bars) or presence (red bars) of the FAAH inhibitor PF3845. The data are normalized to the uninhibited sample for each luciferin (black bars). (B) Relative flux from live luciferase-expressing HeLa cells treated with the same set of substrates after transfection with empty pcDNA3.1 vector (black bars), pcDNA3.1-hFAAH (blue bars), or pcDNA3.1-hFAAH and treatment with the FAAH inhibitor PF3845 (red bars). The data are normalized to the uninhibited hFAAH-transfected sample for each luciferin (blue bars). All assays were performed in triplicate and are represented as the mean ± SEM.
HeLa cells do not express FAAH,16,17 and luciferin amides do not yield bioluminescence in luciferase-expressing HeLa cells (Figure 3B). Transfection of HeLa cells with human FAAH enabled bioluminescence with luciferin amides (Figures 3B and S2). Specific inhibition of the transfected FAAH with PF3845 blocked bioluminescence (Figure 3B) and the potency of FAAH inhibitors could be evaluated in these FAAH-transfected live cells (Figure S3). FAAH has been shown to cleave some fatty acid esters,11 and we find that it indeed also contributes to the cleavage of CycLuc1 ethyl ester (Figure S6). However, unlike CycLuc1 amides, the ethyl ester of CycLuc1 is not exclusively cleaved by FAAH and is hydrolyzed to CycLuc1 in both CHO and HeLa cells (Figure S6).
In mice, FAAH is highly expressed in the brain.9 We thus expected that luciferin amides would result in strong brain bioluminescence in luciferase-expressing mice if able to access this tissue. We used adeno-associated virus 9 (AAV9) to express luciferase only in the brain striatum.18 The amides are less water-soluble than the parent carboxylates, necessitating a lower imaging dose. Nonetheless, CycLuc1-amide yielded dramatically higher photon flux in these mice than the parent luciferin CycLuc1 or the conventional substrate d-luciferin (Figure 4). A 400-fold lower dose of CycLuc1-amide was markedly superior to the standard imaging dose of d-luciferin (Figure 4). Even 1000-fold lower doses yielded higher brain bioluminescence than d-luciferin (Figure S7).
Figure 4.

CycLuc1-amide compared to d-Luciferin for bioluminescence imaging in live mice expressing luciferase in (A) the brain or (B) the heart and leg muscles. Quantification is represented as the mean ± SEM for n = 3 mice.
Pretreatment with PF3845 (Figure 2), which has been demonstrated to inhibit only FAAH in mice,13 completely blocked brain bioluminescence when using luciferin amides (Figure S8). Tail-vein injection of AAV9-CMV-luc2 primarily transduces heart19 and leg muscles (Figure 4), tissues where FAAH activity has been reported to be absent.9 In these mice, luciferin amides yielded dramatically lower photon flux than could be achieved with their parent luciferins (Figure 4). By contrast, CycLuc1 ethyl ester was on par with equal doses of the parent luciferin in the heart and leg muscles, but ineffective in the brain (Figure 4). These differences likely reflect the location of the liberating enzymatic activity and biodistribution of the more hydrophobic ester.
To visualize FAAH activity throughout the mouse, we next turned to transgenic mice that express luciferase in all tissues.20 When d-luciferin or CycLuc1 is introduced into these mice, the weakest light emission is from the head, and bioluminescence is dominated by superficial tissues (Figures 5 and S9). In marked contrast, injection of CycLuc1-amide revealed the strongest bioluminescence signals from the brain and kidneys (Figure 5), tissues known to have high FAAH activity.9 Ventral bioluminescence was less well-defined, which may reflect rapid transit of released luciferin out of FAAH-expressing tissues such as the liver (Figure S9). Pretreatment of mice with PF3845 completely blocked bioluminescence from luciferin amides in the brain and in all peripheral tissues (Figures 5, S10, and S11) but had no effect on bioluminescence from the parent luciferins (Figure S12). The aminoluciferin amides (Figure 1) readily sense FAAH activity in vivo (Figure S9), and can be imaged at extremely low doses (as low as 8 nmol/kg; Figure S13). Although d-luciferin amide senses FAAH activity in vitro and in live cells, it works poorly in live mice and cannot sense FAAH activity in the brain (Figure S9). This is consistent with our contention that the improved biodistribution properties of aminoluciferins and low Km values render them superior for use as luminogenic sensors in vivo.4 Interestingly, CycLuc1-methyl amide did not exhibit an advantage over CycLuc1-amide in the mouse (Figure S9). Presumably, inhibition of luciferase by uncleaved luciferin primary amides is not an issue at the substrate concentrations achieved in vivo.
Figure 5.
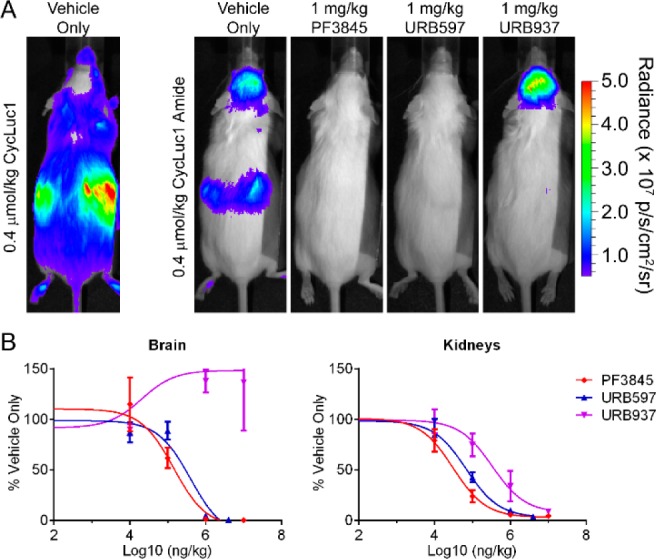
(A) Bioluminescence imaging with CycLuc1 or CycLuc1-amide in ubiquitously expressing transgenic luciferase mice treated with vehicle only or the indicated FAAH inhibitor. (B) Total flux from the brain and kidneys quantitated as a function of inhibitor concentration and normalized to the vehicle-only signal, represented as the mean ± SEM for n = 3 mice. Data were fit by nonlinear regression to determine relative IC50 values in the brain (PF3845, 0.14 mg/kg; URB597, 0.40 mg/kg; URB937, ND) and kidneys (PF3845, 0.03 mg/kg; URB597, 0.07 mg/kg; URB937, 0.33 mg/kg).
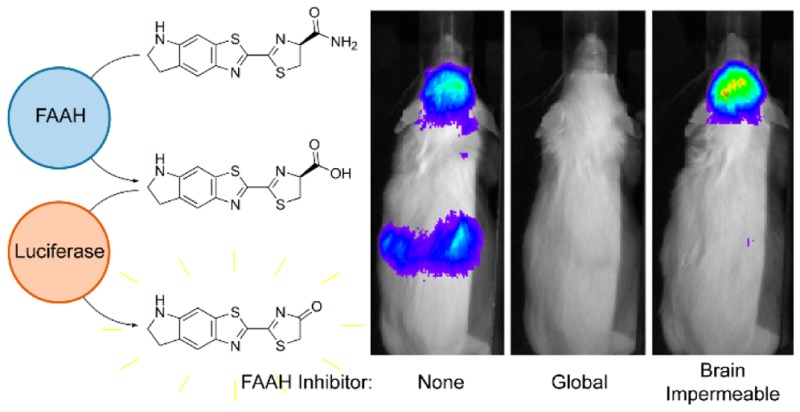
Finally, we sought to determine whether luciferin amides could be used to evaluate the tissue distribution of prospective FAAH inhibitors, which can have important effects on their efficacy.21 URB937 is a brain-impermeable FAAH inhibitor that differs from the global FAAH inhibitor URB597 by a single hydroxyl group (Figure 2).21 Bioluminescence imaging with CycLuc1-amide confirmed that URB597 inhibits FAAH activity in both peripheral and brain tissues (Figures 5, S10, and S11), whereas no inhibition of FAAH activity is detected in the brains of URB937-treated mice (Figure 5).
Many bioluminescent sensors have been described based on “caged” pro-luciferins that can release a luciferin upon the action of an enzyme or reactive small molecule.4,23 The labile moiety is distinct from the luciferin and often separated by a self-immolative linker.24 A limitation of this approach is that the luciferin itself is not contributing to specific recognition or selectivity; the best one can hope for is that its presence is innocuous. Our approach embraces the inherent fatty acid mimetic properties of luciferin analogs to create sensors for enzymes that release fatty acids. The power of this approach is borne out in the exquisite specificity and sensitivity of luciferin amides for FAAH even in vivo, simply by replacing an oxygen atom with nitrogen (Figure 1). Furthermore, we find that luminogenic sensors based on high-affinity, cell-permeable aminoluciferins perform better in mice than those based on d-luciferin. As the number of structurally distinct luciferin analogs grows,4,5 we anticipate there will be additional opportunities to build sensors based on the inherent properties of the luciferin itself.
In summary, we have found that luciferin amides are highly sensitive and selective reporters of FAAH activity. These sensors readily translate from in vitro assays to live cells to in vivo imaging to report on the activity of a single enzyme in its natural context. The bioluminescence approach described herein reveals otherwise invisible endogenous enzymatic activity in live cells and mice and more broadly allows imaging of the biodistribution consequences of subtle modifications to a prospective therapeutic inhibitor in vivo (e.g., ability to cross the blood-brain barrier). Further refinement and modification of the structure of the luciferin4,5 and the scissile bond could potentially allow extension of this bioluminescence detection approach to other enzymes.25,26 Finally, luciferin amides are excellent reagents for increasing the sensitivity of bioluminescence imaging in FAAH-expressing cells and tissues, such as the brain, and allow orders of magnitude lower imaging doses than the natural luciferase substrate.
Acknowledgments
We thank Kathryn Chase for guidance on performing striatal injection, Joanna Chaurette for preliminary imaging studies, and Neil Aronin for the use of a stereotactic frame. We thank Li Zhong and Qin Mao, for tail-vein AAV injections. This work was supported by the US National Institutes of Health (R01EB013270 and R01DA039961).
Supporting Information Available
Experimental procedures, compound characterization, and supplementary figures. The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.5b04357.
The authors declare the following competing financial interest(s): UMass Medical School has filed a patent application on luciferin amides. At present, there is no financial interest.
Supplementary Material
References
- Oba Y.; Ojika M.; Inouye S. FEBS Lett. 2003, 5401–3251. 10.1016/S0014-5793(03)00272-2. [DOI] [PubMed] [Google Scholar]
- Mofford D. M.; Reddy G. R.; Miller S. C. Proc. Natl. Acad. Sci. U. S. A. 2014, 111124443. 10.1073/pnas.1319300111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy G. R.; Thompson W. C.; Miller S. C. J. Am. Chem. Soc. 2010, 1323913586. 10.1021/ja104525m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams S. T.; Miller S. C. Curr. Opin. Chem. Biol. 2014, 21, 112. 10.1016/j.cbpa.2014.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mofford D. M.; Reddy G. R.; Miller S. C. J. Am. Chem. Soc. 2014, 1363813277. 10.1021/ja505795s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blankman J. L.; Cravatt B. F. Pharmacol. Rev. 2013, 652849. 10.1124/pr.112.006387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cravatt B. F.; Giang D. K.; Mayfield S. P.; Boger D. L.; Lerner R. A.; Gilula N. B. Nature 1996, 384660483. 10.1038/384083a0. [DOI] [PubMed] [Google Scholar]
- Shoup T. M.; Bonab A. A.; Wilson A. A.; Vasdev N. Mol. Imaging Biol. 2015, 172257. 10.1007/s11307-014-0789-1. [DOI] [PubMed] [Google Scholar]
- Long J. Z.; LaCava M.; Jin X.; Cravatt B. F. J. Lipid Res. 2011, 522337. 10.1194/jlr.M012153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boger D. L.; Fecik R. A.; Patterson J. E.; Miyauchi H.; Patricelli M. P.; Cravatt B. F. Bioorg. Med. Chem. Lett. 2000, 10232613. 10.1016/S0960-894X(00)00528-X. [DOI] [PubMed] [Google Scholar]
- Patricelli M. P.; Cravatt B. F. Biochemistry 1999, 384314125. 10.1021/bi991876p. [DOI] [PubMed] [Google Scholar]
- Mileni M.; Johnson D. S.; Wang Z.; Everdeen D. S.; Liimatta M.; Pabst B.; Bhattacharya K.; Nugent R. A.; Kamtekar S.; Cravatt B. F.; Ahn K.; Stevens R. C. Proc. Natl. Acad. Sci. U. S. A. 2008, 1053512820. 10.1073/pnas.0806121105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn K.; Johnson D. S.; Mileni M.; Beidler D.; Long J. Z.; McKinney M. K.; Weerapana E.; Sadagopan N.; Liimatta M.; Smith S. E.; Lazerwith S.; Stiff C.; Kamtekar S.; Bhattacharya K.; Zhang Y.; Swaney S.; Van Becelaere K.; Stevens R. C.; Cravatt B. F. Chem. Biol. 2009, 164411. 10.1016/j.chembiol.2009.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto Y.; Morishita J.; Wang J.; Schmid P. C.; Krebsbach R. J.; Schmid H. H. O.; Ueda N. Biochem. J. 2005, 389Pt 1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramarao M. K.; Murphy E. A.; Shen M. W. H.; Wang Y.; Bushell K. N.; Huang N.; Pan N.; Williams C.; Clark J. D. Anal. Biochem. 2005, 3431143. 10.1016/j.ab.2005.04.032. [DOI] [PubMed] [Google Scholar]
- Dickason-Chesterfield A. K.; Kidd S. R.; Moore S. A.; Schaus J. M.; Liu B.; Nomikos G. G.; Felder C. C. Cell. Mol. Neurobiol. 2006, 264–6405. 10.1007/s10571-006-9072-6. [DOI] [PubMed] [Google Scholar]
- Day T. A.; Rakhshan F.; Deutsch D. G.; Barker E. L. Mol. Pharmacol. 2001, 5961369. [DOI] [PubMed] [Google Scholar]
- Evans M. S.; Chaurette J. P.; Adams S. T. Jr; Reddy G. R.; Paley M. A.; Aronin N.; Prescher J. A.; Miller S. C. Nat. Methods 2014, 114393. 10.1038/nmeth.2839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inagaki K.; Fuess S.; Storm T. A.; Gibson G. A.; Mctiernan C. F.; Kay M. A.; Nakai H. Mol. Ther. 2006, 14145. 10.1016/j.ymthe.2006.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Y.-A.; Wagers A. J.; Beilhack A.; Dusich J.; Bachmann M. H.; Negrin R. S.; Weissman I. L.; Contag C. H. Proc. Natl. Acad. Sci. U. S. A. 2004, 1011221. 10.1073/pnas.2637010100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clapper J. R.; Moreno-Sanz G.; Russo R.; Guijarro A.; Vacondio F.; Duranti A.; Tontini A.; Sanchini S.; Sciolino N. R.; Spradley J. M.; Hohmann A. G.; Calignano A.; Mor M.; Tarzia G.; Piomelli D. Nat. Neurosci. 2010, 13101265. 10.1038/nn.2632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van de Bittner G. C.; Bertozzi C. R.; Chang C. J. J. Am. Chem. Soc. 2013, 13551783. 10.1021/ja309078t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carl P. L.; Chakravarty P. K.; Katzenellenbogen J. A. J. Med. Chem. 1981, 245479. 10.1021/jm00137a001. [DOI] [PubMed] [Google Scholar]
- Long J. Z.; Cravatt B. F. Chem. Rev. 2011, 111106022. 10.1021/cr200075y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandiera T.; Ponzano S.; Piomelli D. Pharmacol. Res. 2014, 86, 11. 10.1016/j.phrs.2014.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.