

Abstract
Background
Influenza infection is associated with myocardial infarction (MI), suggesting that respiratory viral infection may induce biologic pathways that contribute to MI. We tested the hypotheses that 1) a validated blood gene expression signature of respiratory viral infection (viral GES) was associated with MI and 2) respiratory viral exposure changes levels of a validated platelet gene expression signature (platelet GES) of platelet function in response to aspirin that is associated with MI.
Methods
A previously defined viral GES was projected into blood RNA data from 594 patients undergoing elective cardiac catheterization and used to classify patients as having evidence of viral infection or not and tested for association with acute MI using logistic regression. A previously defined platelet GES was projected into blood RNA data from 81 healthy subjects before and after exposure to four respiratory viruses: Respiratory Syncytial Virus (RSV) (n=20), Human Rhinovirus (HRV) (n=20), Influenza A virus subtype H1N1 (H1N1) (n=24), Influenza A Virus subtype H3N2 (H3N2) (n=17). We tested for the change in platelet GES with viral exposure using linear mixed-effects regression and by symptom status.
Results
In the catheterization cohort, 32 patients had evidence of viral infection based upon the viral GES, of which 25% (8/32) had MI versus 12.2% (69/567) among those without evidence of viral infection (OR 2.3; CI [1.03-5.5], p=0.04). In the infection cohorts, only H1N1 exposure increased platelet GES over time (time course p-value = 1e-04).
Conclusions
A viral GES of non-specific, respiratory viral infection was associated with acute MI; 18% of the top 49 genes in the viral GES are involved with hemostasis and/or platelet aggregation. Separately, H1N1 exposure, but not exposure to other respiratory viruses, increased a platelet GES previously shown to be associated with MI. Together, these results highlight specific genes and pathways that link viral infection, platelet activation, and MI especially in the case of H1N1 influenza infection.
Introduction
Influenza viral infections are associated with an increased risk of myocardial infarction (MI), in part, due to an association with platelet activation [1][2][3][4]. The H1N1 strain of influenza has been associated with an acute MI in one case report of a young patient without coronary artery disease [5]. Similar associations may exist for other influenza strains (e.g., H3N2) and other respiratory viruses, but are not as frequently reported [6][7][8]. These associations suggest that viral infection (or exposure) may induce biologic pathways that contribute to MI.
Microarray analysis is used as a genome-wide assessment of gene expression. Compared with individual gene expression, the use of gene-expression “signatures”, or groups of genes with similar expression patterns, can be used to represent the activities of certain biological pathways, some of which have yet to be functionally defined. We have recently identified a peripheral blood gene expression signature (GES) of viral infection that can identify individuals with symptomatic, respiratory viral infection with >95% accuracy and classify viral versus non-viral acute respiratory infection with >93% accuracy [9][10][11][12] (viral GES). In a separate study, we used peripheral blood gene expression profiling to identify and to validate a GES correlative of platelet function in response to aspirin (platelet GES). This signature was primarily made up of platelet genes and was predictive of death or MI in patients with cardiovascular disease [13]. Because the platelet GES was not correlative of platelet function in the absence of aspirin, the platelet GES can be thought of as being reflective of aspirin’s effect on platelet function; lower levels are indicative of a greater aspirin effect on platelets and lower risk for death/MI.
Here we test the hypotheses that 1) biological pathways that change in response to viral infection are associated with MI and 2) viral exposure and/or viral infection are associated with an aspirin-responsive platelet pathway that is associated with MI.
Materials and Methods
Overview
We used existing microarray data from two prior studies: 1) gene expression data from patients at the time of cardiac catheterization (Database of Genotypes and Phenotypes accession numbers: phs000548.v1.p1 and phs000551.v1.p1) [9] to test Hypothesis #1 and 2) serial gene expression from prospective cohorts of healthy volunteers exposed to four different respiratory viruses (Gene Expression Omnibus database accession numbers GSE17156 and GSE52428) [11] to test Hypothesis #2.
CATHGEN Cohort
From 2004–2010, the Catheterization Genetics (CATHGEN, http://cathgen.duhs.duke.edu) biorepository banked whole blood RNA in PAXgene blood tubes from Duke University Medical Center patients at the time of cardiac catheterization [10]. Baseline medical history, medication usage, clinical and procedural characteristics are available through the Duke Databank for Cardiovascular Disease. Two previously defined cohorts of CATHGEN participants with available microarray data were combined for analysis, yielding 594 unique patients [13][14]. The primary outcome was MI in the CATHGEN cohort. All potential MI cases were verified by chart and laboratory data review through chart review by an unblinded internist. MI occurring after the initial catheterization was excluded. The following criteria were used to determine MI: 1) MI was the indication for catheterization, 2) myocardial infarction listed in the discharge summary or admission history and physical, and 3) elevated of CK-MB or troponin above the local upper limit of normal. Eighty potential MI cases were identified and selected for chart review, of which 3 were excluded (not considered MI cases), leaving 77 confirmed MI cases. The remaining 517 patients served as non-MI controls. Because ST-segment elevation myocardial infarction (STEMI) is a more homogeneous clinical condition that is less likely to be confused with non-thrombotic causes of MI (such as demand ischemia or myocarditis), we further classified MI as STEMI if the following criteria were met: 1) meeting criteria for MI and 2) STEMI diagnosis listed in the discharge summary and/or admission history. Of the 77 MI cases (which included non-ST segment elevation myocardial infarction (NSTEMI) and STEMI), 12 (15.6%) were determined to be STEMI based on chart review and the remaining classified as NSTEMI.
Viral Infection Cohorts
Four separate healthy volunteer cohorts were each exposed to a different viral strain, and then monitored for symptoms, and RNA was ascertained at different time points as previously described [9][11] (Fig 1). In general, “healthy” was defined as absence of any significant acute or chronic, uncontrolled medical or psychiatric illness, that in was associated with increased risk of complications of respiratory viral illness (subjects with uncomplicated chronic diagnoses stable and treated for three [3] months, e.g., mild hypertension well-controlled with medication, were enrolled—provided the condition and its therapy are not known to be associated with an immunocompromised state or increased risk of complications of respiratory viral illness). Although certain concomitant medications were allowed, none were on aspirin at the time of viral challenge. Subjects were classified as symptomatic vs. asymptomatic based on the Modified Jackson Score [15][16][17].
Fig 1. Design of viral exposure of infection cohort patients.

Four cohorts of healthy volunteers were exposed to different viruses (H1N1—Influenza A (A/Brisbane/59/2007); H3N2—Influenza A A/Wisconsin/67/2005 (H3N2); HRV—Human rhinovirus; RSV—Respiratory Syncytial Virus). Blood RNA data were collected at baseline and at additional timepoints following viral exposure to assess for changes in the platelet gene expression signature (platelet GES) (S1 Table) Median time post exposure for peak symptom of each respective virus is shown (in hours). The point of treatment with Tamiflu (H1N1 and H3N2) or release from quarantine is shown as end of arrow (in hours). The final numbers of symptomatic (symp) or a symptomatic (asymp) status of each cohort is also shown.
Definition of Viral GES
We have previously used two approaches, factor modeling and factor model projection [13][18] to reduce the dimensionality of microarray data and to generate gene expression signatures. Briefly when applied to a microarray datasets, a factor model generates a series of “factors”, which are sets of coexpressed transcripts representative of a potentially unknown biological pathways. Each sample can be assigned a “factor score”, which represents the aggregate expression of each of the transcripts within a factor. The factor scores can then be used for association testing with phenotypes of interest. In order to estimate factor scores in new datasets, factor model projection is used.
In the infection cohorts, we previously identified a factor that discriminated symptomatic (infected) subjects (HRV, RSV or influenza A) from asymptomatic (uninfected) individuals. However, because the normalization procedures in the infection cohorts differed from that in the CATHGEN cohort, we were unable to directly project the factors from the infection cohorts onto the CATHGEN cohort microarray data. To use the same normalization procedure on both the infection and CATHGEN cohorts, we re-derived the infection cohort factors using the same methodology, and projected this re-derived factor model on the CATHGEN datasets. Using this approach we were able to derive a factor based on the RNA data collected at baseline, 72-77h, and 93-96h after exposure that, we have previously shown, clearly discriminated those with/without viral infection (p-value = 3.9e-17 and FDR q-value = 5.28e-16) [9]. This factor included 957 genes, including 38 of the top 40 genes used for factor loading for the RSV, HRV, Influenza, Panviral cohorts in Zaas et al [9]. Therefore, this re-derived factor recapitulates the original Zaas et al signature with respect to gene membership as well as ability to discriminate between symptomatic and asymptomatic viral infection. This factor is defined as the “viral GES” used in the infection and CATHGEN cohorts for all statistical analyses (Table 1).
Table 1. List of 72 Genes in Platelet GES and top 49 genes in the Viral GES.
| Platelet GES | Viral GES | |||||
|---|---|---|---|---|---|---|
| FSTL1 | CPNE5 | TPM1 | CDC14B | IFI35 | COX4I1 | STAT1 |
| CTTN | CLEC1B | MGLL | C6ORF79 | MAK8 | RPl12 | HNRNPC |
| CTDSPL | SELP | CLU | TTC7B | FAM102A | HNRNPU | CALU |
| TREML1 | IGF2BP3 | THBS1 | ARHGAP6 | Elf1 | EIF3A | CTNNA1 |
| SPARC | SH3BGRL2 | MYL9 | PARVB | CD3D | CLTC | ZYX |
| ITGA2B | PROS1 | PF4 | TUBB1 | IFIH1 | WARS | PSME1 |
| CMTM5 | ALOX12 | GP1BB | GNG11 | ELF4 | SOD1 | RPS15 |
| SLC24A3 | JAM3 | TGFB1I1 | PRSS1 | STAT1 | GLUL | RPL29 |
| MPL | LRRC32 | PCSK6 | PRKAR2B | GAS6 | S100A11 | PRDX6 |
| CLU | ITGB3 | CALD1 | MFAP3L | RPL18 | DNAJB1 | H2AFZ |
| TMEM64 | PPBP | GUCY1B3 | ENDOD1 | RpL19 | SPARC | CNOT1 |
| BEND2 | RAB27B | PDE5A | FRMD3 | RPS11 | PPP2R1A | RNF114 |
| MYLK | ELOVL7 | PBX1 | CLEC4D | RPl10a | NPC2 | PSAP |
| C12ORF39 | RHOBTB1 | MMD | SDPR | C22ORF28 | ACADVL | |
| PCGF5 | HIST1H3H | PF4V1 | HIST1H2AG | HSP90AB1 | RPL13A | |
| RAB4A | HIST1H2BG | LGALSL | ARHGAP18 | SNX3 | RPL10 | |
| FSTL1 | CPNE5 | TPM1 | CDC14B | USP22 | ACTR2 | |
| CTTN | CLEC1B | MGLL | C6ORF79 | PTP4A1 | GNB1 | |
Viral GES “cut-off”
To determine a “cut-off” value of viral GES to classify viral infection in the CATHGEN cohort we generated a receiver operating characteristics (ROC) curve using microarray data from the infection cohorts to discriminate between asymptomatic and symptomatic subjects (Fig 2). The area under the ROC curve in the infection cohorts was 0.91 and the optimal cutoff (calculated as the value that achieved the maximum sum of sensitivity and specificity) was a viral GES of ≥ 0.63. The distribution of the viral GES score was approximately the same in the CATHGEN (median viral GES score = -0.56 interquartile range (IQR) [-0.84, -0.23] vs. asymptomatic time points from viral infection cohorts (median viral GES = -0.51, IQR [-0.64, -0.18]. Therefore, we directly applied the cutoff derived in the viral infection cohorts to the CATHGEN data to classify individuals as having molecular evidence of viral infection (viral GES ≥ 0.63, “positive”) or no molecular evidence of viral infection (viral GES < 0.63, “negative”). Sensitivity analyses were performed by varying the cutoff level in the CATHGEN cohort.
Fig 2. Experimental Design for CATHGEN cohort.
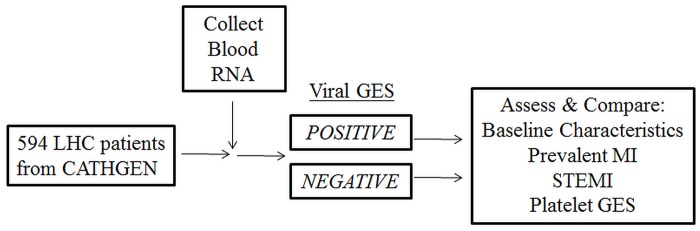
Viral GES is projected on the 594 patients from CATHGEN, then separated into positive or negative viral GES based on a previously defined cutoff (see Methods). Baseline characteristics and MI status were compared between groups. LHC—Left Heart Catheterization
Platelet GES
The platelet GES is a factor containing 62 genes, primarily of platelet origin, that was validated in two independent cohorts as a set of co-expressed genes associated with platelet function in response to aspirin (Table 1)[13]. In the same combined CATHGEN cohort used in the current work, a higher platelet GES was associated with a higher risk of death/MI following cardiac catheterization. We projected the platelet GES into the infection cohorts test for changes following viral exposure.
Statistical Methods
CATHGEN Cohort
Baseline characteristics are presented as medians (25th, 75th percentiles) for continuous variables and frequencies for categorical variables in those positive vs. negative viral GES. The chi-square test was used to identify differences in categorical variables between groups. For continuous data, a student’s t-test was used test for differences in means; a Wilcoxon rank sum test was used for significant deviations from normality. We performed logistic regression in the CATHGEN cohorts to test the association of viral GES with MI or STEMI. Results are reported as odds ratios (OR) with 95% confidence interval (CI) and p-values. Student’s t-test was used to compare mean platelet GES in those with a positive vs. negative viral GES. Pearson correlation was used to test for association between the viral GES and the platelet GES.
Infection cohorts
We used linear mixed-effects regression with random effects for subject to model the platelet GES score over time, modeled as time + time2. To determine the association between platelet GES and viral exposure we compared an intercept only model vs. a model with time. To assess for homogeneity of the effects of viral exposure models with and without interactions between symptom status and time were compared. All model comparisons were performed using an ANOVA likelihood ratio test. The prediction curves included in each plot are predictions made on the fixed effects only.
All analyses were performed using SAS Enterprise Guide 4.3 (SAS Institute Inc, Cary, North Carolina, USA) or R (version 2.8.1). Results were declared significant at a two-sided p-value <0.05. As this was a hypothesis generating study, no adjustments were made for multiple testing.
Institutional Review Board (IRB) Approval
The Influenza challenge (H1N1, H3N2) protocols were approved by the East London and City Research Ethics Committee 1 (London, UK), an independent institutional review board (WIRB: Western Institutional Review Board; Olympia, WA), the IRB of Duke University Medical Center. (Durham, NC), and the SSC-SD IRB (US Department of Defense; Washington, DC) and were conducted in accordance with the Declaration of Helsinki. The other viral challenge studies (HRV, RSV) were approved by the respective site IRB/ethics boards—WIRB (RSV) and the University of Virginia IRB (HRV). All subjects enrolled in viral challenge studies and CATHGEN were provided written informed consent per standard IRB protocol. The current analyses using the databases from these trials were approved by the IRB of Duke University Medical Center.
Results
Hypothesis #1: Viral GES is associated with MI in CATHGEN cohorts
Of the 594 patients in the CATHGEN cohorts, 32 (5.39%) had evidence of viral infection—as determined by the presence of a viral GES score ≥ 0.63. There were no significant differences in baseline, medication, or clinical characteristics between those with positive vs. negative viral GES scores (Table 2). We assessed for the imbalance in aspirin use at the time of cardiac catheterization and found no statistically significant difference in aspirin use between the groups.
Table 2. Baseline characteristics of the CATHGEN cohort.
Continuous variables reported as mean values, categorical variables reported as percentages.
| Positive viral GES (n = 32) | Negative viral GES (n = 562) | p-value | |
|---|---|---|---|
| Characteristic | N or Mean (% or Q1,Q3) | N or Mean (% or Q1,Q3) | |
| Demographics | |||
| Age | 63 (53,71) | 62 (54,71) | 0.59* |
| BMI | 26.4 (25,31) | 28.7 (25,33) | 0.12* |
| Female Gender | 12 (37.5%) | 195 (34.7%) | 0.48 |
| AA Ethnicity | 11(34.3%) | 123 (21.9%) | 0.13 |
| Medical History | |||
| HTN | 27 (84.4%) | 391 (69.6%) | 0.075 |
| CHF | 12 (37.5%) | 169 (30.1%) | 0.41 |
| FH CAD | 10 (31.3%) | 199 (35.4%) | 0.63 |
| DM | 9 (28.1%) | 188 (33.2%) | 0.53 |
| History MI | 12(37.5%) | 173(30.8%) | 0.42 |
| Hyperlipidemia | 17(53.1%) | 348 (61.9%) | 0.32 |
| Smoker | 18 (56.3%) | 278 (49.5%) | 0.46 |
| Medications | |||
| ACEi | 20 (66.7%) | 358 (68.8%) | 0.80 |
| Beta Blocker | 23 (76.7%) | 361 (69.4%) | 0.40 |
| Statin | 18 (60.0%) | 358 (68.8%) | 0.31 |
| Aspirin | 22 (73.3%) | 430 (82.7%) | 0.19 |
| Clopidogrel | 7 (23.3%) | 200 (38.5%) | 0.10 |
| Catheterization data | |||
| EF | 52.4 | 56.14 | 0.47* |
| CAD Index [49] | 32 (0,63) | 32(0,63) | 0.52* |
| # Diseased Vessels | 0.44 | ||
| 0 | 13 (44.8%) | 188 (34.6%) | |
| 1 | 4 (13.8%) | 100 (18.42%) | |
| 2 | 6 (20.7%) | 99 (18.2%) | |
| 3 | 6 (20.7%) | 152 (28.0%) | |
#—Number; AA—African American; ACEi = Angiotensin converting enzyme inhibitor; BMI—body mass index in kg/m^2; CAD—coronary artery disease; HTN—hypertension; CHF—congestive heart failure; EF—Left Ventricular Ejection Fraction; F—female; DM—history of diabetes mellitus; FH CAD—family history of coronary artery disease; MI—myocardial infarction; Angiotensin Converting Enzyme Inhibitor;
* indicates non-parametric, Wilcoxon rank-sum test used to test for differences between groups due to deviations from normality.
To test the hypothesis that prior viral infection was associated with MI, we tested for the association between molecular evidence of viral infection (positive/negative viral GES) and MI in the CATHGEN cohort (Table 3). Among the 32 with a positive viral GES, 8 (25.0%) had MI vs. 69 of 562 (12.3%) in those with a negative viral GES (OR = 2.3 [95% CI = 1.03–5.51], p = 0.04). To determine the extent to which the cut-off value influenced our results, we performed a sensitivity analysis by adjusting the threshold and found similar associations with MI across a range of values. (Table 3) Because STEMI is a more homogenous and readily identified clinical condition, we next limited our analysis to those with STEMI (remaining non-STEMI cases excluded). We found that 3/32 (9.38%) of those with positive viral GES had STEMI compared to 9/562 (1.60%) in those with a negative viral GES (OR = 6.36 [95% CI = 1.63–24.74] p = 0.008).
Table 3. Sensitivity analysis of viral threshold on association with MI.
| Threshold value | Viral GES status* | MI cases | Non-MI controls | Logistic regression p-value for association with MI | Odds Ratio | 95% CI |
|---|---|---|---|---|---|---|
| 0.70 | (+) | 8 | 23 | 0.03 | 2.5 | 1.01–5.6 |
| (-) | 69 | 494 | ||||
| 0.65 | (+) | 8 | 23 | 0.03 | 2.5 | 1.01–5.6 |
| (-) | 69 | 494 | ||||
| 0.63** | (+) | 8 | 24 | 0.04 | 2.3 | 1.03–5.5 |
| (-) | 69 | 493 | ||||
| 0.60 | (+) | 8 | 26 | 0.06 | 2.2 | 0.89–4.8 |
| (-) | 69 | 497 | ||||
| 0.55 | (+) | 11 | 27 | 0.004 | 3.0 | 1.4–6.2 |
| (-) | 66 | 490 |
*Viral GES status was determined by a viral GES greater than or equal to (+) or less than (-) a threshold value (see Methods for more details).
**Predetermined threshold value; GES = Gene expression Signature; MI = myocardial infarction; CI = confidence interval
The CATHGEN cohort has no direct measures of platelet function. However, to further examine the extent to which the association of MI and viral infection was due to a potential platelet-related biological pathway, we compared the platelet GES in those with a positive versus negative viral GES and found no association (p = 0.70). Further, we found no correlation between the platelet GES score and viral GES (p = 0.70).
Hypothesis #2: Platelet GES changes in response to viral exposure in infection cohorts
H1N1
Exposure to HIN1 was associated with an increasing platelet GES over time (time course p-value = 1e-04) (Fig 3). To confirm that the individual genes represented by the platelet GES also changed in response to H1N1 exposure we tested each probe set within the signature and found that, 40 of 50 probe sets were significantly (p < 0.05) up regulated in response to H1N1 viral exposure (S1 Fig). When comparing the symptomatic vs. asymptomatic subsets, we observed that symptomatic subjects differed at baseline with no significant differences across time (Fig 3, symptom by time interaction p-value = 0.1).
Fig 3. Distribution of platelet GES score by time point in the H1N1 exposure cohort.

Individual platelet gene expression signature values (platelet GES, y-axis) are plotted over time (hours, x-axis) following H1N1 viral exposure and by symptom status (symptomatic/dashed thin lines; asymptomatic/solid thin lines). Prediction curves (thick lines) for the symptomatic (dashed) vs. asymptomatic (solid) subsets are plotted based on predictions made from mixed-effects regression model (see Methods). P-values represent the association between platelet GES over time and differences over time between symptomatic vs. asymptomatic subjects.
Remaining Viral Exposures
For the H3N2, RSV, and HRV cohorts we did not observe a significant association of viral exposure with platelet GES (time course p-values = 0.1, 0.2, and 0.09, respectively).
Discussion
Despite the epidemiologic link between respiratory viral infections, including influenza, and myocardial infarction (MI), the biological pathways that underlie these associations are unknown. We hypothesized that patients with molecular evidence of viral infection would be more likely to have an MI in our cohort. We found that those patients with molecular evidence of viral infection were twice as likely (25.0% vs. 12.3%) to present with MI compared to those without. Separately, in an experimental model of viral infection we hypothesized that viral infection would affect a previously described platelet pathway of aspirin response that was associated with MI [13]. We found that H1N1 influenza exposure increased expression of genes in this aspirin response, platelet pathway. Taken, together, these findings suggest that specific biologic pathways that may mechanistically link influenza and MI.
Respiratory viral infections, in general, and influenza infection, specifically H1N1 and H3N2, are associated with an increased risk of MI [2][3][4][6][7][8][19] for up to 2 weeks after infection [20][6]. Remote infection with influenza A and B (i.e., presence of positive IgG antibodies) is also associated with acute MI [21]. These prior observations suggest that there is an initial and continued heightened risk of MI following viral infection, in particular influenza. Although H1N1 and H3N2 have both been associated with MI [7], we only found evidence that H1N1 changed platelet GES. Prior studies demonstrated that different influenza strains can produce differential effects on gene expression and cytokine induction [22][23][24]; therefore, this may not be unexpected. Alternatively, given the small numbers in each cohort we may have been underpowered to detect more subtle changes in gene expression in the non-H1N1 cohorts. Further validation will be required to confirm the H1N1 specific effects we observed in this study.
It is well known that platelets play a critical role in the development of MI. Viral infection, particularly with influenza, results in platelet hyperreactivity and activation in both human and animal models [1][25][26]. The introduction of inactivated influenza vaccine itself has been associated with platelet activation [27]. Influenza has also been shown to influence hemostasis and endothelial activation/dysfunction [28][29]. Of the 62 genes in the platelet GES up to 31 overlapped with platelet or megakaryocyte specific genes [13]. Therefore, our findings that H1N1 influenza exposure alters a platelet GES add to existing data linking influenza and platelet activation by highlighting specific platelet genes/proteins connected to this response.
The viral GES contains a large number of genes from biologically plausible gene networks involved in host viral response [30]. Of the top 49 genes, 9 (18%) are related to platelets or hemostasis (Table 4). Of particular interest is growth arrest-specific 6 (GAS6), which appears to link viral infection and MI. GAS6 plays a key role in platelet aggregation and vascular homeostasis [31][32] by amplifying endothelial cell activation in response to inflammatory stimuli [33]. In our study, we observed that H1N1 viral infection increases GAS6 expression and higher GAS6 expression is associated with a higher risk of MI (Fig 4) which is consistent prior work demonstrating that GAS6 deficient mice are protected from thrombosis [31]. Infection then theoretically increases GAS6 which may increase thrombosis risk versus lower levels of GAS6; this could potentially be a target pathway for future study [34]. Inflammation has long been postulated to be associated with atherosclerosis, and the host inflammatory response to influenza infection may represent an alternative biological pathway by which influenza leads to MI [35]. In a case control series, patients with influenza antibodies indicative of prior influenza infection had a higher risk of MI in addition to increased levels of multiple inflammatory cytokines [36]. Therefore, the viral GES represents multiple inflammatory, coagulation, and platelet pathways that together may contribute to contribute to the development of MI after viral infection.
Table 4. Potential role of viral GES genes in platelet activation, thrombosis or hemostasis.
| Gene | Description |
|---|---|
| GNB1 | Gene involved in platelet activation pathway, thrombin signaling and hemostasis [50][51] |
| CALU | Released by activated platelets, Expressed in atherosclerotic lesions but not normal vasculature [52][53] |
| PRDX6 | Protective versus oxidant injury, Decreased in Influenza [54][55] |
| PPP2R1A | Involved in thromboxane A2 synthesis (thrombin activated platelets) [56] |
| PSAP | Sphingolipid metabolism, sphingolipids are involved with ischemia/reperfusion injury of the heart, found in platelets and plasma, metabolism altered in MI models [57][58] |
| SPARC | Glycoprotein secreted by platelets, maintains cardiac extracellular matrix after MI, down regulated in ACS patients versus controls [59][60] |
| ACTR2 | Gene involved in hemostasis [51] |
| ZYX | Thrombin signaling via interaction with PAR-1 receptor, upregulated in ACS versus non-ACS patients [61][62] |
| GAS6 | Elevated in septicemia and general inflammation, involved in vascular homeostasis and platelet aggregation, deficient mice are protected against thrombosis [31][32][33][34] |
Fig 4. Association of selected viral gene expression signature genes with myocardial infarction.

Genes from the viral gene expression signature (viral GES) were selected based on their role in platelet activation, thrombosis, and hemostasis (Table 4 and Discussion). The association between gene expression and myocardial infarction (MI) is plotted as the standardized odds ratio (y-axis) for each gene (x-axis). Higher odds ratio imply that higher gene expression is associated with higher risk of MI. * indicate genes that are significantly (p-value < 0.05) associated with MI.
There are several, potential clinical implications if our findings are confirmed by others. Because of the strength of evidence surrounding H1N1 and H3N2 vaccination and a reduced risk of MI, vaccination is recommended in patients with coronary or atherosclerotic vascular disease [37][38][39]. Our findings provide additional, complementary data, to support current recommendations for vaccination, although vaccination itself may increase platelet activation [27]. Further, in the H1N1 infection cohort, we observed that those that did vs. did not develop symptoms after H1N1 viral exposure had similar increases in the platelet GES (Fig 3), suggesting that viral exposure even in the absence of infection may be associated with increased cardiovascular risk. Therefore in addition to vaccination programs that prevent viral infection, our findings suggest that prevention of viral exposure (i.e. though infection control programs) may additionally prevent MI in patients at risk for cardiovascular disease. Second, several studies have shown statins reduce morbidity and mortality in patients with influenza infections [40][41]. Statins have acute effects in reducing inflammation and platelet activation—two pathways represented by viral GES genes—and stabilizing atherosclerotic plaque. Therefore, there may be role for statin therapy following influenza infection for CVD prevention. Last, prior work by others links platelet activation with influenza infection [42]. Our findings link an aspirin-responsive pathway to the heightened platelet activation observed with H1N1 exposure. Therefore, a potential therapeutic strategy reduce the burden on influenza-related CVD may be to prescribe aspirin. There is an aversion to starting new aspirin therapy in influenza patients with the classic association of aspirin, influenza and Reye’s Syndrome (which does occur in adults) [43]. Retrospective evidence suggests patients who use aspirin have higher mortality compared to those who do not [44][45]; however, these associations may be confounded by concomitant risk factors associated with aspirin use. Recent studies have proposed mechanisms by which aspirin could have anti-viral and anti-inflammatory effects in influenza infections [46][47]. Therefore the effect of starting aspirin therapy for CVD risk reduction as well as influenza-related outcomes in the peri-influenza period is not known and warrants further study.
Limitations
The primary limitation in the CATHGEN cohort is that we have no laboratory or clinical information of viral exposure or infection. As a consequence, we cannot confirm the molecular evidence of viral infection by viral GES with any laboratory/clinical data. Further, we do not know what type of respiratory virus patients may have been exposed to. This additional information could help explain the lack of correlation between the viral and platelet GES in the CATHGEN cohorts. We found evidence for primarily an H1N1 effect on the platelet GES in the infection cohorts. In contrast the viral GES is a non-specific respiratory viral infection classifier. Therefore, if there were a predominance of non-H1N1 viral exposure in CATHGEN patients then we would not expect to find a correlation between the viral and platelet GES. Second, the CATHGEN cohort study was performed as a cross sectional study; hence, we cannot know if the MI led to an increased viral GES or vice versa. Longitudinal studies could help provide evidence of causality. Third, it is possible that the changes in the viral GES could be, in part, due to 1) vaccination because 57% (n = 28/49) of genes in the viral GES do overlap with genes that change in response to influenza vaccination [48] or 2) concomitant medications used to prevent MI. Because the viral GES is an acute response signature, vaccination would have had to occur within days of catheterization and thus it is unlikely to confound the association we found between viral GES and MI. We assessed for an imbalance in concomitant medication use at the time of cardiac catheterization and found none between those with a positive vs. negative viral GES (Table 3). Last, influenza is associated with peri-/myocarditis, which clinically may mimic MI. Therefore a portion of our MI cases may have represented myocarditis and not thrombosis of a coronary artery. However, when we analyzed the STEMI subgroup, which is a clinical condition more clearly linked to thrombosis and less likely to be confused with pericarditis or myocarditis, we found a similar association with viral GES.
Among the viral exposure studies, the main limitation is that we did not measure traditional measures platelet function such as platelet aggregometry; however, we have previously shown that the platelet GES is a reproducible biomarker for platelet aggregation in response to aspirin [13].
In both the CATHGEN and infection cohorts, the samples sizes were relatively small, therefore independent validation in other cohorts is critical to confirm (or refute) our observations.
Conclusions
A blood GES of viral infection was associated with MI. Separately, H1N1 exposure was associated with changes in a platelet GES that reflects higher levels of platelet function and an associated risk for MI. Together, these results highlight specific genes and pathways that explain the known relationship between viral infection, platelet activation, and MI especially in the case of H1N1 influenza infection.
.
Supporting Information
(PDF)
(PDF)
Acknowledgments
We would like to thank the CATHGEN (Catheterization Genetics) Research Project, MURDOCK (Measurement to Understand the Reclassification of Disease Of Cabarrus / Kannapolis) Study Group, and Z. Elaine Dowdy for their contributions to our project.
Data Availability
The raw and normalized microarray data for the CATHGEN cohort are available through the database of Genotypes and Phenotypes (phs000548.v1.p1 and phs000551.v1.p1) and for the viral infection cohorts on the Gene Expression Omnibus database (GSE17156 and GSE52428).
Funding Statement
Funding sources are the following grants: Defense Advanced Research Projects Agency (DARPA)-N66001-09-C-2082 and National Institutes of Health (NIH) 5RC1-GM091083-03.
References
- 1. Rondina MT, Brewster B, Grissom CK, Zimmerman GA, Kastendieck DH, Harris ES, et al. In vivo platelet activation in critically ill patients with primary 2009 influenza A (H1N1). Chest. 2012. June;141(6):1490–5. 10.1378/chest.11-2860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Guan XR, Li X, Xin XM, Jiang LX, Cui LY, Wang LF, et al. Influenza virus infection and risk of acute myocardial infarction. Inflammation. 2008. August;31(4):266–72. 10.1007/s10753-008-9074-2 [DOI] [PubMed] [Google Scholar]
- 3. Warren-Gash C, Bhaskaran K, Hayward A, Leung GM, Lo SV, Wong CM, et al. Circulating influenza virus, climatic factors, and acute myocardial infarction: a time series study in England and Wales and Hong Kong. J Infect Dis. 2011. June 15;203(12):1710–8. 10.1093/infdis/jir171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lichenstein R, Magder LS, King RE, King JC Jr. The relationship between influenza outbreaks and acute ischemic heart disease in Maryland residents over a 7-year period. J Infect Dis. 2012. September 15;206(6):821–7. 10.1093/infdis/jis435 [DOI] [PubMed] [Google Scholar]
- 5. Arbit B, Gaultier CR, Schwarz ER. H1N1 virus infection associated with acute myocardial infarction in a young patient without coronary artery disease—first reported case. Acta Cardiol. 2011. December;66(6):807–10. [DOI] [PubMed] [Google Scholar]
- 6. Meier CR, Jick SS, Derby LE, Vasilakis C, Jick H. Acute respiratory-tract infections and risk of first-time acute myocardial infarction. Lancet. 1998. May 16;351(9114):1467–71. [DOI] [PubMed] [Google Scholar]
- 7. Wiwanitkit V. Cardiology concern on new H3N2 influenza outbreak. Anadolu Kardiyol Derg. 2013. December;13(8):821. [DOI] [PubMed] [Google Scholar]
- 8. Guan XR, Jiang LX, Ma XH, Wang LF, Quan H, Li HY. Respiratory syncytial virus infection and risk of acute myocardial infarction. Am J Med Sci. 2010. November;340(5):356–9. [DOI] [PubMed] [Google Scholar]
- 9. Zaas AK, Chen M, Varkey J, Veldman T, Hero AO 3rd, Lucas J, et al. Gene expression signatures diagnose influenza and other symptomatic respiratory viral infections in humans. Cell Host Microbe. 2009. September 17;6(3):207–17. 10.1016/j.chom.2009.07.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wang L, Hauser ER, Shah SH, Pericak-Vance MA, Haynes C, Crosslin D, et al. Peakwide mapping on chromosome 3q13 identifies the kalirin gene as a novel candidate gene for coronary artery disease. Am J Hum Genet. 2007. April;80(4):650–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Woods CW, McClain MT, Chen M, Zaas AK, Nicholson BP, Varkey J, et al. A host transcriptional signature for presymptomatic detection of infection in humans exposed to influenza H1N1 or H3N2. PLoS One. 2013;8(1):e52198 10.1371/journal.pone.0052198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zaas A. K., Burke T., Chen M., McClain M., Nicholson B., Veldman T, et al. A Host-Based RT-PCR Gene Expression Signature to Identify Acute Respiratory Viral Infection. Sci. Transl. Med. 5, 203ra126 (2013). 10.1126/scitranslmed.3006280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Voora D, Cyr D, Lucas J, Chi JT, Dungan J, McCaffrey TA, et al. Aspirin Exposure Reveals Novel Genes Associated with Platelet Function and Cardiovascular Events. J Am Coll Cardiol. 2013. October 1;62(14):1267–76. 10.1016/j.jacc.2013.05.073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Shah SH, Granger CB, Hauser ER, Kraus WE, Sun JL, Pieper K, et al. ; MURDOCK Horizon 1 Cardiovascular Disease Investigators. Reclassification of cardiovascular risk using integrated clinical and molecular biosignatures: Design of and rationale for the Measurement to Understand the Reclassification of Disease of Cabarrus and Kannapolis (MURDOCK) Horizon 1 Cardiovascular Disease Study. Am Heart J. 2010. September;160(3):371–379.e2. 10.1016/j.ahj.2010.06.051 [DOI] [PubMed] [Google Scholar]
- 15. Jackson GG, Dowling HF, Spiesman IG, Boand AV. Transmission of the common cold to volunteers under controlled conditions. I. The common cold as a clinical entity. AMA Arch Intern Med 1958;101:267–278. [DOI] [PubMed] [Google Scholar]
- 16. Barrett B, Brown R, Voland R, Maberry R, Turner R. Relations among questionnaire and laboratory measures of rhinovirus infection. Eur Respir J 2006;28:358–363. [DOI] [PubMed] [Google Scholar]
- 17. Turner RB. Ineffectiveness of intranasal zinc gluconate for prevention of experimental rhinovirus colds. Clin Infect Dis 2001;33:1865–1870. [DOI] [PubMed] [Google Scholar]
- 18. Lucas JE, Kung HN, Chi JT. Latent factor analysis to discover pathway-associated putative segmental aneuploidies in human cancers. PLoS Comput Biol. 2010;6:e1000920 10.1371/journal.pcbi.1000920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Erden I, Erden EC, Ozhan H, Basar C. Acute myocarditis mimicking acute myocardial infarction associated with pandemic 2009 (H1N1) influenza A virus. Cardiol J. 2011;18(5):552–5. [PubMed] [Google Scholar]
- 20. Warren-Gash C, Hayward AC, Hemingway H, Denaxas S, Thomas SL, Timmis AD, et al. Influenza infection and risk of acute myocardial infarction in England and Wales: a CALIBER self-controlled case series study. J Infect Dis. 2012. December 1;206(11):1652–9. 10.1093/infdis/jis597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Guan X, Yang W, Sun X, Wang L, Ma B, Li H, et al. Association of influenza virus infection and inflammatory cytokines with acute myocardial infarction. Inflamm Res. 2012. June;61(6):591–8. 10.1007/s00011-012-0449-3 [DOI] [PubMed] [Google Scholar]
- 22. Kuo RL, Zhao C, Malur M, Krug RM. Influenza A virus strains that circulate in humans differ in the ability of their NS1 proteins to block the activation of IRF3 and interferon-β transcription. Virology. 2010. December 20;408(2):146–58. 10.1016/j.virol.2010.09.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Geiler J, Michaelis M, Sithisarn P, Cinatl J Jr. Comparison of pro-inflammatory cytokine expression and cellular signal transduction in human macrophages infected with different influenza A viruses. Med Microbiol Immunol. 2011. February;200(1):53–60. 10.1007/s00430-010-0173-y [DOI] [PubMed] [Google Scholar]
- 24. Mussá T, Ballester M, Silva-Campa E, Baratelli M, Busquets N, Lecours MP, et al. Swine, human or avian influenza viruses differentially activates porcine dendritic cells cytokine profile. Vet Immunol Immunopathol. 2013. July 15;154(1–2):25–35. 10.1016/j.vetimm.2013.04.004 [DOI] [PubMed] [Google Scholar]
- 25. Seki M, Kosai K, Hara A, Imamura Y, Nakamura S, Kurihara S, et al. Expression and DNA microarray analysis of a platelet activating factor-related molecule in severe pneumonia in mice due to influenza virus and bacterial co-infection. Jpn J Infect Dis. 2009. January;62(1):6–10. [PubMed] [Google Scholar]
- 26. Garcia CC, Russo RC, Guabiraba R, Fagundes CT, Polidoro RB, Tavares LP, et al. Platelet-activating factor receptor plays a role in lung injury and death caused by Influenza A in mice. PLoS Pathog. 2010. November 4;6(11):e1001171 10.1371/journal.ppat.1001171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lanza GA, Barone L, Scalone G, Pitocco D, Sgueglia GA, Mollo R, et al. Inflammation-related effects of adjuvant influenza A vaccination on platelet activation and cardiac autonomic function. J Intern Med. 2011. January;269(1):118–25. 10.1111/j.1365-2796.2010.02285.x [DOI] [PubMed] [Google Scholar]
- 28. Armstrong SM, Darwish I, Lee WL. Endothelial activation and dysfunction in the pathogenesis of influenza A virus infection. Virulence. 2013. August 15;4(6):537–42. 10.4161/viru.25779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Berri F, Rimmelzwaan GF, Hanss M, Albina E, Foucault-Grunenwald ML, Lê VB, et al. Plasminogen controls inflammation and pathogenesis of influenza virus infections via fibrinolysis. PLoS Pathog. 2013. March;9(3):e1003229 10.1371/journal.ppat.1003229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Huang Y, Zaas AK, Rao A, Dobigeon N, Woolf PJ, Veldman T, et al. Temporal dynamics of host molecular responses differentiate symptomatic and asymptomatic influenza a infection. PLoS Genet. 2011. August;7(8):e1002234 10.1371/journal.pgen.1002234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Angelillo-Scherrer A, de Frutos P, Aparicio C, Melis E, Savi P, Lupu F, et al. Deficiency or inhibition of Gas6 causes platelet dysfunction and protects mice against thrombosis. Nat Med. 2001. February;7(2):215–21. Hurtado B, de Frutos PG. GAS6 in systemic inflammatory diseases: with and without infection. Crit Care. 2010;14(5):1003. [DOI] [PubMed] [Google Scholar]
- 32. Laurance S, Lemarié CA, Blostein MD. Growth arrest-specific gene 6 (gas6) and vascular hemostasis. Adv Nutr. 2012. March 1;3(2):196–203. 10.3945/an.111.001826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tjwa M, Bellido-Martin L, Lin Y, Lutgens E, Plaisance S, Bono F, et al. Gas6 promotes inflammation by enhancing interactions between endothelial cells, platelets, and leukocytes. Blood. 2008. April 15;111(8):4096–105. [DOI] [PubMed] [Google Scholar]
- 34. Maree AO, Jneid H, Palacios IF, Rosenfield K, MacRae CA, Fitzgerald DJ. Growth arrest specific gene (GAS) 6 modulates platelet thrombus formation and vascular wall homeostasis and represents an attractive drug target. Curr Pharm Des. 2007;13(26):2656–61. [DOI] [PubMed] [Google Scholar]
- 35. Naghavi M, Barlas Z, Siadaty S, Naguib S, Madjid M, Casscells W. Association of influenza vaccination and reduced risk of recurrent myocardial infarction. Circulation. 2000. December 19;102(25):3039–45. [DOI] [PubMed] [Google Scholar]
- 36. Guan X, Yang W, Sun X, Wang L, Ma B, Li H, et al. Association of influenza virus infection and inflammatory cytokines with acute myocardial infarction. Inflamm Res. 2012. June;61(6):591–8. 10.1007/s00011-012-0449-3 [DOI] [PubMed] [Google Scholar]
- 37. Loomba RS, Aggarwal S, Shah PH, Arora RR. Influenza vaccination and cardiovascular morbidity and mortality: analysis of 292,383 patients. J Cardiovasc Pharmacol Ther. 2012. September;17(3):277–83. 10.1177/1074248411429965 [DOI] [PubMed] [Google Scholar]
- 38. Davis MM, Taubert K, Benin AL, Brown DW, Mensah GA, Baddour LM, et al. ; American Heart Association; American College of Cardiology. Influenza vaccination as secondary prevention for cardiovascular disease: a science advisory from the American Heart Association/American College of Cardiology. Circulation. 2006. October 3;114(14):1549–53. [DOI] [PubMed] [Google Scholar]
- 39. Udell JA, Zawi R, Bhatt DL, Keshtkar-Jahromi M, Gaughran F, Phrommintikul A, et al. Association between influenza vaccination and cardiovascular outcomes in high-risk patients: a meta-analysis. JAMA. 2013. October 23;310(16):1711–20. 10.1001/jama.2013.279206 [DOI] [PubMed] [Google Scholar]
- 40. Brett SJ, Myles P, Lim WS, Enstone JE, Bannister B, Semple MG, et al. ; Influenza Clinical Information Network (FLU-CIN). Pre-admission statin use and in-hospital severity of 2009 pandemic influenza A(H1N1) disease. PLoS One. 2011. April 25;6(4):e18120 10.1371/journal.pone.0018120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Vandermeer ML, Thomas AR, Kamimoto L, Reingold A, Gershman K, Meek J, et al. Association between use of statins and mortality among patients hospitalized with laboratory-confirmed influenza virus infections: a multistate study. J Infect Dis. 2012. January 1;205(1):13–9. 10.1093/infdis/jir695 [DOI] [PubMed] [Google Scholar]
- 42. Bogomol'tsev BP, Deviatkin AV. Clinical implications of impaired microcirculation and hemodynamics in acute respiratory viral infections and their pharmacological correction. Klin Med (Mosk). 2003;81(5):9–15. Review. [PubMed] [Google Scholar]
- 43. Link A, Kaplan BT, Böhm M. 21-year-old woman with Reye's syndrome after influenza. Dtsch Med Wochenschr. 2012. September;137(38):1853–6. 10.1055/s-0032-1305311 [DOI] [PubMed] [Google Scholar]
- 44. Eyers S, Weatherall M, Shirtcliffe P, Perrin K, Beasley R. The effect on mortality of antipyretics in the treatment of influenza infection: systematic review and meta-analysis. J R Soc Med. 2010. October;103(10):403–11. Review. 10.1258/jrsm.2010.090441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Starko KM. Salicylates and pandemic influenza mortality, 1918–1919 pharmacology, pathology, and historic evidence. Clin Infect Dis. 2009. November 1;49(9):1405–10. 10.1086/606060 [DOI] [PubMed] [Google Scholar]
- 46. Mazur I, Wurzer WJ, Ehrhardt C, Pleschka S, Puthavathana P, Silberzahn T, et al. Acetylsalicylic acid (ASA) blocks influenza virus propagation via its NF-kappaB-inhibiting activity. Cell Microbiol. 2007. July;9(7):1683–94. [DOI] [PubMed] [Google Scholar]
- 47. Li W, Liu Y, Mukhtar MM, Gong R, Pan Y, Rasool ST, et al. Activation of interleukin-32 pro-inflammatory pathway in response to influenza A virus infection. PLoS One. 2008. April 16;3(4):e1985 10.1371/journal.pone.0001985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Bucasas KL, Franco LM, Shaw CA, Bray MS, Wells JM, Niño D, et al. Early patterns of gene expression correlate with the humoral immune response to influenza vaccination in humans. J Infect Dis. 2011. April 1;203(7):921–9. 10.1093/infdis/jiq156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Mark DB, Nelson CL, Califf RM, Harrell FE Jr., Lee KL, Jones RH, et al. Continuing evolution of therapy for coronary artery disease. Initial results from the era of coronary angioplasty. Circulation. 1994;89:2015–2025. [DOI] [PubMed] [Google Scholar]
- 50. Liberzon A, Subramanian A, Pinchback R, Thorvaldsdóttir H, Tamayo P, Mesirov JP. Molecular signatures database (MSigDB) 3.0. Bioinformatics. 2011. June 15;27(12):1739–40. Gene Sets: REACTOME_PLATELET_ACTIVATION_SIGNALING_A, RAGHAVACHARI_PLATELET_SPECIFIC_GE. 10.1093/bioinformatics/btr260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Liberzon A, Subramanian A, Pinchback R, Thorvaldsdóttir H, Tamayo P, Mesirov JP. Molecular signatures database (MSigDB) 3.0. Bioinformatics. 2011. June 15;27(12):1739–40. Gene Sets: PID_THROMBIN_PAR1_PATHWAY, BIOCARTA_PAR1_PATHWAY, REACTOME_HEMOSTASIS. 10.1093/bioinformatics/btr260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Coppinger JA, Cagney G, Toomey S, Kislinger T, Belton O, McRedmond JP, et al. Characterization of the proteins released from activated platelets leads to localization of novel platelet proteins in human atherosclerotic lesions. Blood. 2004. March 15;103(6):2096–104. [DOI] [PubMed] [Google Scholar]
- 53. Hansen GA, Vorum H, Jacobsen C, Honoré B. Calumenin but not reticulocalbin forms a Ca2+-dependent complex with thrombospondin-1. A potential role in haemostasis and thrombosis. Mol Cell Biochem. 2009. January;320(1–2):25–33. 10.1007/s11010-008-9895-1 [DOI] [PubMed] [Google Scholar]
- 54. Yamada Y, Limmon GV, Zheng D, Li N, Li L, Yin L, et al. Major shifts in the spatio-temporal distribution of lung antioxidant enzymes during influenza pneumonia. PLoS One. 2012;7(2):e31494 10.1371/journal.pone.0031494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Schremmer B, Manevich Y, Feinstein SI, Fisher AB. Peroxiredoxins in the lung with emphasis on peroxiredoxin VI. Sub-cellular Biochemistry. 2007;44:317–344. [DOI] [PubMed] [Google Scholar]
- 56. Moscardó A, Vallés J, Piñón M, Aznar J, Martínez-Sales V, Santos MT. Regulation of cytosolic PlA2 activity by PP1/PP2A serine/threonine phosphatases in human platelets. Platelets. 2006. September;17(6):405–15. [DOI] [PubMed] [Google Scholar]
- 57. Knapp M, Zendzian-Piotrowska M, Błachnio-Zabielska A, Zabielski P, Kurek K, Górski J. Myocardial infarction differentially alters sphingolipid levels in plasma, erythrocytes and platelets of the rat. Basic Res Cardiol. 2012;107(6):294 10.1007/s00395-012-0294-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Sano A, Hineno T, Mizuno T, Kondoh K, Ueno S, Kakimoto Y, et al. Sphingolipid hydrolase activator proteins and their precursors. Biochem Biophys Res Commun. 1989. December 29;165(3):1191–7 [DOI] [PubMed] [Google Scholar]
- 59. Parguiña AF, Grigorian-Shamajian L, Agra RM, Teijeira-Fernández E, Rosa I, Alonso J, et al. Proteins involved in platelet signaling are differentially regulated in acute coronary syndrome: a proteomic study. PLoS One. 2010. October 14;5(10):e13404 10.1371/journal.pone.0013404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Schellings MW, Vanhoutte D, Swinnen M, Cleutjens JP, Debets J, van Leeuwen RE, et al. Absence of SPARC results in increased cardiac rupture and dysfunction after acute myocardial infarction. J Exp Med. 2009. January 16;206(1):113–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Han J, Liu G, Profirovic J, Niu J, Voyno-Yasenetskaya T. Zyxin is involved in thrombin signaling via interaction with PAR-1 receptor. FASEB J. 2009. December;23(12):4193–206. 10.1096/fj.09-131862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Dabek J, Ligus J, Szota J. Oligonucleotide microarray and QRT-PCR study of adhesion protein gene expression in acute coronary syndrome patients. Inflammation. 2010. December;33(6):398–407. 10.1007/s10753-010-9198-z [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(PDF)
(PDF)
Data Availability Statement
The raw and normalized microarray data for the CATHGEN cohort are available through the database of Genotypes and Phenotypes (phs000548.v1.p1 and phs000551.v1.p1) and for the viral infection cohorts on the Gene Expression Omnibus database (GSE17156 and GSE52428).