

Abstract
Superparamagnetic iron oxide nanoparticles (SPIOs) have the potential for use as a multimodal cancer therapy agent due to their ability to carry anticancer drugs and generate localized heat when exposed to an alternating magnetic field, resulting in combined chemotherapy and hyperthermia. To explore this potential, we synthesized SPIOs with a phospholipid-polyethylene glycol (PEG) coating, and loaded Doxorubicin (DOX) with 30.8% w/w loading capacity when the PEG length is optimized. We found that DOX-loaded SPIOs exhibited a sustained DOX release over 72 hours where the release kinetics could be altered by PEG length. In contrast, the heating efficiency of the SPIOs showed minimal change with PEG length. With a core size of 14 nm, the SPIOs could generate sufficient heat to raise the local temperature to 43°C, enough to trigger apoptosis in cancer cells. Further, we found that DOX-loaded SPIOs resulted in cell death comparable to free DOX, and that the combined effect of DOX and SPIO-induced hyperthermia enhanced cancer cell death in vitro. This study demonstrates the potential of using phospholipid-PEG coated SPIOs for chemotherapy-hyperthermia combinatorial cancer treatment with increased efficacy.
Introduction
Conventional cancer chemotherapy treatments are often compromised by systemic toxicity which stems from a lack of tumor specificity when anticancer drugs are delivered. The side effects limit the dose of the drug used, rendering effective cancer treatment difficult. Nanoparticle-based drug delivery has the potential to overcome this challenge by targeted delivery to tumor and taking the advantage of tumor’s natural leaky vasculature to increase the accumulation of drug-loaded nanoparticles within the tumor interstitium.1, 2 By loading anticancer drug into nanoparticles, the more favorable pharmacokinetics and tunable biodistribution of nanoparticles can increase the efficacy of the drug.3, 4 Drug molecules loaded into a nanoparticle may also be protected from degradation and oxidation while in circulation.5, 6 A similar approach has been used with liposomal carriers in the form of Doxil; however, while having some reduction in cardiotoxicity, the efficacy in reducing tumor burden was not improved over free Doxorubicin.7 Specifically, the use of liposomes as a drug carrier is hindered by their large size (>100 nm) which makes deep penetration into the tumor tissue difficult following extravasation from the blood vessel.8, 9 Despite its shortcomings, the use of Doxil has stimulated the development of more advanced multi-functional nanoparticles.
Superparamagnetic iron oxide nanoparticles (SPIOs) have the potential to improve cancer treatment by generating local heat when exposed to an alternating magnetic field (AMF). Cancer cells are susceptible to hyperthermia, as raising the temperature to ~43°C for 30–60 minutes can trigger apoptosis.10–12 It is also possible that the cell membrane, cytoskeleton, and proteins involved in DNA damage repair could be affected by the increased temperature which further destabilizes the cell and offsets homeostasis, making cancer cells more vulnerable to chemotherapy.13–15 It has been shown that tumors are especially susceptible to hyperthermia compared to normal tissues because of their faster cell division, increased hypoxia, low pH, and limited temperature regulation due to poor fluid transfer.16, 17 However, hyperthermia alone may not be sufficient for cancer treatment and thus is often used as an adjuvant to other forms of therapy such as radiotherapy and chemotherapy.18, 19 Further, the application of hyperthermia through high-intensity focused ultrasound, radio-frequency interstitial tumor ablation, or laser induced thermal therapy may pose risks of damaging the surrounding tissue, thus preventing the widespread use of hyperthermia for cancer therapies.20 Although previous studies have utilized SPIOs for hyperthermia21–23 and drug delivery24–26 separately, with different core synthesis methods and coating compositions, only very limited efforts have been made to optimize the SPIO-based approach for a combinatorial hyperthermia and chemotherapy for cancer treatment.27
Here we report the development and optimization of phospholipid-PEG coated SPIOs for simultaneous local heat generation and delivery of chemotherapeutic agents. The SPIO nanoparticle is composed of an iron oxide core of ~14 nm coated with a phospholipid-PEG layer which renders the SPIO water soluble and enables a high degree of drug loading through hydrophobic/hydrophilic and/or electrostatic interactions (Schematic 1). The SPIOs can generate a high amount of heat when an alternating magnetic field (AMF) is applied.28 We quantified the effect of PEG length on drug loading and release, as well as temperature change, and identified PEG 2000 to be the optimal length. We demonstrate that SPIOs coated with 1, 2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000] (DSPE-PEG 2000) copolymer can enhance cancer cell death through the concurrent delivery of local heat and chemotherapeutic drugs. Using the SPIOs as both drug carrier and heat source may have the potential advantage of ensuring a synergy between chemotherapy and hyperthermia for more effective cancer treatment.
Schematic 1.
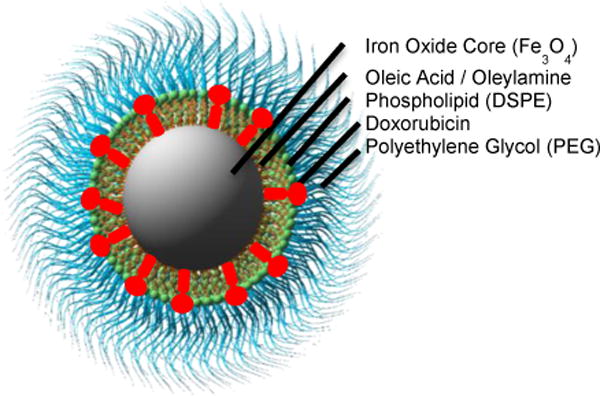
Phospholipid PEG-coated iron oxide nanoparticle loaded with doxorubicin. The nanoparticle consists of an inner iron oxide core, covered with a surfactant layer of oleic acid and oleylamine, and an outer coating of phospholipid-PEG.
Materials and methods
Chemicals
Iron acetylacetonate (Fe(acac)3 99%), 1,2-hexadecanediol (technical grade, 90%), oleic acid (technical grade, 90%), and oleylamine(technical grade, 70%), toluene (≥99.9%), chloroform (99%), dimethyl sulfoxide (DMSO 99%) benzyl ether (98%), hydrochloric acid, hydroxylamine HCl, sodium hydroxide, ammonium acetate, ferrozine, and thioglycolic acid (≥98%) were purchased from Sigma-Aldrich and used as received; DSPE-PEG 2000/5000 was purchased from Avanti Polar Lipids. Doxorubicin hydrochloride (98.0–102.0% HPLC) was purchased from Sigma-Aldrich and dissolved in an aqueous solution at 1 mg/ml.
Synthesis of iron oxide nanoparticles
Iron oxide nanoparticle cores of 14 nm diameter were synthesized by an adapted published procedure.29 Iron acetylacetonate (12 mmol), 1,2-hexadecanediol (60 mmol), oleic acid (72 mmol), and oleylamine (72 mmol) were mixed in benzyl ether (60 ml) in a 500 ml round bottom flask and magnetically stirred. A vacuum was applied for 40 minutes, then the solution was raised to 120°C by a heating mantle attached to a temperature controller. After another 40 minutes, the vacuum was removed and replaced by a flow of Nitrogen. The solution was then raised to 200°C and held for 2 hours before ramping to 300°C and holding for 1 hour. The solution was then cooled to room temperature and ethanol (240 ml) was added to separate and precipitate the nanoparticle cores via three rounds of centrifugation. Finally, the cores were redispersed in toluene.
Dual solvent exchange coating method
To coat 1 mg Fe of SPIOs, 200 μl of the ferrofluid at 5 mg/ml Fe concentration was mixed with DSPE-PEG (2000, 5000) in 400 μl of chloroform in a 100 ml round bottom flask. The amount of DSPE-PEG was determined based on a ratio of 8 DSPE-PEG molecules per nm2 SPIO surface area. Then, 4 ml of DMSO was incrementally added followed by a 30-minute incubation at room temperature. The toluene and chloroform was removed via vaporization under vacuum. Afterwards, 20 ml of deionized water was then slowly added to the solution. DMSO was removed and replaced with deionized water through three rounds of centrifugation in Vivaspin 20 centrifugal filter tubes with molecular weight cut-off 100 kDa (Sartorius, Goettingen, Germany). Finally, the solution was passed through a 0.2 μm HT Tuffryn syringe filter membrane (Pall Life Sciences, Ann Arbor, MI) and stored in deionized water at 4°C.
Characterization
Transmission electron microscopy (TEM)
The average core diameter of the SPIOs was measured via transmission electron microscopy. The polymer coating layer on the SPIOs was visualized by negative staining with phosphotungstic acid on glow discharged copper grids prior to imaging. TEM images were recorded with a transmission electron microscope (Hitachi H-7500, Tokyo, Japan) connected to a CCD camera. The negative staining and TEM procedures were conducted by the Robert P. Apkarian Integrated Electron Microscopy Core at Emory University. The images were analyzed using ImageProPlus® software.
SQUID magnetization measurements
Magnetization measurements on uncoated iron oxide cores were carried out using a superconducting quantum interference device (SQUID) magnetometer (Quantum Design MPMS-5S, San Diego, CA). Field-dependent magnetization curves were measured at 300 K as a function of the external magnetic field in the range of 0–5 × 104 Gauss.
Dynamic light scattering
The hydrodynamic diameter of the coated SPIOs was measured using a dynamic light scattering device (DynaPro Nanostar, Wyatt Technology, Santa Barbara, CA). The SPIOs were dispersed in deionized water at 100 μg/ml Fe and measured at 25°C. The mass weighted size distribution was reported.
Iron concentration measurement
Iron content of samples was determined using a ferrozine assay. Briefly, 50 μl of sample was mixed with 50 μl of 12 M HCl and incubated at room temperature for 30 minutes. Then, 240 μl of 2 M NaOH, 50 μl of 4 M ammonium acetate, 110 μl 5% hydroxylamine HCl, and 500 μl water were added to the solution sequentially. After 30 minutes of incubation, 50 μl of the solution were mixed with 0.02% ferrozine solution in a 384 well plate. Light absorption was read at 562 nm with 810 nm as the reference wavelength using a microplate reader (Safire2, Tecan Group Ltd., Männedorf, Switzerland). The absorption was compared to a molecular iron standard to determine the concentration. The number of SPIOs per gram of iron was estimated to be 1.35E17 based on the volume of a spherical 14 nm diameter SPIO and the density of magnetite (5.17 g/cm3) assuming the cores were made up of Fe3O4.
Drug delivery
Drug loading
The chemotherapeutic drug Doxorubicin was loaded into the SPIOs by incubating the drug with nanoparticles at a 1:1 mass ratio in deionized water for 24 hours undisturbed at room temperature. The free drug was then removed via three rounds of ultracentrifugation for 1 hour at 40,000 g. The amount of Doxorubicin loaded was quantified by first incubating the SPIOs in thiogylcolic acid for 3 hours. Then the light absorption of Doxorubicin was measured using a microplate reader (Safire2, Tecan Group Ltd., Männedorf, Switzerland) at 480 nm with 810 nm as the reference wavelength. The absorption was compared to a standard to determine the concentration. The DOX concentration was then divided by the iron concentration measured by a ferrozine assay to determine the DOX loading as % w/w DOX/Fe.
Drug release
The drug release profile was characterized by injecting 1 mg of SPIOs into a 20 kDa MWCO dialysis cassette (Slide-A-Lyzer, Pierce, Rockford, Ill). The cassette was placed in a release buffer of 80 ml PBS with 5% BSA kept at 37°C and stirred. At the given time points, 100 μl aliquots were removed and replaced with fresh buffer to ensure sink conditions during the release period. The DOX fluorescence intensity of the aliquots was measured at 595 nm via UV-Vis spectrophotometer and the concentration of the aliquots was determined with a known standard. The amount released was compared to the initial amount loaded into the SPIOs to estimate the percent released.
Magnetic fluid hyperthermia
A 1 ml volume of coated SPIOs at 0.4 mg/ml Fe was placed inside a polystyrene-insulated 7.5-turn inductor coil (EasyHeat 2.4kW, Ameritherm, Scottsville, NY). The field strength within the coil was estimated according to the Biot-Savart law and the profile was calculated using Matlab (Figure S1). An alternating magnetic field (23.77 kA/m, 355 kHz) was generated within the coil, causing the SPIOs to produce heat. The resulting temperature rise in the ferrofluid was measured over three minutes with a fiber optic temperature probe (FLUOTEMP, Photon Control Inc., Burnaby BC, Canada) and recorded in real time. The slope of the linear region of the temperature vs. time plot was normalized to a water sample and used to calculate the SAR using the equation below:
where is the mass of the iron in the sample, is the specific heat of water, is the mass of the water, and dT/dt is the temperature rise of the fluid. For the cellular experiments, the water circulating through the coil was adjusted to maintain a 37°C temperature in a 1 ml sample of media without SPIO during the AMF treatment. This baseline was chosen to mimic body temperature.
Cell culture
HeLa cells were obtained from ATCC, USA and cultured in T75 cell culture dishes following ATCC instructions. Cells were grown in Dulbecco’s Modified Eagle’s Medium (DMEM) (Sigma-Aldrich) supplemented with 10% fetal bovine serum (ATCC). Cultures were replenished with fresh media every other day and passaged upon reaching 70% confluence.
Dose response
HeLa cells were seeded in 96-well plates at 3000 cells per well in 100 μl of media and allowed to attach overnight. The media was replaced with either free DOX or DOX-loaded SPIOs (DOX-SPIOs) at a range of DOX concentrations from 0.1–50 μg/ml and incubated for 48 hours. An MTT assay was performed to assess cell viability. Briefly, the media in the wells was replaced with phenol red-free DMEM with 0.5 mg/ml MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide, Invitrogen) and incubated at 37°C for 2 h. The formazan crystals were dissolved in 200 μl of DMSO and the absorbance was read at 570 nm with 810 nm as the reference. The cell viability was compared to the control, and a dose response curve was generated to determine the IC50 by curve fitting.
Cell internalization
HeLa cells were seeded onto eight chamber glass slides at 5 × 104 cells per chamber and allowed to attach overnight. The media was replaced with either free DOX or DOX-SPIOs at 1 μg/ml DOX. At 2 and 8 hours of incubation, cells were washed with PBS, fixed with paraformaldehyde, and stained with DAPI. Cells were imaged with a DeltaVision deconvolution fluorescent microscope equipped with DAPI, TRITC, and Cy5 filters (Applied Precision, Issaquah, WA). Confocal image processing was done using the DeltaVision Softworx program (Applied Precision, Issaquah, WA). The fluorescent intensity of the images was normalized using ImageJ (NIH, Bethesda, MD). The iron internalization was measured by trypsinizing the cells, performing a cell count, and measuring the iron content of the cell population with a ferrozine assay and subtracting the endogenous iron measured from an untreated sample.
Evaluation of combinatorial response to magnetic fluid hyperthermia and Doxorubicin delivery
HeLa cells were grown to 80% confluency in a T-75 cell culture flask. The cells were detached with 0.05% trypsin/EDTA and resuspended in cell culture media with 10 μM HEPES. The cells were counted and 5×104 cells were pipetted into eight different cryovials. The vials were then filled to 1 ml of either media, media with 100 μg/ml Fe SPIO (without DOX), 100 μg/ml Fe DOX-loaded SPIO (16.6 μg/ml DOX), or an equivalent free DOX solution. Each of these groups had a +AMF/−AMF sample with the +AMF samples undergoing a 1 hour AMF treatment (23.77 kA/m, 355 kHz) and the −AMF samples kept in a cell culture incubator at 37°C. An additional sample was heated in a water bath at 43°C for 1 hour. Two hours after the AMF treatment, the media in each sample was then replaced with fresh media and seeded into a 96-well plate at 5000 cells per well so that the total incubation time with the SPIO/SPIO-DOX/DOX was 3 hours. After 24 hours, an MTT assay was performed to compare the cell viabilities between each of the groups.
Results and discussion
Nanoparticle synthesis and coating
Superparamagnetic iron oxide cores were synthesized using a thermal decomposition technique which produces monodisperse iron oxide cores of 14 nm in diameter (Fig. S2†) stabilized with an oleic acid oleylamine surfactant layer (Fig. 1a).29 To confer aqueous solubility to the nanoparticle cores, the SPIOs were coated with a layer of DSPE-PEG using a dual solvent exchange method.30 Phospholipid-PEG is an amphiphilic molecule that enables simultaneous interdigitation of its lipid tails with the carbon chains of the oleic acid surfactant layer and exposure of the PEG chains to the aqueous environment to prevent protein opsonization and macrophage recognition.31, 32 The coated SPIOs can be stored in an aqueous solution for months without any aggregation. The presence of the coating layer was confirmed through TEM imaging of the coated SPIOs negatively stained with phosphotungstic acid, as demonstrated by the white halos surrounding the dark iron oxide cores (Fig. 1B). Two different DSPE-PEG lengths were used for coating with molecular weights of 2000 and 5000 kDa. The coated SPIOs exhibited narrow size distributions (Fig. 1C) with peak hydrodynamic diameters were found to be 28.12 nm and 30.83 nm for the PEG 2000 and 5000 SPIOs respectively. Although the conformation of the PEG molecules has some effect, the size increase over the core diameter (14 nm) roughly correlates with the estimated size of the phospholipid-PEG molecules. It is important to note that the size shown on the negative stain TEM image in Fig. 1B does not accurately reflect the hydrodynamic diameter of the SPIOs, since the drying process results in contraction of the PEG chains.
Fig. 1.
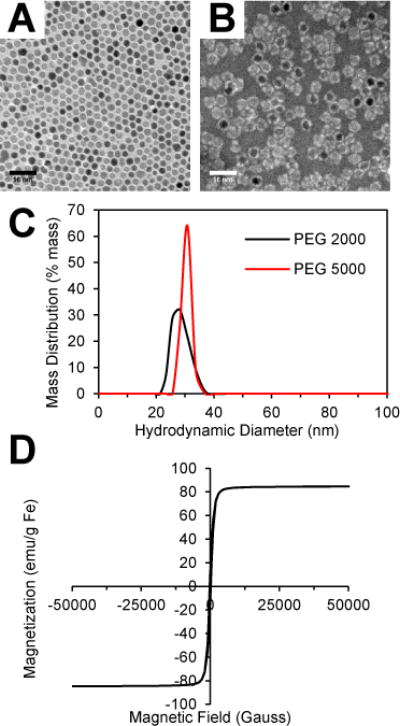
Characterization of iron oxide nanoparticles. (A) TEM image of SPIO cores and (B) PEG 2000 SPIOs negatively stained with phosphotungstic acid. (C) Hydrodynamic size distribution of SPIOs coated with DSPE-PEG 2000 and 5000 measured by dynamic light scattering. (D) Room temperature magnetization curve of SPIO cores.
Heat generation by SPIOs
When SPIOs are exposed to an alternating magnetic field, they generate heat, which can be used for hyperthermia-based cancer treatment. The two main factors that influence the amount of local heat generation are: (1) the specific absorption rate (SAR), a measure of the heat generation per unit mass of SPIO and (2) the local SPIO concentration. The SAR is governed by the physical properties of the SPIOs and the applied magnetic field. Thus, the magnetization of the SPIOs was measured using a superconducting quantum interference device (SQUID) magnetometer, and the magnetic saturation was found to be 84.6 emu/g Fe, which approaches the theoretical limit of bulk magnetite (Fig. 1D). The magnetization curve also displays the on/off behavior indicative of superparamagnetism, and the lack of hysteresis suggests that the primary mechanism of heat generation can be attributed to Néelian and Brownian relaxation rather than hysteretic losses.28, 33
The SAR of the SPIOs was calculated to evaluate the effect of PEG length on the efficiency of heat generation (see details in the Methods section). Aqueous solutions containing coated SPIOs with 0.4 mg/ml Fe were exposed to an alternating magnetic field (23.77 kA/m, 355 kHz), and the temperature of the ferrofluid was measured as a function of time (Fig. S3†). The SAR did not show significant differences between the PEG lengths and was determined to be 371 ± 15 and 425 ± 20 W/g Fe for SPIOs with PEG 2000 and 5000 respectively (Fig. 2A), as described in the Methods section. These results are consistent with the theoretical prediction that for SPIOs with a 14 nm core, Neélian relaxation is thought to be the predominant mechanism which is unaffected by the size of the coating.28, 33
Fig. 2.
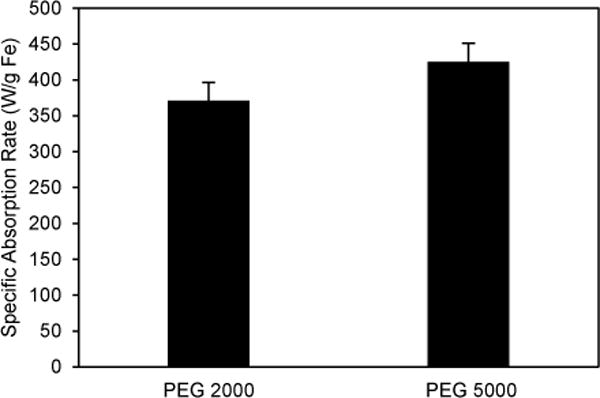
PEG length dependence of heat generation of SPIOs. The specific absorption rates of SPIOs coated with DSPE-PEG 2000 and 5000 exposed to an alternating magnetic field (23.77 kA/m, 355 kHz). Error bars represent standard error.
Doxorubicin loading into SPIOs
The amphiphilic nature of the SPIO coating layer facilitates loading of hydrophobic and amphiphilic drugs into the nanoparticles. The anthracycline antibiotic Doxorubicin (DOX) was chosen as a model drug in this study since it is a common chemotherapeutic that suffers from poor solubility in physiological buffers and exhibits significant cardiotoxicity.7 Further, the fluorescence of DOX enables quantification of loading and release as well as analysis of cellular localization using microscopy. DOX was loaded into SPIOs by incubation with coated SPIOs at room temperature for 72 hours. This drug loading method does not require complex conjugation steps and can achieve high payloads similar to micelle-based drug delivery.6, 34, 35 Specifically, DOX loading into phospholipid-PEG micelles has previously been found to be highly efficient due to hydrophobic and electrostatic interactions between DOX and phospholipid-PEG.34 To characterize the effect of PEG length on DOX loading into the SPIOs, the loading capacity of the PEG 2000 and 5000 SPIOs was compared. We found that the drug loading capacity of SPIOs with PEG 2000 was significantly higher (30.8 ± 2.2% w/w (DOX/iron)) than the PEG 5000 (14.5 ± 3.3%) (Fig. 3A). We also found that loading DOX into the coated SPIOs did not significantly alter the zeta potential of the SPIOs (Fig. S4†). We believe that the negatively charged phosphate group in the phospholipid-PEG layer may have aided drug loading through electrostatic interactions with the positively charged amine group on the DOX. The results shown in Fig. 3A and Fig. S4† suggests that PEG 5000 shielded the negatively charged phosphate group more than that of PEG 2000, demonstrating that the high loading capacity is driven by a combination of electrostatic and hydrophilic/hydrophobic interactions between DOX and the SPIO coating layer. This is consistent with previous results on DOX loading into lipid-PEG micelles.34
Fig. 3.
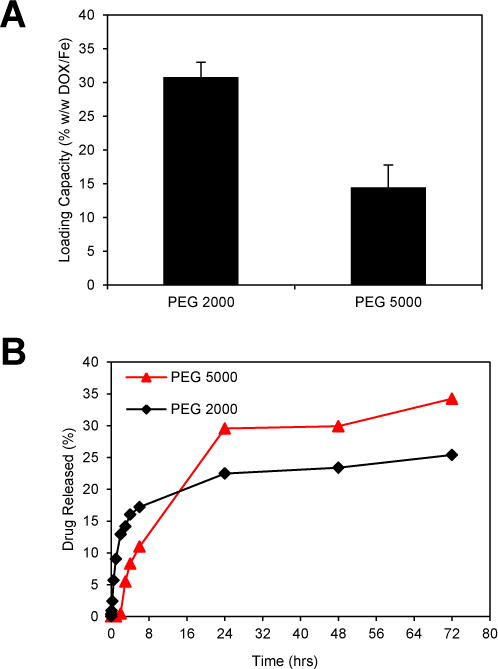
Effect of PEG length on drug loading and release from SPIOs. (A) DOX loading capacity of SPIOs coated with DSPE-PEG 2000 and 5000. Loading capacity was determined by the mass ratio (w/w) of loaded DOX compared to the mass of the SPIOs. Error bars represent standard error. * = significant difference (Tukey test p<0.01) (B) DOX release from DOX-SPIOs as a function of time in a PBS buffer containing 5% BSA. Error bars represent standard error.
Doxorubicin release
The drug release kinetics was assessed to ensure that the time scale of release is sufficient to allow for systemic circulation and subsequent sustained release in the tumor. The release profile was quantified using a dialysis cassette with a release medium consisting of PBS + 5% BSA, since serum albumin has been shown to bind anticancer drugs in circulation.36 As shown in Fig. 3B, there were moderate differences in the release profiles between SPIOs with different PEG lengths though each was characterized by an initial burst release within the first six hours followed by a period of slow sustained release. The sustained release helps maintain effective drug concentrations at a tumor site over time to achieve efficacy and reduce dosing frequency. The SPIOs with PEG 2000 had the highest initial burst release, but SPIOs with PEG 5000 had a similar initial release rate after a two-hour lag period. The slower release rate for SPIOs with PEG 5000 suggests the longer PEG length inhibits diffusion of DOX out of the SPIOs. We believe the much higher loading capacity of SPIOs coated with PEG 2000 has a clear advantage in overall effectiveness as a drug delivery agent, although in some cases a slower, more sustained release would be favorable as well. The incomplete drug release found with both PEG lengths suggests that some DOX molecules were buried deep in the coating layer thus difficult to be released due to their interactions with the hydrophobic lipid layer. Using similar SPIOs loaded with hydrophobic molecules, we did not observe a significant change in the release profile when temperature was increased to 43 °C (data not shown).
Intracellular DOX-SPIO localization
Based on the results of magnetic fluid hyperthermia and drug loading and release, in all the subsequent cellular studies, SPIOs with PEG 2000 were used (hereafter referred to as SPIOs). To compare the cellular delivery efficiency of free DOX and SPIO-based DOX, HeLa cells were incubated with DOX-loaded SPIOs (DOX-SPIOs) or free DOX respectively and fluorescence microscopy was performed. DOX was observed in the cytoplasm and nucleus of the DOX-SPIO treated cells after 2 and 8 hours of incubation respectively. In contrast, the free DOX entered the cell nucleus after only 2 hours of incubation (Fig. 4). The slower accumulation of DOX in the cell nucleus when delivered via SPIOs may be attributed to the fact that DOX needs to be released from SPIOs prior to translocation, and that burst release takes ~6 hours. Internalization of the DOX-SPIOs provides an alternative form of drug entry into the cell instead of relying on diffusion of drug molecules through the cell membrane. To assess SPIO internalization, the iron content of the cells was analyzed and found to be 1.14 pg iron per cell after subtracting endogenous iron in control cells, equivalent to ~153,000 SPIOs per cell. Estimating a cell volume of 2.5 pL, this correlates to an intracellular iron concentration of 455 μg/ml Fe, which should be sufficient for strong intracellular MFH based on the SAR measurement. These results suggest that the DOX-SPIO based approach enables efficient delivery of DOX into cells and a sufficient amount of the SPIOs are internalized to achieve both drug delivery and hyperthermia.
Fig. 4.
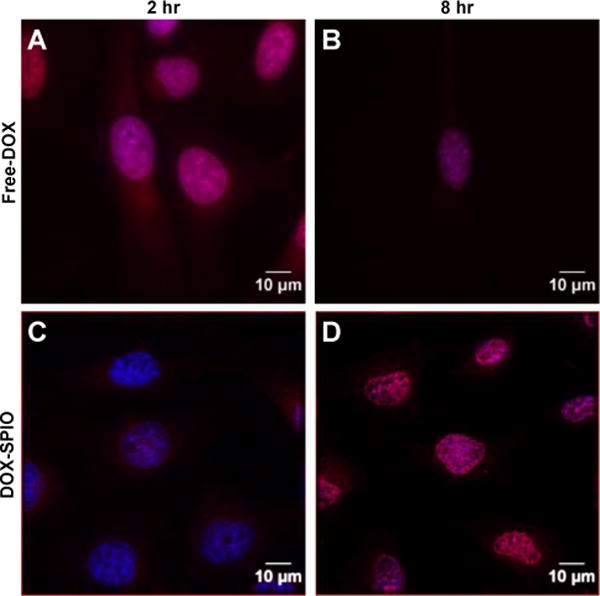
DOX-SPIO internalization into HeLa cells. Confocal images of HeLa cells incubated with free DOX, (A) and (B), or DOX-loaded PEG 2000 SPIOs, (C) and (D), for 2 or 8 hours, respectively. The images displayed in (A–D) are overlays of two channels where cell nuclei are labeled in blue with Hoescht and DOX fluorescence is in red.
Dose response and synergy with hyperthermia
The cytotoxic effect of the DOX-SPIOs was assessed and compared to free DOX in a dose response experiment. HeLa cells were incubated with either free DOX or DOX-SPIOs for 48 hours at a range of equivalent DOX concentrations and subsequently assessed for viability to generate a dose response curve. As shown in Fig. 5, the DOX-SPIOs had a slightly higher IC50 (2.42 μg/ml DOX, determined via curve fitting) compared to the free DOX (1.40 μg/ml DOX). The higher IC50 observed for the DOX-SPIOs may be due to the slower and incomplete release of DOX from DOX-SPIOs. These results suggest that loading DOX into SPIOs does not have an effect on DOX activity following release. The combinatorial effect of hyperthermia and Doxorubicin was tested using water bath heating over a range of DOX concentrations as shown in Fig. S5† as this is a conventional method for applying hyperthermia. We found that with 1 μg/ml free DOX concentration, HeLa cells subjected to hyperthermia exhibited an enhanced combinatorial effect compared with the predicted additive effect of the individual treatments.
Fig. 5.
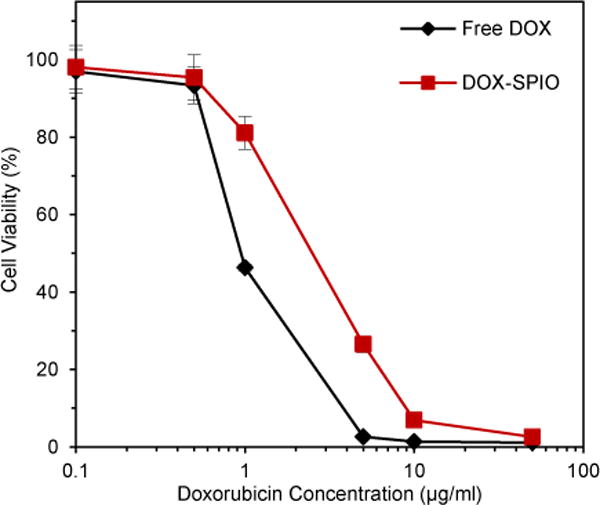
Dose response. Cell viability of HeLa cells incubated with equivalent DOX concentrations of either free DOX or DOX-loaded PEG 2000 SPIOs for 48 hours. Measurements were taken with an MTT assay normalized to an untreated control. Error bars represent standard error.
Combinatorial MFH and DOX delivery
In order to demonstrate the potential of SPIOs for combined cancer chemotherapy and hyperthermia, the effect of SPIO-induced MFH together with SPIO-based DOX delivery was determined through the assessment of cell viability. To observe a combinatorial effect, a mild thermal dose and short DOX-SPIO incubation with cells were chosen such that the combined effect would not be concealed by the death of the entire cell population by either treatment individually. By varying the treatment parameters, it was found that with a 100 μg Fe/ml (16.6 μg DOX/ml) concentration of DOX-SPIOs, neither a 1-hour MFH nor 3-hour DOX-SPIO incubation resulted in total cell death. Fig. 6A shows the temperature profile for the media, SPIO, and DOX-SPIO samples when exposed to the AMF for one hour. The SPIO and DOX-SPIO solutions reached 43°C within 20 minutes while the media held a temperature of ~37°C (Fig. 6A). The oscillation in the temperature curves is due to the water running through the inductor coil used to maintain the baseline temperature of 37°C. Thus, applying AMF for 1 hour should subject HeLa cells to the apoptotic temperature regime (43–46°C) with the necessary duration (~30 minutes) to trigger apoptosis.37
Fig. 6.
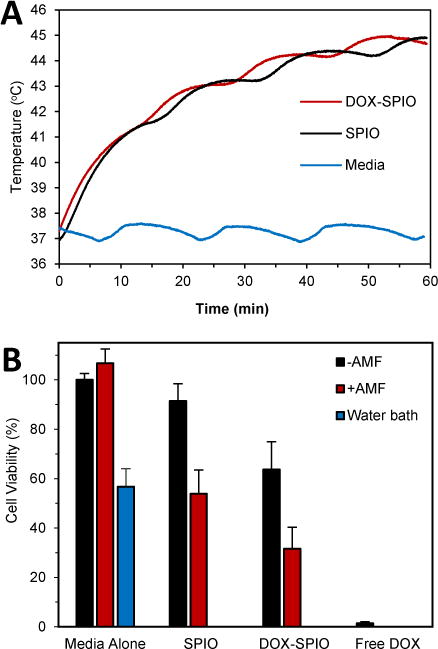
Combined SPIO-based DOX delivery and SPIO-induced MFH. The viability of HeLa cells was determined following treatment with media alone, water bath heating (43°C), just PEG 2000 SPIO (without DOX), DOX-loaded PEG 2000 SPIOs, or free DOX. Each of the indicated groups had samples with or without a 1 hour exposure to an AMF (23.77 kA/m, 355 kHz). (A) Temperature profiles during AMF treatment of media, SPIO, DOX-SPIO samples. (B) Cell viability measured 24 hours after treatment using an MTT assay. Error bars represent standard error.
As shown in Fig. 6B, incubation with SPIOs alone (i.e., without DOX loading) or the presence of an AMF in the absence of SPIOs did not cause a significant drop in cell viability, indicating that SPIO incubation or magnetic field exposure alone does not affect cell viability. We found that water bath heating of HeLa cells at 43°C for 1 hour and SPIO-based MFH for 1 hour decreased cell viability to 57 ± 7% and 54 ± 10%, respectively, showing a comparable effect. SPIO-based DOX delivery decreased cell viability to 64 ± 11% after 3 hours of incubation, although it was not as dramatic as the free DOX, which is in agreement with the results from the dose response experiment. Based on the results of the release study shown in Fig. 3B, only ~15% of the loaded DOX was released from the SPIOs after 3 hours, equivalent to a 2.49 μg/mL DOX concentration available to affect the cells (in comparison with 16.6 μg/mL in the free DOX case). Using the dose response curve shown in Fig. 5, a DOX concentration of ~2.5 μg/mL correlates to a reduction in cell viability by ~50%, consistent with the result shown in Fig. 6B.
HeLa cells subjected to the multimodal approach of having both the DOX-SPIO delivery and SPIO-based MFH exhibited a lower cell viability (32 ± 9%) than either of the single modality treatments, namely drug delivery with DOX-SPIOs (64 ± 11%) or SPIO-based MFH (54 ± 10%) alone (Fig. 6). As shown in Fig. S6†, we observed a reduction in cell attachment and an altered cell morphology of the DOX, MFH or DOX/MFH combinatorial treated cells (c, d, f, g) compared to the control cells (a, b, e), further confirming the effect of DOX-SPIO delivery and SPIO-based MFH. The combinatorial effect was consistent with the calculated additive response based on the decrease in cell viability observed in the individual treatments (Fig. 6). The drug delivery kinetics coupled with the narrow window of synergistic behavior seen in Fig. S5† could mask the potential for a synergistic response. The exact interaction between DOX and elevated temperature remains elusive, since there are conflicting reports suggesting that the combined effect could be additive, superadditive, or threshold-like when free DOX is used in conjunction with hyperthermia.38–41 Nevertheless, our results suggest that a combined DOX delivery and MFH using SPIOs is more effective than using either treatment alone.
Conclusions
Conventional chemotherapy of cancer is often compromised by systemic toxicity, and hyperthermia alone does not have the required efficacy in cancer treatment. To address this important clinical challenge, in this work, we demonstrate that phospholipid-PEG coated SPIOs have the potential to concurrently deliver Doxorubicin and generate heat for an enhanced multimodal cancer treatment. We investigated the role of PEG length in both treatment modalities, and identified phospholipid-PEG2000 as the optimal coating that gives a high drug loading capacity of 30.8 ± 2.2% w/w (DOX/iron). We found that the phospholipid-PEG coated SPIOs were capable of raising the local temperature to apoptotic levels and a sustained DOX release without affecting drug activity. The multimodal hyperthermia-DOX treatment delivered by these SPIOs was more effective in causing cell death over the individual modalities. This SPIO-based approach offers many potential benefits for more effective cancer treatment. In addition to the ability to have combined chemotherapy and hyperthermia, the superparamagnetic property of the SPIOs provides T2 contrast for magnetic resonance imaging (MRI), as well as the ability to accumulate in specific tissues or organs by applying an external magnetic field locally.42–46 The phospholipid-PEG coating also facilitates specific cell targeting by covalently attaching targeting ligands to the PEG chain, and fluorescence imaging by loading a lipophilic dye into the coating layer.43, 47 Therefore, by utilizing these multifunctional abilities, it is possible to integrate diagnostic imaging with targeted drug delivery and hyperthermia, to track drug-loaded SPIOs in vivo, and to further enhance the efficacy of cancer treatment by increasing specific accumulation of SPIOs in tumor. These possibilities will be explored in subsequent studies.
Supplementary Material
Acknowledgments
This work was supported by the National Heart Lung and Blood Institute of the NIH as a Program of Excellence in Nanotechnology award (HHSN268201000043C to GB).
Footnotes
Electronic Supplementary Information (ESI) available: Core size distribution; temperature increase for specific absorption rate calculations; effect of DOX loading on zeta potential; combined effect of hyperthermia and free DOX; cell morphology following DOX/hyperthermia treatment.
Notes and references
- 1.Matsumura Y, Maeda H. Cancer Research. 1986;46:6387–6392. [PubMed] [Google Scholar]
- 2.Davis ME, Chen ZG, Shin DM. Nature reviews Drug Discovery. 2008;7:771–782. doi: 10.1038/nrd2614. [DOI] [PubMed] [Google Scholar]
- 3.Rahman A, Carmichael D, Harris M, Roh JK. Cancer Research. 1986;46:2295–2299. [PubMed] [Google Scholar]
- 4.Gabizon A, Shmeeda H, Barenholz Y. Clinical Pharmacokinetics. 2003;42:419–436. doi: 10.2165/00003088-200342050-00002. [DOI] [PubMed] [Google Scholar]
- 5.Kwon MNG, Yokoyama M, Okano T, Sakurai Y, Kataoka K. Journal of Controlled Release. 1999;48:195–201. [Google Scholar]
- 6.Tang N, Du G, Wang N, Liu C, Hang H, Liang W. Journal of the National Cancer Institute. 2007;99:1004–1015. doi: 10.1093/jnci/djm027. [DOI] [PubMed] [Google Scholar]
- 7.O’Brien ME, Wigler N, Inbar M, Rosso R, Grischke E, Santoro A, Catane R, Kieback DG, Tomczak P, Ackland SP, Orlandi F, Mellars L, Alland L, Tendler C. Annals of oncology: official journal of the European Society for Medical Oncology/ESMO. 2004;15:440–449. doi: 10.1093/annonc/mdh097. [DOI] [PubMed] [Google Scholar]
- 8.Weissig V, Whiteman KR, Torchilin VP. Pharmaceutical Research. 1998;15:1552–1556. doi: 10.1023/a:1011951016118. [DOI] [PubMed] [Google Scholar]
- 9.Fang C, Shi B, Pei YY, Hong MH, Wu J, Chen HZ. European Journal of Pharmaceutical Sciences. 2006;27:27–36. doi: 10.1016/j.ejps.2005.08.002. [DOI] [PubMed] [Google Scholar]
- 10.Harmon BV, Takano YS, Winterford CM, Gobe GC. International Journal of Radiation Biology. 1991;59:489–501. doi: 10.1080/09553009114550441. [DOI] [PubMed] [Google Scholar]
- 11.Vorotnikova E, Ivkov R, Foreman A, Tries M, Braunhut SJ. International Journal of Radiation Biology. 2006;82:549–559. doi: 10.1080/09553000600876678. [DOI] [PubMed] [Google Scholar]
- 12.Sakaguchi Y, Stephens LC, Makino M, Kaneko T, Strebel FR, Danhauser LL, Jenkins GN, Bull JM. Cancer Research. 1995;55:5459–5464. [PubMed] [Google Scholar]
- 13.Sellins KS, Cohen JJ. Radiation Research. 1991;126:88–95. [PubMed] [Google Scholar]
- 14.Chu KF, Dupuy DE. Nature Reviews Cancer. 2014;14:199–208. doi: 10.1038/nrc3672. [DOI] [PubMed] [Google Scholar]
- 15.Roti Roti JL, Kampinga HH, Malyapa RS, Wright WD, vanderWaal RP, Xu M. Cell Stress & Chaperones. 1998;3:245–255. doi: 10.1379/1466-1268(1998)003<0245:nmaatf>2.3.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Issels RD. European Journal of Cancer. 2008;44:2546–2554. doi: 10.1016/j.ejca.2008.07.038. [DOI] [PubMed] [Google Scholar]
- 17.Otte J. European journal of pediatrics. 1988;147:560–569. doi: 10.1007/BF00442463. [DOI] [PubMed] [Google Scholar]
- 18.Hildebrandt B, Wust P, Ahlers O, Dieing A, Sreenivasa G, Kerner T, Felix R, Riess H. Critical Reviews in Oncology Hematology. 2002;43:33–56. doi: 10.1016/s1040-8428(01)00179-2. [DOI] [PubMed] [Google Scholar]
- 19.Wust P, Stahl H, Dieckmann K, Scheller S, Loffel J, Riess H, Bier J, Jahnke V, Felix R. Int J Radiation Oncology Biol Phys. 1996;34:635–646. doi: 10.1016/0360-3016(95)02151-5. [DOI] [PubMed] [Google Scholar]
- 20.Wust P, Hildebrandt B, Sreenivasa G, Rau B, Gellermann J, Riess H, Felix R, Schlag PM. The Lancet Oncology. 2002;3:487–497. doi: 10.1016/s1470-2045(02)00818-5. [DOI] [PubMed] [Google Scholar]
- 21.Chandrasekharan P, Maity D, Yong CX, Chuang KH, Ding J, Feng SS. Biomaterials. 2011;32:5663–5672. doi: 10.1016/j.biomaterials.2011.04.037. [DOI] [PubMed] [Google Scholar]
- 22.Sadhukha T, Wiedmann TS, Panyam J. Biomaterials. 2014;35:7860–7869. doi: 10.1016/j.biomaterials.2014.05.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pala K, Serwotka A, Jelen F, Jakimowicz P, Otlewski J. International Journal of Nanomedicine. 2014;9:67–76. doi: 10.2147/IJN.S52539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jain TK, Morales MA, Sahoo SK, Leslie-Pelecky DL, Labhasetwar V. Molecular Pharmaceutics. 2005;2:194–205. doi: 10.1021/mp0500014. [DOI] [PubMed] [Google Scholar]
- 25.Liong M, Lu J, Kovochich M, Xia T, Ruehm SG, Nel AE, Tamanoi F, Zink JI. ACS Nano. 2008;2:889–896. doi: 10.1021/nn800072t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dilnawaz F, Singh A, Mohanty C, Sahoo SK. Biomaterials. 2010;31:3694–3706. doi: 10.1016/j.biomaterials.2010.01.057. [DOI] [PubMed] [Google Scholar]
- 27.Taratula O, Dani RK, Schumann C, Xu H, Wang A, Song H, Dhagat P, Taratula O. International Journal of Pharmaceutics. 2013;458:169–180. doi: 10.1016/j.ijpharm.2013.09.032. [DOI] [PubMed] [Google Scholar]
- 28.Rosensweig RE. Journal of Magnetism and Magnetic Materials. 2002;252:370–374. [Google Scholar]
- 29.Xie J, Peng S, Brower N, Pourmand N, Wang SX, Sun S. Pure and Applied Chemistry. 2006;78 [Google Scholar]
- 30.Tong S, Hou S, Ren B, Zheng Z, Bao G. Nano letters. 2011;11:3720–3726. doi: 10.1021/nl201978c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Owens DE, 3rd, Peppas NA. International Journal of Pharmaceutics. 2006;307:93–102. doi: 10.1016/j.ijpharm.2005.10.010. [DOI] [PubMed] [Google Scholar]
- 32.Gref R, Luck M, Quellec P, Marchand M, Dellacherie E, Harnisch S, Blunk T, Muller RH. Colloids and Surfaces B, Biointerfaces. 2000;18:301–313. doi: 10.1016/s0927-7765(99)00156-3. [DOI] [PubMed] [Google Scholar]
- 33.Lartigue L, Innocenti C, Kalaivani T, Awwad A, Sanchez Duque Mdel M, Guari Y, Larionova J, Guerin C, Montero JL, Barragan-Montero V, Arosio P, Lascialfari A, Gatteschi D, Sangregorio C. Journal of the American Chemical Society. 2011;133:10459–10472. doi: 10.1021/ja111448t. [DOI] [PubMed] [Google Scholar]
- 34.Wang Y, Wang R, Lu X, Lu W, Zhang C, Liang W. Pharmaceutical Research. 2010;27:361–370. doi: 10.1007/s11095-009-0029-6. [DOI] [PubMed] [Google Scholar]
- 35.Gao ZG, Lukyanov AN, Singhal A, Torchilin VP. Nano Letters. 2002;2:979–982. [Google Scholar]
- 36.Ghuman J, Zunszain PA, Petitpas I, Bhattacharya AA, Otagiri M, Curry S. Journal of Molecular Biology. 2005;353:38–52. doi: 10.1016/j.jmb.2005.07.075. [DOI] [PubMed] [Google Scholar]
- 37.Palzer RJ, Heidelberger C. Cancer Research. 1973;33:415–321. [PubMed] [Google Scholar]
- 38.Hahn G, Braun J, Har-Kedar I. Proceedings of the National Academy of Sciences. 1975;72:937–940. doi: 10.1073/pnas.72.3.937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kawai H, Minamiya Y, Kitamura M, Matsuzaki I, Hashimoto M, Suzuki H, Abo S. Cancer Cell. 1997;79:214–219. doi: 10.1002/(sici)1097-0142(19970115)79:2<214::aid-cncr3>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 40.Marmor J, Hilerio F, Hahn G. Cancer Research. 1979;39:2166–2171. [PubMed] [Google Scholar]
- 41.Bull JMC. Cancer Research. 1984;44:4853s–4856s. [PubMed] [Google Scholar]
- 42.Kalambur VS, Han B, Hammer BE, Shield TW, Bischof JC. Nanotechnology. 2005;16:1221–1233. [Google Scholar]
- 43.Landazuri N, Tong S, Suo J, Joseph G, Weiss D, Sutcliffe DJ, Giddens DP, Bao G, Taylor WR. Small. 2013;9:4017–4026. doi: 10.1002/smll.201300570. [DOI] [PubMed] [Google Scholar]
- 44.Sathe TR, Agrawal A, Nie S. Analytical Chemistry. 2006;78:5627–5632. doi: 10.1021/ac0610309. [DOI] [PubMed] [Google Scholar]
- 45.Yavuz CT, Mayo JT, Yu WW, Prakash A, Falkner JC, Yean S, Cong L, Shipley HJ, Kan A, Tomson M, Natelson D, Colvin VL. Science. 2006;314:964–967. doi: 10.1126/science.1131475. [DOI] [PubMed] [Google Scholar]
- 46.Chertok B, Moffat BA, David AE, Yu F, Bergemann C, Ross BD, Yang VC. Biomaterials. 2008;29:487–496. doi: 10.1016/j.biomaterials.2007.08.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tong S, Hou S, Zheng Z, Zhou J, Bao G. Nano letters. 2010;10:4607–4613. doi: 10.1021/nl102623x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.