

Abstract
Adaptive immune systems have recently been recognized in prokaryotic organisms where, in response to viral infection, they incorporate short fragments of invader-derived DNA into loci called Clustered Regularly Interspaced Short Palindromic Repeats (CRISPRs). In subsequent infections, the CRISPR loci are transcribed and processed into guide sequences for the neutralization of the invading RNA or DNA. The CRISPR-associated protein machinery (Cas) lies at the heart of this process, yet many of the molecular details of the CRISPR/Cas system remain to be elucidated. Here we report the first structure of Csa3, a CRISPR-associated protein from Sulfolobus solfataricus (Sso1445), which reveals a dimeric two-domain protein. The N-terminal domain is a unique variation on the di-nucleotide binding-domain that orchestrates dimer formation. In addition, it utilizes two conserved sequence motifs (Thr-h-Gly-Phe-(Asn/Asp)-Glu-X4-Arg and Leu-X2-Gly-h-Arg) to construct a 2-fold symmetric pocket on the dimer axis. This pocket is likely to represent a regulatory ligand-binding site. The N-terminal domain is fused to a C-terminal MarR-like winged helix-turn-helix domain that is expected to be involved in DNA recognition. Overall, the unique domain architecture of Csa3 suggests a transcriptional regulator under allosteric control of the N-terminal domain. Alternatively, Csa3 may function in a larger complex, with the conserved cleft participating in protein-protein or protein-nucleic acid interactions. A similar N-terminal domain is also identified in Csx1, a second CRISPR associated protein family of unknown function.
Keywords: Prokaryotic RNAi, Transcriptional regulation, COG0640, COG4006, COG1517
INTRODUCTION
All organisms are targets for infection by selfish genetic elements. Accordingly, they have evolved defensive systems to counteract these pathogens. In higher eukaryotes, defenses include both the innate and adaptive immune systems 1. Systems of innate immunity are also found in prokaryotes, including processes leading to abortive infections and the well-known restriction-modification system. Recently, however, prokaryotic adaptive immune systems have also been recognized; for recent reviews see 1; 2; 3; 4;5. Many prokaryotes can modify their genomic sequence at loci called Clustered Regularly Interspaced Short Palindromic Repeats (CRISPRs) 1 to store information that allows recognition and clearance of pathogens during subsequent infections. Recent surveys have identified CRISPR loci in 39% of the sequenced bacterial genomes and 88% of the sequenced archaeal genomes6.
CRISPR loci consist of a series of small direct repeats (24–47 base pairs) separated by short “spacer” sequences (20–50 base pairs) derived from mobile genetic elements such as plasmids and viruses6; 7; 8; 9; 10; 11; 12. These invader derived sequences are transcribed and processed into short RNA molecules known as CRISPR-RNA or prokaryotic silencing RNA that serve as guide sequences for the recognition and neutralization of invading nucleic acid13; 14.
A significant number of gene families associated with CRISPR loci have been identified15; 16; 17. Some of these CRISPR-associated (cas) genes encode the machinery used to process the CRISPR-transcripts, while others are thought to function in the recognition and neutralization of foreign genetic elements or the incorporation of new spacers 1. While the process is reminiscent of eukaryotic RNAi, there are significant differences. The prokaryotic CRISPR/Cas machinery lacks apparent homology to the RNAi protein machinery in both primary sequence and three dimensional structure 18; 19; 20, and CRISPR/Cas appears to target both DNA and RNA 21; 22. Cas genes are generally organized in putative operons, which are physically close to the CRISPR loci. Co-occurrence patterns for cas genes within genomes and gene clusters suggest the Cas machinery takes several different forms. These are alternatively called subtypes by Haft et al. 16 or CRISPR-associated systems (CASS) by Makarova et al. 17.
A comprehensive study by Haft et al. identified 45 CRISPR-associated gene families 16. These include a set of “core” cas genes, (cas1-6) and 8 groups of subtype-specific gene families (cse1-4, csy1-4, csn1-2, csd1-2, cst1-2, csh1-2, csa1-5 and csm1-5). A given CRISPR/Cas system will encode several of the core Cas proteins plus at least one of these eight subtypes. In addition, several CRISPR/Cas systems include a third cluster of genes that belong to the Repeat Associated Mysterious Protein (RAMP) superfamily and are named cmr1-6. Finally, Haft et al. also identify seven additional CRISPR-associated gene families (csx1-7), that lack an identified contextual pattern 16.
Similarly, Makarova et al. have categorized the CRISPR-associated proteins into 25 families with names loosely based on clusters of orthologous groups (COGs). Subsets of these protein families are then grouped into 7 CRISPR-associated systems (CASS1-7) based on the presence and genomic organization of these COG-based families 17. Functions were predicted for several CRISPR-associated proteins; these include nucleases, helicases and nucleic acid-binding proteins 16; 17. Nuclease activities have now been experimentally confirmed for Cas1, Cas2, Cas3b, Cas6 and Cse3 14; 18; 19; 20; 23. In E. coli Cse1-4 and Cas5e form a complex which processes CRISPR transcripts and is thought to participate in a subsequent neutralization of invading DNA 14. Similarly, the proteins of the RAMP module (Cmr1-6) in Pyrococcus furiosus form a complex with crRNA; which displays an activity analogous to the RNA-Induced Silencing Complex in eukayotic RNAi 22.
Sulfolobus solfataricus, a model system for the Crenarchaeota, has played a significant role in studies of CRISPR/Cas17; 20; 23; 24; 25; 26; 27. S. solfataricus utilizes CRISPR-associated system 7a of Makarova et al. and the Aeropyrum pernix or Apern CRISPR/Cas subtype, designated csa, of Haft et al. 16; 17. S. solfataricus strain P2 contains six CRISPR loci designated CRISPRs A-F. CRISPRs A-D are localized to an 80 kbp region that, except for csa4, contains one or more copies of each of the Cas and csa genes. These include sso1444 and sso1445 which encode Csa3 proteins16.
Csa3 proteins are predicted to have an N-terminal domain of unknown function fused to a C-terminal winged helix-turn-helix domain, and are thus predicted to be involved in regulation of CRISPR-Cas16; 17. Csa3 proteins are members of COG064016 and are predicted to share their domain of unknown function with COG1517-like proteins17, a large family identified by Makarova et al., that includes both CRISPR-associated and non-CRISPR-associated proteins17. Here, using X-ray crystallography and solution-state small angle X-ray scattering (SAXS), we report the structure of Sso1445, the first structure of a Csa3 protein.
RESULTS
S. solfataricus
Csa3 (Sso1445) was expressed in E. coli with a minimal, N-terminal His6–tag, giving a total calculated mass of 27,769 Da. The purified protein migrates as a 60 kDa particle on a Superdex 75 column, suggesting a dimer in solution. Csa3 was crystallized in space group P212121 with two chains per asymmetric unit, and the structure was solved by multi-wavelength anomalous dispersion (MAD) at the selenium edge. Detailed statistics on data collection and structure refinement are presented in Tables 1 and 2. The coordinates and structure factors have been deposited with the protein databank (PDB ID 2WTE).
Table 1.
Data collection
| Data Set | Se-Edge | Se-Peak | Se-Remote |
|---|---|---|---|
| Wavelength (Å) | 0.97925 | 0.97869 | 0.91737 |
| Space Group | P212121 | ||
| Cell Constants (Å) | a = 53.400, b = 89.810, c= 102.501 | ||
| (°) | α = β = γ = 90.00 | ||
| Resolution Rangea (Å) | 50–1.90 (1.94–1.90) | 50–1.90 (1.94–1.90) | 50–1.80 (1.84–1.80) |
| Unique Reflectionsa | 39,389 | 74,809 | 46,357 |
| Average Redundancya | 4.0(4.0) | 4.2(4.1) | 4.0(4.1) |
| I/σa | 30.0(3.2) | 34.0(4.0) | 27.5(1.9) |
| Completeness (%) | 99.1(95.0) | 99.6(98.0) | 99.5 (100) |
| Rsyma,b (%) | 3.6 (25.8) | 3.6 (25.8) | 4.4 (45.7) |
Numbers in parenthesis refer to the highest resolution shell.
Rsym=100*ΣhΣi|Ii(h)−<I(h)>|/ ΣhI(h) where Ii(h) is the ith measurement of reflection h and <I(h)> is the average value of the reflection intensity.
Table 2.
Model Refinement
| Rworkc (%) | 18.0 (23.4) |
| Rfreec (%) | 22.0 (32.9) |
| Real Space CCd (%) | 96.1 |
| Mean B Value (overall; Å2) | 16.3 |
| Coordinate Error (base on maximum likelihood, Å) | 0.074 |
| RMSD from ideality: | |
| Bonds (Å) | 0.015 |
| Angles (°) | 1.287 |
| Ramachandran Plote: | |
| Most Favored (%) | 99.5 |
| Additional Allowed (%) | 0.5 |
| PDB Accession Code | 2WTE |
Rwork = Σ||Fo|−Fc||/ΣFo| where Fo and Fc are the observed and calculated structure factor amplitudes used in refinement. Rfree is calculated as Rcryst, but using the “test” set of structure factor amplitudes that were withheld from refinement (4.9%).
Correlation coefficient (CC) is agreement between the model and 2mFo−DFc density map.
Calculated using Molprobity 63
Structure of the Csa3 Protomer
The structure reveals a two domain protein. The N-terminal domain, comprised of residues 1–132, consists of a six-stranded, doubly wound, mixed β-sheet with flanking α-helices (Fig. 1A). β-strands N1 through N5 run parallel to each other with β-strand N6 running antiparallel to the other 5. The β-sheet displays 3-2-1-4-5-6 topology with strands βN1 through βN5 connected to each other by right handed helical crossovers. Strand βN5 is then connected to strand βN6 by an 8-residue reverse turn.
Figure 1.

Csa3 is a dimer with two domains per subunit (Embedded 3D content, see below for instructions). (A) Stereo ribbon diagram of the Csa3 protomer. The N-terminal domain is colored cyan, the linker in blue and the C-terminal domain in deep teal. Secondary structural elements are labeled by domain and in ascending order from the N- to C-termini. (B) Stereo ribbon diagram of the Csa3 dimer. The N-terminal domain, linker and C-terminal domains are colored cyan, light blue and deep teal in chain A and light orange, tan and orange in chain B. The recognition helices in both chains are colored pale blue. (C) Stereo view of the Csa3 dimer looking down upon the winged helix-turn helix domains. Relative to panel B, the dimer has been rotated 90° about a horizontal axis in the plane of the page. Embedded interactive content requires the free Adobe Reader software, version 9 or later and can be activated by clicking on any part of the figure. The model can be manipulated interactively using the mouse. Options for selecting, rotating, panning and zooming are available in the toolbar or contextual menu. Parts of the model can be individually accessed and toggled on or off using the model tree. Preset views can be accessed using the dropdown “views” menu. To end -D viewing, right-click on the model and select “disable content”;for MAC users, Ctrl+click.
The overall fold is thus similar to that of the classic dinucleotide binding domain composed of six parallel β-stands connected by right handed helical crossovers 28, but with several critical modifications. These include the use of the reverse turn connecting strands βN5 and βN6, as opposed to the α-helical crossover, resulting in an antiparallel βN6, rather than the more common parallel sixth strand. In addition, Csa3 also lacks the Gly-X-Gly-X-X-(Gly/Ala) signature motif found in the α1-β1 loop of the dinucleotide binding domain. Instead, Sso1445 and other Csa3 sequences contain a conserved Gly-(Phe/Ile) sequence in their βN1-αN1 loops (Figs. 4 and 5).
Figure 4.

Two conserved sequence motifs are found in the Csa3 N-terminal domain. The genome properties tool 43 and CRISPR database 6 were used to identify csa3 genes that are near clusters of cas genes and at least one CRISPR locus. The Csa3 sequences were aligned using 3D-COFFEE 44. The two sequence motifs which are conserved among Csa3 orthologs are outlined with boxes. Csa3 residues contributing to the dimer interface are marked with asterisks’ (*). Structure-based pairwise alignments of the VC1899 and Sso1389 (Csx1) N-terminal domains and the OhrR wHTH domain with Sso1445 were calculated using Dali 39. Secondary structural elements of Sso1445 (Csa3), VC1899, Sso1389 (Csx1) and OhrR were identified using DSSP. The sequence motifs, which are conserved among Csa3 orthologs are not present in VC1899 or Csx1. Csx1 has a 16-residue insertion at the site corresponding to Motif 1 in Csa3. GI numbers associated with the listed Csa3 proteins are presented in Table S1. The figure was generated using ALINE 70.
Figure 5.
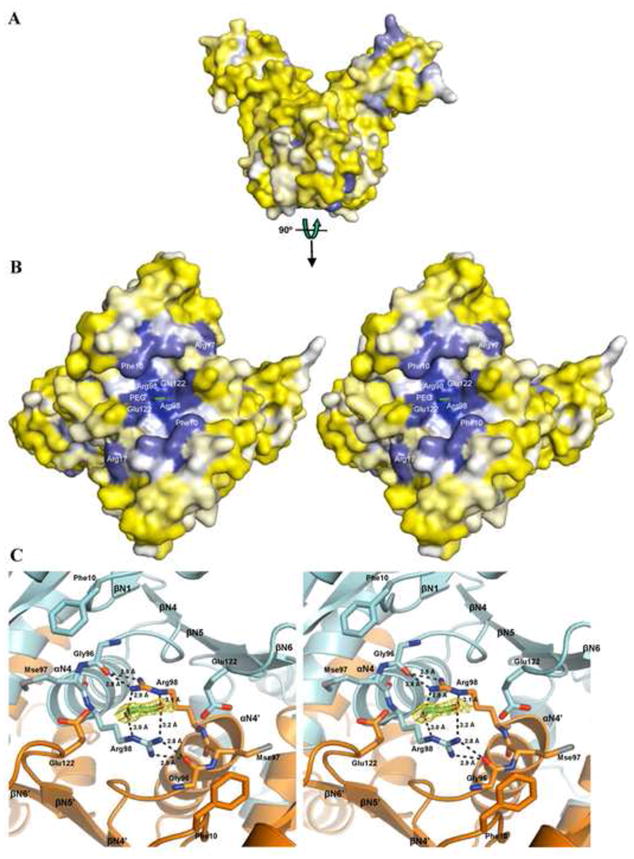
Putative ligand-binding pocket on the Csa3 N-terminal domain. (A) Surface conservation of Csa3. Residues are colored based on their relative conservation among the Csa3 orthologs using Consurf 71. Blue residues are more conserved and yellow residues are less conserved. (B) Stereo image of the conserved pocket on the N-terminal domain. The most highly conserved residues are labeled. Because the pocket spans the dimer interface, there are two copies of each residue. A four-atom length of ordered PEG is shown in stick rendering. The oxygen atoms are colored red and the carbon atoms are colored green. (C) Close-up view of the putative ligand-binding pocket. Chain A is colored cyan and chain B is colored orange. Phe10, Glu122, and the Gly-h-Arg motif (Gly96, selenomethionine (Mse) 97, and Arg98) are shown in stick rendering with nitrogen atoms in blue and oxygen atoms in red. The four atoms of ordered PEG are shown in stick rendering with the carbon atoms colored green and oxygen atoms red. The yellow mesh depicts the 2Fo-Fc omit map for the PEG contoured at 1 σ. The black dotted lines depict inter-protomer hydrogen bonds between Arg98 and Gly96, and hydrogen bonds between Arg98 and the ordered PEG.
The polypeptide then exits the N-terminal domain, and connects to the C-terminal domain via a 12 residue linker. The linker is present as two turns of α-helix, followed by 4 additional residues in an extended conformation. Following the linker, we find a C-terminal domain comprised of residues 145–212 that form a MarR-like, winged helix-turn-helix (wHTH) DNA binding domain (Fig. 1A). Finally, electron density is lacking for the remaining 23 to 25 residues of chains B and A, respectively, and these residues are apparently disordered.
This MarR-like wHTH fold, which is commonly found in bacteria and archaea, is part of the larger superfamily of helix-turn-helix proteins that are frequently involved in DNA recognition 29. In the C-terminal domain of Csa3, helices αC1, αC2 and αC3 form a right-handed three-helix bundle where helices αC2 and αC3 constitute the “helix-turn-helix” motif and helix αC3 (residues 173–786) corresponds to the DNA-recognition helix (Figs. 1 and 3). This tri-helical bundle is followed by a β-hairpin or “wing” comprised of strands βC1 and βC2 and a fourth α-helix (αC4) that helps to differentiate Mar-R like proteins from other members of the wHTH family 29; 30. In chain B, residues 192–196 at tip of the wing are disordered. Consistent with nucleic acid recognition, the overall pI of Csa3 is 9.2, and the pI of the winged HTH domain alone is 9.6, with a basic patch on the putative DNA-binding surface (Suppl. Fig. S3).
Figure 3.

Superpositional docking of DNA to Csa3. (A) The Csa3 dimer is docked to the ohrR promoter. Csa3 is colored as in Fig. 1B except that the recognition helix is colored light teal and the additional putative DNA-interacting regions are colored blue. (B) Close-up view of the chain A wHTH domain docked to DNA. Structural features predicted to interact with the DNA are labeled.
Small-angle X-ray scattering identifies the Csa3 solution state dimer
The behavior of Csa3 on the size exclusion column suggested that Csa3 is present as a dimer in solution. To further investigate the structure of this putative solution-state dimer, we undertook small-angle X-ray scattering (SAXS) studies using the SIBYLS beamline at the Advanced Light Source 31; 32. The resultant small angle scattering was consistent with a globular protein (Fig. 2A) and the Guinier plots were linear, indicating that the samples were monodisperse with a radius of gyration of 28 Å (Suppl. Fig. S2).
Figure 2.

Validation of the biological unit by SAXS. (A) The parabolic nature of the Kratky plot at low values of q indicates that Csa3 is well folded. (B) The solution dimer corresponds to the crystallographic dimer present in a single asymmetric unit. The observed scattering curve of Csa3 (black) was compared with hypothetical scattering curves for the asymmetric unit (red), a potential monomer (light blue), and four alternative dimers (blue, violet, orange and green). Only the hypothetical scattering curve for the dimer corresponding to the crystallographic asymmetric unit agrees well with the observed scattering.
In order to identify the solution state dimer, the experimental scattering curve was compared to the hypothetical scattering curves of a Csa3 monomer and all potential dimers found in the crystal (Fig. 2B). Only the dimer comprised of chains A and B from a single asymmetric unit (Fig. 1B) gave a hypothetical scattering curve in agreement with the experimental scattering curve (χ= 3.223). Comparison of the other potential dimers with the experimental scattering yielded χ values of 11.566, 12.221, 10.424 and 24.739. Small discrepancies between the curves are likely due to the presence of the disordered His6-tag and C-termini in the crystal structure 33 or slight changes in the Csa3 conformation due to crystal packing. Visual examination of the Csa3 crystal structure and an analysis of crystal packing by the PISA server 34 also supported assignment of the A/B pair as the solution state dimer.
Structure of the Csa3 dimer
Dimer formation buries ~2,200 Å2 of solvent accessible surface area per subunit, or 18.5%. The dimer interface includes 24 hydrophobic residues per subunit, complemented by 9 inter-subunit salt-bridges and 21 inter-subunit hydrogen bonds. One principal component is the interaction between the N-terminal domains (NA-NB), which accounts for approximately half the interface. The remainder comes from interactions between the N- and C-terminal domains in symmetry related subunits. Thus, an additional one quarter of the buried surface area per subunit is found on the N-terminal domain, where it interacts with the C-terminal domain of a symmetry related chain (NA-CB), and in a mirror image, another quarter is on the C-terminal domain, where it interacts with the symmetry related N-terminal domain (CA-NB). When analyzed individually by the PISA server 34, both the NA-NB and the NA-CB interfaces give complexation significance scores of 1.000, implying that each interface plays an essential role in complex formation.
An interesting facet of the NA-NB interface is the role of helix αN4. In the traditional dinucleotide binding domain, strands βN5 and βN6 are covered by an additional helix that runs parallel to αN4, and connects strands βN5 and βN6. Because of the reverse turn connecting βN5 and βN6 (discussed above), this covering helix is lost in the Csa3 subunit. However, dimer formation restores this structural element, placing αN4′ of the symmetry related subunit in an equivalent position, where it runs parallel to helix αN4, while covering βN5 and βN6 (Suppl. Fig. S1). Thus, one function of the noncanonical βN5-βN6 reverse turn is the construction of the Csa3 dimer interface.
The role of αC4, the last helix in the wHTH domain is also noteworthy. In Mar-R like proteins this helix frequently serves to orient the wHTH domain with respect to other domains or subunits 29; 30; 35; 36; 37;38. Accordingly, in Csa3 αC4 is found at the domain interface where it contacts the linker helix in the same subunit and αN3′ in the N-terminal domain of the symmetry related subunit (Fig. 1). These contacts appear to anchor the position of the C-terminal wHTH domain with respect to the N-terminal domains of the Csa3 dimer, and thus, also fix the position of the wHTH domains with respect to each other, at least in the crystal.
Potential Csa3/DNA Interactions
A Dali search 39 identified the MexR repressor from the multidrug efflux operon of Pseudomonas aeruginosa as the nearest structural homologue to the Csa3 wHTH domain [PDB ID 1LNW, Z=12.5, 1.1 Å RMSD for 68 equivalent residues, 16% identity 40]. However, the best match to a protein-DNA complex was OhrR, a transcription factor from Bacillus subtillis [PDB ID 1Z9C, Z=10.8, 1.5 Å RMSD for 68 equivalent residues, 18% identity 41]. Like Csa3, MexR and OhrR are dimeric 2-domain proteins whose structures consist of a dimerization domain that is unrelated to the Csa3 N-terminal domain, and a MarR-family winged HTH DNA-binding domain. For OhrR, the dimerization domain includes an allosteric effector site where, under oxidative stress, Cys15 of OhrR is oxidized to Cys-sulphenic acid, resulting in derepression of the ohrA promoter 41.
Superposition of the OhrR/DNA complex on the wHTH domain of Csa3 shows that many features responsible for DNA recognition in OhrR are also present in Csa3 (Fig. 3). These include insertion of the recognition helix (αC3) into the major groove, with opportunities for residues 172–184 to make base specific interactions or contact with the ribose-phosphate backbone. Among residues making potential base-specific contacts, Thr175 is the best conserved among Csa3 orthologs (Fig. 4). Like most wHTH proteins, OhrR uses the wing to interact with the ribose-phosphate backbone. Likewise, the Csa3 wing includes four basic residues, Lys192, Lys194, Arg196 and Lys197 that likely serve a similar function (Fig. 3B and 4). Relative to many wHTH proteins, however, the wing in OhrR is quite long, allowing access to the minor grove for base specific contacts. In contrast, the Csa3 wing, which is 5 residues shorter than the wing in OhrR, is more typical in length and appears too short to access the minor grove (Figs. 3 and 4).
The similarity to OhrR suggests two additional sets of protein/DNA interactions. First, like OhrR, it appears that helix αC2 will contribute to DNA recognition by utilizing the N-terminal end of the helix dipole to interact with the DNA backbone. Second, OhrR utilizes a “helix-helix” motif in DNA recognition 41, which in Csa3 corresponds to the linker helix and helix αC1. Superpositional docking suggests these helices may play a minor role in DNA recognition, utilizing Arg141 in the connecting loop, and Arg145 at the N-terminus of helix C1 to facilitate interactions with the DNA backbone (Fig. 3).
The dimeric structure of Csa3 suggests that the two wHTH domains will simultaneously bind to DNA, recognizing a palindromic or pseudo-palindromic sequence. However, while the OhrR DNA can be docked by superposition to a single chain of Csa3, it could not be simultaneously docked to both chains of Csa3 without significant steric clash. Similarly, we were unable to satisfactorily dock generic B-form DNA to the Csa3 dimer. This suggests the crystallized conformation of Csa3 is unfavorable for DNA binding, or that Csa3 will recognize distorted B-form DNA. Indeed, the analogy to OhrR supports both possibilities. DNA binding by OhrR was observed to cause a 25° rotation of the winged HTH subunit and the OhrR DNA is bent by 10° and unwound by 1.4°.
We thus asked whether the small angle X-ray scattering data might indicate movement of the wHTH domains relative to each other within the Csa3 dimer. The small angle X-ray scattering data were thus reexamined with BILBO-MD, a program that generates a series of models by molecular dynamics and identifies models that agree with the experimental scattering data 42. However, no apparent domain movement was identified. While this does not rule out interdomain movement in DNA recognition, the SAXS data analysis suggests the anchoring N-C interface restricts conformational heterogeneity in Csa3, at least in the absence of DNA or some regulatory ligand.
A putative ligand binding site in the N-terminal domain
As mentioned above, the Gly-X-Gly-X-X-Ala/Gly motif that is a hallmark of the dinucleotide binding domain is absent in Csa3. In addition, superpositional docking with NAD(P)H or FADH2 results in severe steric clashes within a single subunit, and even greater clashes when considering the Csa3 dimer. Thus, it seems Csa3 is unlikely to bind these dinucleotides.
To identify surface features that may be functionally important, we examined the conserved residues in the N-terminal domain of Csa3. Because many members of COG0640 are not CRISPR associated, that is, they are not Csa3 proteins 16, the genome properties tool 43 was used to query the Comprehensive Microbial Resource Database for specific csa3 genes, with the additional stipulation that they were encoded in genomes that also contained cas1, cas2 and at least one CRISPR locus. All the Csa3 orthologs identified are within 9 kb of the nearest CRISPR locus and adjacent to, or part of, putative Cas operons. Ten sequences were retrieved and aligned using 3D-COFFEE 44. The multiple sequence alignments reveal two conserved sequence motifs in the N-terminal domain. The first motif is Thr-h-Gly-Phe-(Asn/Asp)-Glu-X4-Arg, where h represents a hydrophobic residue, and is present in the βN1-αN1 loop (Figs. 4 and 5). The second motif, Leu-X2-Gly-h-Arg, is found within the βN4-αN4 loop. In addition to these two larger motifs, Glu122 in the reverse turn connecting stands βN5 and βN6 is also strongly conserved. Interestingly, the residues in motifs 1 and 2, and the βN5-βN6 loop harboring Glu122 are spatially close to each other on the surface of the N-terminal domain, where they form a single, prominent cleft that spans the dimer interface. Thus, in addition to contributing to the architecture of the subunit interface, the reverse turn connecting βN5 and βN6 also defines two symmetry related walls of this cleft. Two additional walls are formed by the 2 copies of the βN1-αN1 loop, while the βN4-αN4 loop forms the floor. This conserved cleft, which is distal to the winged HTH domain, is approximately 35 Å long (Cα ofGlu67 chain A to Cα of Glu67 chain B), 9 Å deep (CZ of Phe10 to main-chain N of Mse97), and varies between 11 (Cα of Gly132 chain A to Cβ of Val39 chain B) and 18 Å (Cβ of Phe10 chain A to Cβ of Phe10 chain B) in width. The side-chain of Phe10 (motif 1) extends out over the cleft, while the Arg98 side-chains (motif 2) lie on the floor of the cleft (Fig. 5).
In all, this cleft has the hallmarks of a conserved ligand binding site 45. Because the cleft spans the 2-fold symmetric dimer interface, a potential ligand could mirror this symmetry. Alternatively, the cleft might accommodate an asymmetric ligand in either of two equivalent orientations. Interestingly, difference maps identified four-sigma difference electron density within the cleft, centered on the twofold axis. This density most likely represents a component of the crystallization mother liquor or a series of several overlapping partial-occupancy water sites. We have tentatively modeled this density as a fragment of polyethylene glycol (PEG), a component of the crystal mother liquor, at 50% occupancy. Weaker electron density is present in other regions of the pocket, suggestive of additional ordered interactions with PEG, water or other mother liquor components, but has not been modeled.
Structural homology to the Csx1 family of CRISPR associated proteins
The N-terminal domain of Csa3 was used to query the protein databank using Dali 39. It was found to be structurally homologous to a similar domain in members of the COG1517-like and Csx1 families of CRISPR associated proteins. As anticipated by Makarova et al. 17, the N-terminal domain of Csa3 superposes on the N-terminal domain of Vibrio cholera VC1899 with a 2.3 Å RMSD (PDB ID: 1XMX, Z = 14.2, 124 equivalent residues, 12% identity, Midwest Center for Structural Genomics, unpublished). The Csa3 N-terminal domain also superposes on Sso1389 (Csx1) from S. solfataricus with a 3.9 Å RMSD (PDB ID: 2I71, Z =8.8, 128 equivalent residues, 14% identity, Midwest Center for Structural Genomics, unpublished). The VC1899 and Csx1 N-terminal domains (residues 1–122 and 1–186 respectively) were realigned with the Csa3 N-terminal domain plus the linker (residues 1–144) using the SSM68 server yielding an RMSD of 2.05 over 118 residues for the Csa3-VC1899 alignment and an rmsd of 3.18 Å over 102 residues for the Csa3-Csx1 alignment (Suppl. Fig. S4). However, these are distant relationships, which are not detected by a conserved domain database (CDD) search 46, or an HHpred 47 query of the COG database. Although Csa3, VC1899 and Csx1 share a common fold for their N-terminal domains, they are dissimilar with regard to their C-terminal domains and overall domain architecture. Indeed, relative to Csa3 and Csx1, the C-terminal domains of VC1899 are found on the opposite face of the dimeric N-terminal domains (Fig. 6). While specific functions for Csa3, VC1899 and Sso1389 remain to be determined, collectively, their structures suggest functions related to nucleic acid binding and/or nucleic acid metabolism.
Figure 6.
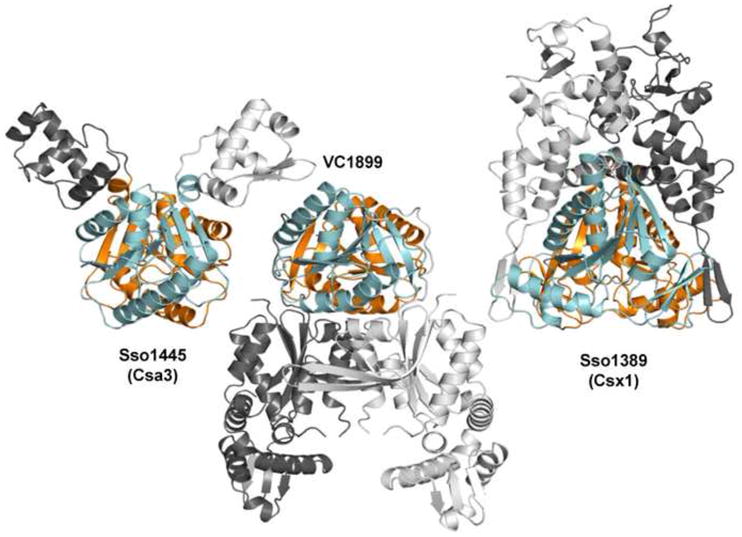
Csa3, VC1899 and Csx1 share similar N-terminal domains but differ in overall domain architecture. Csa3, VC1899, and Sso1389 (Csx1) are shown in equivalent orientations based on structural superposition of their N-terminal domains (cyan and orange). The structurally dissimilar C-terminal domains are shown in shades of grey. Note that the C-terminal domains in VC1899 are found on the opposite side of the Csa3-like N-terminal domain.
As noted above, not all proteins containing the Csa3-like N-terminal domain are expected to be CRISPR associated proteins 16; 17, and this appears to be the case for VC1899. The O1 biovar El Tor strain N16961, from which VC1899 hails, does not harbor a CRISPR. In addition, for Vibrio genomes that do harbor CRISPRs, they are far from any VC1899 orthologs. For these reasons, the relevance of VC1899 to CRISPR/Cas biology is uncertain.
In contrast, Sso1389, which contains a C-terminal domain of unknown function, is a clear member of the Csx1 and COG1517 families of CRISPR associated proteins. Relative to the Csa3 N-terminal domain, the N-terminal domain of Sso1389 contains an extra helix inserted between βN2 and αN2 (Csa3 numbering), and two additional β-strands running along each edge of the central beta sheet, yielding a 10 stranded mixed β-sheet. Two of these strands, connected by a reverse turn, are inserted into the βN1-αN1 loop and run along βN3. The other two strands run along βN6, and are provided by an extension from the C-terminal domain of unknown function (Fig. 4 and 6).
Visual examination of the Sso1389 crystal structure reveals a crystallographic dimer that is mediated in large part by the N-terminal domain of Csx1. The interdomain interactions are strikingly similar to those in the Csa3 N-terminal domain, although there are fewer hydrophobic residues at the interfaces, particularly on helix αN4. This suggests Sso1389 might also form a Csa3-like dimer in solution, though the association may not be as tight as that seen for Csa3. This conclusion is supported by the PISA server, which gives a complexation significance score of 0.639, suggesting this interface may play an important role in dimer formation. VC1899 also appears to form a dimer with an interface similar to Csa3 and Csx1 (Fig. 6). Thus, the dimer interface observed in Csa3 and Csx1 appears to be a common feature of the Csa3 and COG1517 N-terminal domains.
While the conserved sequence motifs in the N-terminal domain of Csa3 are not found in Sso1389/Csx1, this putative Csx1 dimer does contain a large cleft in the N-terminal domain that spans the dimer interface. In addition, a sequence alignment of CRISPR associated Csx1 proteins identifies three conserved sequence motifs in the Csx1 family that line this cleft (Suppl. Figs. S5 and S6), suggesting that this may be a functional ligand binding site in Csx1 that might be involved in formation of a ligand induced dimer. However, the lack of sequence similarity between Csx1 and Csa3 families suggests that if these are functional ligand binding sites, the identity of the ligand may differ. Finally, a fourth motif is found in the Csx1 C-terminal domain of unknown function, suggesting a second possible ligand binding site in this protein family (Suppl. Figs. S5 and S6).
DISCUSSION
While the molecular mechanisms of CRISPR/Cas systems are an area of active research, little is known regarding the transcriptional regulation of CRISPR-Cas in most organisms. Present insight comes from E. coli K12, where transcription of CRISPR-I and the Cas operon encoding Cse1-4, Cas5e and Cas1-2 is suppressed by heat-stable nucleoid-structuring (H-NS) protein48 and derepressed by the transcriptional activator LeuO49. In addition, some elements of the T. thermophilus CRIPR-Cas system appear to be controlled by the cAMP receptor protein 50. However, it’s clear that other regulatory factors remain to be identified in T. thermophils 50, and perhaps in E. coli as well.
The first structure of a Csa3 protein, determined using X-ray crystallography and solution state small-angle X-ray scattering, suggests that Csa3 is a transcriptional regulator with a novel binding site for an allosteric effector in the conserved cleft of the N-terminal domain. The presence of this conserved ligand-binding site in the N-terminal domain of Csa3 suggests the existence of a small-molecule regulator of archaeal CRISPR-Cas systems. In this light, the identity of a controlling allosteric effector for Csa3 is of significant interest. The putative Csa3 ligand-binding site is two-fold symmetric and includes a strictly conserved positively charged residue (Arg98), and a strictly conserved solvent-accessible aromatic sidechain (Phe10). While we cannot rule out the possibility that the conserved cleft binds one or two copies of an asymmetric small-molecule regulator, the structure of Csa3 suggests the ligand may be a two-fold symmetric molecule with negatively charged and hydrophobic or aromatic moieties. Known small-molecule regulators that exhibit these characteristics include a number of dinucleoside polyphosphates 51; 52.
Alternatively, Csa3 might function as part of a larger complex during adaptation or interference, perhaps in the recognition of foreign DNA, or the generation and integration of new spacers. In either case, the conserved cleft in the N-terminal domain might serve to recruit Csa3 into a larger complex. We thus attempted to identify proteins that interact with Csa3, both by incubating immobilized Csa3 with Sulfolobus solfataricus lysate and by purifying tandem-affinity tagged Csa3 expressed in Sulfolobus solfataricus. Neither approach was successful in identifying partners for Csa3 (data not shown). While the negative result may be due to the presence of the affinity tag, a loose interaction, or low levels of the partner protein, it is also consistent with our first hypothesis, that the conserved cleft represents a small-molecule ligand binding site.
Also of interest are the identities of any transcriptional targets that might be controlled by Csa3. One possibility is that Csa3 might regulate expression of the neighboring CRISPRs (CRISPR C and D). However, we were unable to demonstrate recognition of the putative promoters using an electromobility shift assay (data not shown). Whether DNA recognition requires the presence of an unidentified regulatory ligand, or Csa3 recognizes DNA in some other context, remains to be determined. Efforts to identify conserved (pseudo-)palindromic motifs by in silico analysis of the csa3 genomic neighborhood in S. Solfataricus, or in the immediate neighborhoods surrounding csa3 orthologs, were also unsuccessful.
Importantly, the structural findings in regard to Csa3 can be extended to Csx1, a second CRISPR-associated protein, and the non-CRISPR-associated protein, VC1899. Both are proteins of unknown function whose structures were solved in structural genomics efforts. While Csa3 (COG0640), Csx1 (COG1517) and VC1899 (predicted to be similar to COG1517 17) have different overall structures, they share a common N-terminal domain. The fold of this domain is similar to that of a dinucleotide binding domain with the critical modification that strand 6 of the 6-stranded β-sheet (Csa3 numbering) runs antiparallel to strands 1–5. In all three structures this antiparallel sixth strand negates the need for one helical crossover and facilitates formation of a common dimer interface. The presence of a similar dimer interface in all three crystal structures, and in solution for Csa3, suggests that dimerization is one function of this unique fold. Accurate determination of the biological unit from crystal structures can sometimes be a non-trivial issue. Thus, the biological unit may be incorrectly assigned for VC1899 (monomer) and ambiguously assigned for Csx1 (two possible dimers) in their respective protein databank entries (PDB IDs: 1XMX and 2I71 www.rcsb.org). This underscores the inherent synergy between X-ray crystallography, which provides an accurate atomic model of Csa3, and small-angle X-ray scattering, which discriminates the oligomeric state of Csa3 in solution. Together, as evidenced by recent results on such combined methods53, the crystallographic and SAXS data provide significant new insight into the prokaryotic adaptive immune system, as well as an accurate structural foundation for further investigation of the roles of Csa3 and Csx1 in CRISPR-Cas.
MATERIALS AND METHODS
The Genome Properties tool 43 was used to identify gene families associated with the Apern-subtype CRISPR/Cas systems in the S. solfataricus using Hidden Markov Model (HMM)-based searches 43. The S. solfataricus genome encodes two copies of csa3, sso1444 and sso1445. Sso1445 was amplified using a nested Polymerase Chain Reaction (PCR) from purified Sulfolobus solfataricus P2 genomic DNA. The PCR primers introduced a Shine-Dalgarno sequence, an N-terminal His6-tag and attB sites to facilitate ligase free subcloning (Gateway-Invitrogen). The internal forward and reverse primers were CCATGCATCACCATCACCATCACATGAAGTCCTATTTTGTCACCATGGGA and CACTTTGTACAAGAAAGCTGGGTCCTAATAAGGTATGTTAACTTCTTTTCCCTT, while the external forward and reverse primers were GGGGACAAGTTTGTACAAAAAAGCAGGCTTCGAAGGAGATAGAACCATGCATCACCATCAT CAC and GGGGACCACTTTGTACAAGAAAGCTGGGTCCTA, respectively. The PCR product was inserted into pDONR201 using the Gateway site-specific recombination reaction (Invitrogen) 54 and the resulting entry clone was sequence verified (Nevada Genomics). The entry clone was then transferred into pDEST14 (Invitrogen) with a second site-specific recombination reaction, yielding the expression vector pEXP14-6xHis-Sso1445.
Expression and purification
For Sso1445 protein expression, BL21(DE3)-pLysS E.coli (Stratagene) were transformed with pEXP14-6xHis-Sso1445. Typically a single colony was used to inoculate 25 ml of ZYP-0.8G 55 with 100 μg/ml ampicillin and 34 μg/ml chloramphenicol and grown for 6–8 hours. The resulting starter culture was stored at 4 °C for up to two months. Native protein was expressed in 500–750 mL of ZYP-5025 autoinducing media 55 containing 100 μg/ml ampicillin and 34 μg/ml chloramphenicol, inoculated with starter culture at a 1,000-fold dilution, and grown at 37 °C for 16–20 hours. For expression of selenomethionine-incorporated protein, 500 mL of PASM-5025 medium 55 were inoculated with starter culture at a 1,000-fold dilution and grown at 37 °C for 30 hours. Cells were harvested by centrifugation at 5,500 × g (Sorvall Superspeed RC-2) for 10 minutes and the pellets were stored at −80 °C.
Cell pellets were thawed and resuspended at 5 ml/g of cell pellet in lysis buffer (20 mM Tris, 400 mM NaCl, pH 8.0). Phenylmethylsulfonyl fluoride (PMSF, 0.1 mM) was added to the cell suspension and cells were lysed by passage through a French Press (American Instrument Co., Inc., Silver Springs, MD). The lysate was incubated at 65 °C for 20 minutes to denature E. coli proteins, and clarified by centrifugation at 22,000 × g for 30 minutes. The supernatant was then applied to a gravity-flow column containing a 1–3 ml bed volume of Ni-NTA Agarose (Qiagen). The column was washed with 8 column volumes of wash buffer (20 mM Tris, 400 mM NaCl, 10 mM imidizole, pH 8.0) and Sso1445 was eluted in 10 mM Tris (pH 8.0), 50 mM NaCl and 200 mM imidizole. Sso1445 was then applied to a calibrated Superdex 75 (GE Healthcare) column equilibrated with 10 mM Tris (pH 8.0) and 50 mM NaCl. Protein concentrations were determined by Bradford assay 56 using Protein Assay Reagent (Bio-Rad) and BSA as a standard. The purity and molecular weight of Sso1445 were confirmed by SDS-PAGE.
Crystallization and data collection
Purified selenomethionine-incorporated Sso1445 was concentrated to 15 mg/ml with 5,000 Da MWCO Amicon Ultra™ spin concentrators (Millipore). The protein was crystallized using sitting drop vapor diffusion. Drops were setup at 22 °C using 4 μl of selenomethionine-incorporated Sso1445 and 4 μl of well solution consisting of 37.5–42.5% MPD, 5% PEG-8000, and 0.1 M sodium cacodylate at pH 7.0–7.5. Crystals up to 0.20 × 0.05 × 0.05 mm in size were obtained in 5–7 days time. A three-wavelength anomalous diffraction dataset centered on the Se-K edge was collected at the Stanford Synchrotron Radiation Laboratory (SSRL beamline 9–1). Data were indexed, integrated and scaled in space group P212121 using the HKL2000 software package 57. Crystal parameters and data quality are presented in Table 1.
Structure determination and refinement
SOLVE 58 was used to determine the positions of the 16 selenium atom substructure and to calculate initial phases. RESOLVE 59 was used for density modification and initial model-building. The asymmetric unit was found to contain two chains of Sso1445 with a 43% solvent content. Iterative model building with Coot 60 and refinement with REFMAC5 61 against the remote data set (λ = 0.91737 Å) led to the final model. Temperature/Libration/Screw (TLS) parameters 62 were included in the refinement, with each of the two Sso1445 chains divided into 9 TLS groups (Chain A: 1:1-24, 2:25-49, 3:50-68, 4:69-93, 5:94-133, 6:134-143, 7:144-163, 8:164-187, 9:188-212, Chain B: 10:1-24, 11:25-51, 12:52-68, 13:69-93, 14:94-133, 15:134-143, 16:144-160, 17:161-186, 18:187-215). The final R factors for the model were 18.1% and 22.0% (Rwork/Rfree). Molprobity 63 was used for model validation, indicating 99.5% of the residues fall in the most favored regions of the Ramachandran plot and none in disallowed regions. The overall Molprobity score 63 was 1.52, placing Csa3 in the 93rd percentile for overall geometric quality among protein crystal structures of similar resolution (1.55–2.05 Å). Residue numbers in the model are consistent with the native, non-tagged protein sequence. Residues 213–237 of chain A, and residues 192–196 and 216–237 of chain B were not modelled due to lack of interpretable electron density. Coordinates and structure factors have been deposited in the Protein Data Bank under accession code 2WTE. Additional details on model refinement and model quality are presented in Table 2. Three dimensional structural homology searches were carried out using the DALI 39 and SSM 64 servers, and structural figures were generated with PYMOL 65. The embedded 3D PDF was prepared with Adobe Acrobat Pro IX Extended as described previously 66; 67; 68.
Small angle X-ray scattering data collection and processing
SAXS data were collected on purified Csa3 at the SIBYLS beamline of the Advanced Light Source (Lawrence Berkeley National Laboratory) with a Mar165 CCD detector. Scattering data for consecutive 0.5, 0.5, 5 and 0.5 sec exposures were collected with 1.03320 Å wavelength X-ray radiation at room temperature, and processed with the ATSAS 2.1 suite 69. Prior to data collection Csa3 was repurified using a 24 ml Superdex 200 column equilibrated with 10 mM Tris pH 8.0 and 50 mM NaCl. Sample volumes were 15 μl. Data were collected from both the peak and tail fractions from the Superdex 200 column, and on samples concentrated from each fraction. The peak fraction and concentrated peak samples were at 2.5, 4.7, 6.2, and 7.2 mg/ml respectively, while the tail and concentrated tail samples were 1.8, 3.1, 6.3, and 7.6 mg/ml. Protein concentrations were calculated from UV absorbance at 280 nm. Data were also collected on buffer blanks consisting of column fractions prior to the void volume for the unconcentrated samples. Flow-though from the spin concentrators was used for background subtraction from the concentrated fractions. Samples were radiation sensitive as monitored by an increase in slope at low scattering angle. However, data from the first and second 0.5 second exposures overlapped, indicating that radiation damage was minimal, at least in the first 0.5 seconds.
The data were processed with PRIMUS of the ATSAS 2.1 suite 69 for the Guinier and Porod analyses. In the Guinier plot (Fig. S2), the data were linear and no aggregation was detected in the gel filtration fractions or any of the concentrated samples. Data from the most concentrated sample was further processed using the regularization technique in GNOM, in which the pairwise electron distribution function (Patterson function) was obtained from the transform of the scattering function with a Dmax of 115 Å. The molecular mass in Da. was calculated by dividing the Porod volume by 1.67. Ab initio shape predictions using the programs DAMMIN and GASBOR were run on the scattering data, and 10 independent runs were averaged. Although the averaged envelope was similar to the crystallographic dimer, the envelope contour did not follow the sharp contours of the crystal structure (data not shown). While this discrepancy could be due to the presence of the His-tag which can contribute to the scattering 33, it is more likely due to the irregular shape, as control shape predictions run on small angle scattering data calculated from the crystallographic dimer also failed to match the crystal structure.
Supplementary Material
Acknowledgments
This work was supported the National Science Foundation (MCB-0628732 and MCB-0920312). Portions of this research were carried out at the Stanford Synchrotron Radiation Laboratory, whose Structural Molecular Biology Program is supported by the Department of Energy, Office of Biological and Environmental Research, the National Institutes of Health, National Center for Research Resources, Biomedical Technology Program, and the National Institute of General Medical Sciences. The Macromolecular Diffraction Laboratory at Montana State University received support from the Murdock Foundation. The SIBYLS beamline and Tainer group at the Advanced Light Source, Lawrence Berkeley National Laboratory, is supported in part by the DOE program Integrated Diffraction Analysis Technologies (IDAT) and the DOE program Molecular Assemblies Genes and Genomics Integrated Efficiently (MAGGIE) under Contract Number DE-AC02-05CH11231 with the U.S. Department of Energy for Sulfolobus and microbial complexes. NGL was supported by fellowships from the Molecular Biosciences Program and from the NSF IGERT Program in Geobiological Systems (DGE 0654336) at Montana State University.
ABBREVIATIONS USED
- CRISPR
Clustered regularly interspaced short palindromic repeats
- Cas
CRISPR associated genes/proteins
- HTH
helix-turn-helix
- wHTH
winged helix-turn-helix
- PEG
polyethylene glycol
- RNAi
RNA interference
- COG
Cluster of orthologous groups
- CASS
CRISPR-associated System
- RAMP
Repeat associated mysterious protein
- CASCADE
CRISPR-associated complex for antiviral defense
- HMM
Hidden Markov model
- TLS
Temperature/Liberation/Screw
- SAXS
Small-angle X-ray scattering
Footnotes
ACCESSION NUMBERS
Coordinates and structure factors have been deposited in the protein databank with accession number 2WTE.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.van der Oost J, Jore MM, Westra ER, Lundgren M, Brouns SJ. CRISPR-based adaptive and heritable immunity in prokaryotes. Trends Biochem Sci. 2009;34:401–7. doi: 10.1016/j.tibs.2009.05.002. [DOI] [PubMed] [Google Scholar]
- 2.Horvath P, Barrangou R. CRISPR/Cas, the immune system of bacteria and archaea. Science. 2010;327:167–70. doi: 10.1126/science.1179555. [DOI] [PubMed] [Google Scholar]
- 3.Karginov FV, Hannon GJ. The CRISPR system: small RNA-guided defense in Bacteria and Archaea. Mol Cell. 2010;37:7–19. doi: 10.1016/j.molcel.2009.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Banfield JF, Young M. Microbiology. Variety--the splice of life--in microbial communities. Science. 2009;326:1198–9. doi: 10.1126/science.1181501. [DOI] [PubMed] [Google Scholar]
- 5.Sorek R, Kunin V, Hugenholtz P. CRISPR--a widespread system that provides acquired resistance against phages in bacteria and archaea. Nature reviews Microbiology. 2008;6:181–6. doi: 10.1038/nrmicro1793. [DOI] [PubMed] [Google Scholar]
- 6.Grissa I, Vergnaud G, Pourcel C. The CRISPRdb database and tools to display CRISPRs and to generate dictionaries of spacers and repeats. BMC Bioinformatics. 2007;8:172. doi: 10.1186/1471-2105-8-172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Andersson AF, Banfield JF. Virus population dynamics and acquired virus resistance in natural microbial communities. Science. 2008;320:1047–50. doi: 10.1126/science.1157358. [DOI] [PubMed] [Google Scholar]
- 8.Mojica FJ, Diez-Villasenor C, Garcia-Martinez J, Soria E. Intervening sequences of regularly spaced prokaryotic repeats derive from foreign genetic elements. J Mol Evol. 2005;60:174–82. doi: 10.1007/s00239-004-0046-3. [DOI] [PubMed] [Google Scholar]
- 9.Bolotin A, Quinquis B, Sorokin A, Ehrlich SD. Clustered regularly interspaced short palindrome repeats (CRISPRs) have spacers of extrachromosomal origin. Microbiology. 2005;151:2551–61. doi: 10.1099/mic.0.28048-0. [DOI] [PubMed] [Google Scholar]
- 10.Pourcel C, Salvignol G, Vergnaud G. CRISPR elements in Yersinia pestis acquire new repeats by preferential uptake of bacteriophage DNA, and provide additional tools for evolutionary studies. Microbiology. 2005;151:653–63. doi: 10.1099/mic.0.27437-0. [DOI] [PubMed] [Google Scholar]
- 11.Shah SA, Hansen NR, Garrett RA. Distribution of CRISPR spacer matches in viruses and plasmids of crenarchaeal acidothermophiles and implications for their inhibitory mechanism. Biochemical Society transactions. 2009;37:23–8. doi: 10.1042/BST0370023. [DOI] [PubMed] [Google Scholar]
- 12.Tyson GW, Banfield JF. Rapidly evolving CRISPRs implicated in acquired resistance of microorganisms to viruses. Environmental microbiology. 2008;10:200. doi: 10.1111/j.1462-2920.2007.01444.x. [DOI] [PubMed] [Google Scholar]
- 13.Barrangou R, Fremaux C, Deveau H, Richards M, Boyaval P, Moineau S, Romero DA, Horvath P. CRISPR provides acquired resistance against viruses in prokaryotes. Science (New York, NY) 2007;315:1709–12. doi: 10.1126/science.1138140. [DOI] [PubMed] [Google Scholar]
- 14.Brouns SJ, Jore MM, Lundgren M, Westra ER, Slijkhuis RJ, Snijders AP, Dickman MJ, Makarova KS, Koonin EV, van der Oost J. Small CRISPR RNAs guide antiviral defense in prokaryotes. Science (New York, NY) 2008;321:960. doi: 10.1126/science.1159689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jansen R, Embden JD, Gaastra W, Schouls LM. Identification of genes that are associated with DNA repeats in prokaryotes. Mol Microbiol. 2002;43:1565–75. doi: 10.1046/j.1365-2958.2002.02839.x. [DOI] [PubMed] [Google Scholar]
- 16.Haft D, Selengut J, Mongodin E, Nelson K. A Guild of 45 CRISPR-Associated (Cas) Protien Families and Multiple CRISPR/Cas Subtypes Exist in Prokaryotic Genomes. Plos Computational Biology. 2005;1:474–483. doi: 10.1371/journal.pcbi.0010060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Makarova KS, Grishin NV, Shabalina SA, Wolf YI, Koonin EV. A putative RNA-interference-based immune system in prokaryotes: computational analysis of the predicted enzymatic machinery, functional analogies with eukaryotic RNAi, and hypothetical mechanisms of action. Biology direct. 2006;1:7. doi: 10.1186/1745-6150-1-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Carte J, Wang R, Li H, Terns RM, Terns MP. Cas6 is an endoribonuclease that generates guide RNAs for invader defense in prokaryotes. Genes & development. 2008;22:3489–96. doi: 10.1101/gad.1742908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wiedenheft B, Zhou K, Jinek M, Coyle SM, Ma W, Doudna JA. Structural basis for DNase activity of a conserved protein implicated in CRISPR-mediated genome defense. Structure. 2009;17:904–12. doi: 10.1016/j.str.2009.03.019. [DOI] [PubMed] [Google Scholar]
- 20.Beloglazova N, Brown G, Zimmerman MD, Proudfoot M, Makarova KS, Kudritska M, Kochinyan S, Wang S, Chruszcz M, Minor W, Koonin EV, Edwards AM, Savchenko A, Yakunin AF. A novel family of sequence-specific endoribonucleases associated with the Clustered Regularly Interspaced Short Palindromic Repeats. The Journal of biological chemistry. 2008 doi: 10.1074/jbc.M803225200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Marraffini LA, Sontheimer EJ. CRISPR interference limits horizontal gene transfer in staphylococci by targeting DNA. Science (New York, NY) 2008;322:1843–5. doi: 10.1126/science.1165771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hale CR, Zhao P, Olson S, Duff MO, Graveley BR, Wells L, Terns RM, Terns MP. RNA-guided RNA cleavage by a CRISPR RNA-Cas protein complex. Cell. 2009;139:945–56. doi: 10.1016/j.cell.2009.07.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Han D, Krauss G. Characterization of the endonuclease SSO2001 from Sulfolobus solfataricus P2. FEBS Lett. 2009;583:771–6. doi: 10.1016/j.febslet.2009.01.024. [DOI] [PubMed] [Google Scholar]
- 24.Han D, Lehmann K, Krauss G. SSO1450--a CAS1 protein from Sulfolobus solfataricus P2 with high affinity for RNA and DNA. FEBS Lett. 2009;583:1928–32. doi: 10.1016/j.febslet.2009.04.047. [DOI] [PubMed] [Google Scholar]
- 25.Lillestol RK, Shah SA, Brugger K, Redder P, Phan H, Christiansen J, Garrett RA. CRISPR families of the crenarchaeal genus Sulfolobus: bidirectional transcription and dynamic properties. Mol Microbiol. 2009;72:259–72. doi: 10.1111/j.1365-2958.2009.06641.x. [DOI] [PubMed] [Google Scholar]
- 26.Tang TH, Polacek N, Zywicki M, Huber H, Brugger K, Garrett R, Bachellerie JP, Huttenhofer A. Identification of novel non-coding RNAs as potential antisense regulators in the archaeon Sulfolobus solfataricus. Mol Microbiol. 2005;55:469–81. doi: 10.1111/j.1365-2958.2004.04428.x. [DOI] [PubMed] [Google Scholar]
- 27.Peng X, Brugger K, Shen B, Chen L, She Q, Garrett RA. Genus-specific protein binding to the large clusters of DNA repeats (short regularly spaced repeats) present in Sulfolobus genomes. J Bacteriol. 2003;185:2410–7. doi: 10.1128/JB.185.8.2410-2417.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rossmann MG, Liljas A, Branden C, Banaszak LJ, Boyer PD, editors. Evolutionary and structural relationships among dehydrogenases. Vol. XI. The Enzymes. New Your: Academic Press; 1975. [Google Scholar]
- 29.Aravind L, Anantharaman V, Balaji S, Babu MM, Iyer LM. The many faces of the helix-turn-helix domain: transcription regulation and beyond. FEMS Microbiol Rev. 2005;29:231–62. doi: 10.1016/j.femsre.2004.12.008. [DOI] [PubMed] [Google Scholar]
- 30.Alekshun MN, Levy SB, Mealy TR, Seaton BA, Head JF. The crystal structure of MarR, a regulator of multiple antibiotic resistance, at 2.3 A resolution. Nat Struct Biol. 2001;8:710–4. doi: 10.1038/90429. [DOI] [PubMed] [Google Scholar]
- 31.Putnam CD, Hammel M, Hura GL, Tainer JA. X-ray solution scattering (SAXS) combined with crystallography and computation: defining accurate macromolecular structures, conformations and assemblies in solution. Q Rev Biophys. 2007;40:191–285. doi: 10.1017/S0033583507004635. [DOI] [PubMed] [Google Scholar]
- 32.Tsutakawa SE, Hura GL, Frankel KA, Cooper PK, Tainer JA. Structural analysis of flexible proteins in solution by small angle X-ray scattering combined with crystallography. J Struct Biol. 2007;158:214–23. doi: 10.1016/j.jsb.2006.09.008. [DOI] [PubMed] [Google Scholar]
- 33.Hura GL, Menon AL, Hammel M, Rambo RP, Poole FL, 2nd, Tsutakawa SE, Jenney FE, Jr, Classen S, Frankel KA, Hopkins RC, Yang SJ, Scott JW, Dillard BD, Adams MW, Tainer JA. Robust, high-throughput solution structural analyses by small angle X-ray scattering (SAXS) Nat Methods. 2009;6:606–12. doi: 10.1038/nmeth.1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Krissinel E, Henrick K. Inference of macromolecular assemblies from crystalline state. J Mol Biol. 2007;372:774–97. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- 35.Kraft P, Oeckinghaus A, Kummel D, Gauss GH, Gilmore J, Wiedenheft B, Young M, Lawrence CM. Crystal structure of F-93 from Sulfolobus spindle-shaped virus 1, a winged-helix DNA binding protein. J Virol. 2004;78:11544–50. doi: 10.1128/JVI.78.21.11544-11550.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Larson ET, Eilers B, Menon S, Reiter D, Ortmann A, Young MJ, Lawrence CM. A winged-helix protein from Sulfolobus turreted icosahedral virus points toward stabilizing disulfide bonds in the intracellular proteins of a hyperthermophilic virus. Virology. 2007;368:249–61. doi: 10.1016/j.virol.2007.06.040. [DOI] [PubMed] [Google Scholar]
- 37.Chen CS, White A, Love J, Murphy JR, Ringe D. Methyl groups of thymine bases are important for nucleic acid recognition by DtxR. Biochemistry. 2000;39:10397–407. doi: 10.1021/bi0009284. [DOI] [PubMed] [Google Scholar]
- 38.Wu RY, Zhang RG, Zagnitko O, Dementieva I, Maltzev N, Watson JD, Laskowski R, Gornicki P, Joachimiak A. Crystal structure of Enterococcus faecalis SlyA-like transcriptional factor. J Biol Chem. 2003;278:20240–4. doi: 10.1074/jbc.M300292200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Holm L, Sander C. Protein structure comparison by alignment of distance matrices. J Mol Biol. 1993;233:123–38. doi: 10.1006/jmbi.1993.1489. [DOI] [PubMed] [Google Scholar]
- 40.Lim D, Poole K, Strynadka NC. Crystal structure of the MexR repressor of the mexRAB-oprM multidrug efflux operon of Pseudomonas aeruginosa. J Biol Chem. 2002;277:29253–9. doi: 10.1074/jbc.M111381200. [DOI] [PubMed] [Google Scholar]
- 41.Hong M, Fuangthong M, Helmann JD, Brennan RG. Structure of an OhrR-ohrA operator complex reveals the DNA binding mechanism of the MarR family. Mol Cell. 2005;20:131–41. doi: 10.1016/j.molcel.2005.09.013. [DOI] [PubMed] [Google Scholar]
- 42.Pelikan M, Hura GL, Hammel M. Structure and flexibility within proteins as identified through small angle X-ray scattering. Gen Physiol Biophys. 2009;28:174–89. doi: 10.4149/gpb_2009_02_174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Haft DH, Selengut JD, Brinkac LM, Zafar N, White O. Genome Properties: a system for the investigation of prokaryotic genetic content for microbiology, genome annotation and comparative genomics. Bioinformatics. 2005;21:293–306. doi: 10.1093/bioinformatics/bti015. [DOI] [PubMed] [Google Scholar]
- 44.O’Sullivan O, Suhre K, Abergel C, Higgins DG, Notredame C. 3DCoffee: combining protein sequences and structures within multiple sequence alignments. J Mol Biol. 2004;340:385–95. doi: 10.1016/j.jmb.2004.04.058. [DOI] [PubMed] [Google Scholar]
- 45.Liang J, Edelsbrunner H, Woodward C. Anatomy of protein pockets and cavities: measurement of binding site geometry and implications for ligand design. Protein Sci. 1998;7:1884–97. doi: 10.1002/pro.5560070905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Marchler-Bauer A, Anderson JB, Chitsaz F, Derbyshire MK, DeWeese-Scott C, Fong JH, Geer LY, Geer RC, Gonzales NR, Gwadz M, He S, Hurwitz DI, Jackson JD, Ke Z, Lanczycki CJ, Liebert CA, Liu C, Lu F, Lu S, Marchler GH, Mullokandov M, Song JS, Tasneem A, Thanki N, Yamashita RA, Zhang D, Zhang N, Bryant SH. CDD: specific functional annotation with the Conserved Domain Database. Nucleic Acids Res. 2009;37:D205–10. doi: 10.1093/nar/gkn845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Soding J. Protein homology detection by HMM-HMM comparison. Bioinformatics. 2005;21:951–60. doi: 10.1093/bioinformatics/bti125. [DOI] [PubMed] [Google Scholar]
- 48.Pul U, Wurm R, Arslan Z, Geissen R, Hofmann N, Wagner R. Identification and characterization of E. coli CRISPR-cas promoters and their silencing by H-NS. Mol Microbiol. 2010;75:1495–512. doi: 10.1111/j.1365-2958.2010.07073.x. [DOI] [PubMed] [Google Scholar]
- 49.Westra ER, Pul U, Heidrich N, Jore MM, Lundgren M, Stratmann T, Wurm R, Raine A, Mescher M, Van Heereveld L, Mastop M, Wagner EG, Schnetz K, Van Der Oost J, Wagner R, Brouns SJ. H-NS-mediated repression of CRISPR-based immunity in Escherichia coli K12 can be relieved by the transcription activator LeuO. Mol Microbiol. 2010;77:1380–93. doi: 10.1111/j.1365-2958.2010.07315.x. [DOI] [PubMed] [Google Scholar]
- 50.Agari Y, Sakamoto K, Tamakoshi M, Oshima T, Kuramitsu S, Shinkai A. Transcription profile of Thermus thermophilus CRISPR systems after phage infection. J Mol Biol. 2010;395:270–81. doi: 10.1016/j.jmb.2009.10.057. [DOI] [PubMed] [Google Scholar]
- 51.Christen M, Kulasekara HD, Christen B, Kulasekara BR, Hoffman LR, Miller SI. Asymmetrical distribution of the second messenger c-di-GMP upon bacterial cell division. Science. 2010;328:1295–7. doi: 10.1126/science.1188658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Monds RD, Newell PD, Wagner JC, Schwartzman JA, Lu W, Rabinowitz JD, O’Toole GA. Di-adenosine tetraphosphate (Ap4A) metabolism impacts biofilm formation by Pseudomonas fluorescens via modulation of c-di-GMP-dependent pathways. J Bacteriol. 2010;192:3011–23. doi: 10.1128/JB.01571-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rambo RP, Tainer JA. Bridging the solution divide: comprehensive structural analyses of dynamic RNA, DNA, and protein assemblies by small-angle X-ray scattering. Curr Opin Struct Biol. 2010;20:128–37. doi: 10.1016/j.sbi.2009.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kraft P, Kummel D, Oeckinghaus A, Gauss GH, Wiedenheft B, Young M, Lawrence CM. Structure of D-63 from sulfolobus spindle-shaped virus 1: surface properties of the dimeric four-helix bundle suggest an adaptor protein function. J Virol. 2004;78:7438–42. doi: 10.1128/JVI.78.14.7438-7442.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Studier FW. Protein production by auto-induction in high density shaking cultures. Protein expression and purification. 2005;41:207. doi: 10.1016/j.pep.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 56.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–54. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 57.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. In: Carter C, Sweet R, editors. Macromolecular Crystallography, Part A. Academic Press; 1997. pp. 276pp. 307–326. [DOI] [PubMed] [Google Scholar]
- 58.Terwilliger TC, Berendzen J. Automated MAD and MIR structure solution. Acta Crystallogr D Biol Crystallogr. 1999;55:849–61. doi: 10.1107/S0907444999000839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Terwilliger TC. Maximum-likelihood density modification. Acta Crystallogr D Biol Crystallogr. 2000;56:965–72. doi: 10.1107/S0907444900005072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–32. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 61.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Cryst. 1997;D53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 62.Painter J, Merritt EA. Optimal description of a protein structure in terms of multiple groups undergoing TLS motion. Acta Crystallogr D Biol Crystallogr. 2006;62:439–50. doi: 10.1107/S0907444906005270. [DOI] [PubMed] [Google Scholar]
- 63.Lovell SC, Davis IW, Arendall WB, 3rd, de Bakker PI, Word JM, Prisant MG, Richardson JS, Richardson DC. Structure validation by Calpha geometry: phi, psi and Cbeta deviation. Proteins. 2003;50:437–50. doi: 10.1002/prot.10286. [DOI] [PubMed] [Google Scholar]
- 64.Krissinel E, Henrick K. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta crystallographica Section D, Biological crystallography. 2004;60:2256–68. doi: 10.1107/S0907444904026460. [DOI] [PubMed] [Google Scholar]
- 65.DeLano WL. The PyMOL Molecular Graphics System. 2002 http://www.pymol.org.
- 66.Kumar P, Ziegler A, Grahn A, Hee CS, Ziegler A. Leaving the structural ivory tower, assisted by interactive 3D PDF. Trends Biochem Sci. 2010 doi: 10.1016/j.tibs.2010.03.008. In Press. [DOI] [PubMed] [Google Scholar]
- 67.Kumar P, Ziegler A, Ziegler J, Uchanska-Ziegler B, Ziegler A. Grasping molecular structures through publication-integrated 3D models. Trends Biochem Sci. 2008;33:408–12. doi: 10.1016/j.tibs.2008.06.004. [DOI] [PubMed] [Google Scholar]
- 68.3D presentation of structural and image data. J Biol Chem. 2009;284:21101. [PMC free article] [PubMed] [Google Scholar]
- 69.Svergun DI, Petoukhov MV, Koch MH. Determination of domain structure of proteins from X-ray solution scattering. Biophys J. 2001;80:2946–53. doi: 10.1016/S0006-3495(01)76260-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bond CS, Schuttelkopf AW. ALINE: a WYSIWYG protein-sequence alignment editor for publication-quality alignments. Acta Crystallogr D Biol Crystallogr. 2009;65:510–2. doi: 10.1107/S0907444909007835. [DOI] [PubMed] [Google Scholar]
- 71.Landau M, Mayrose I, Rosenberg Y, Glaser F, Martz E, Pupko T, Ben-Tal N. ConSurf 2005: the projection of evolutionary conservation scores of residues on protein structures. Nucleic Acids Res. 2005;33:W299–302. doi: 10.1093/nar/gki370. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.