

Abstract
Spinal cord stimulation (SCS) is a therapy used to treat intractable pain with a putative mechanism of action based on the Gate Control Theory. We hypothesized that sensory projection neuron responses to SCS would follow a single stereotyped response curve as a function of SCS frequency, as predicted by the Gate Control circuit. We recorded the responses of antidromically identified sensory projection neurons in the lumbar spinal cord during 1- to 150-Hz SCS in both healthy rats and neuropathic rats following chronic constriction injury (CCI). The relationship between SCS frequency and projection neuron activity predicted by the Gate Control circuit accounted for a subset of neuronal responses to SCS but could not account for the full range of observed responses. Heterogeneous responses were classifiable into three additional groups and were reproduced using computational models of spinal microcircuits representing other interactions between nociceptive and nonnociceptive sensory inputs. Intrathecal administration of bicuculline, a GABAA receptor antagonist, increased spontaneous and evoked activity in projection neurons, enhanced excitatory responses to SCS, and reduced inhibitory responses to SCS, suggesting that GABAA neurotransmission plays a broad role in regulating projection neuron activity. These in vivo and computational results challenge the Gate Control Theory as the only mechanism underlying SCS and refine our understanding of the effects of SCS on spinal sensory neurons within the framework of contemporary understanding of dorsal horn circuitry.
Keywords: chronic pain, Gate Control Theory, spinal cord stimulation, computational modeling, spinal microcircuits
spinal cord stimulation (SCS) is an effective clinical therapy for neuropathic pain. The putative mechanism underlying neuronal responses to SCS, based on the Gate Control Theory (Melzack and Wall 1965; Shealy et al. 1972), predicts that SCS-mediated inhibition of spinal sensory projection neurons depends on stimulation frequency, and a computational modeling study of SCS predicted that 30- to 80-Hz SCS maximally suppresses wide dynamic range (WDR) dorsal horn neuron activity (Zhang et al. 2014b). As sensory projection neuron activity may represent a proxy for perceived pain (Simone et al. 1991), SCS frequency-dependent inhibition of sensory projection neurons may underlie 50- to 80-Hz SCS as being the “optimal” range for clinical SCS (Oakley and Prager 2002). However, prior studies of neuronal responses to SCS largely neglected stimulation frequency dependence, and although the analgesic outcomes of SCS may be correlated with the activity of projection neurons representing the output of spinal nociceptive circuits (Foreman et al. 1976; Hillman and Wall 1969; Simone et al. 1991; Todd 2010), recent studies did not confirm that recorded neurons were projection neurons (Guan et al. 2010; Yakhnitsa et al. 1999). An incomplete understanding of the frequency-dependent effects of SCS on spinal projection neurons contributes to the limited improvement in the state of knowledge regarding the mechanisms underlying SCS (Zhang et al. 2014a), and this lack of knowledge may underlie the lack of improvement in SCS efficacy since its inception (Taylor et al. 2013).
As well, the specific contribution of SCS-mediated inhibition to the frequency-dependent effects of SCS is poorly understood. GABAergic mechanisms may modulate neuronal responses to SCS, as intrathecal application of bicuculline (BIC), a GABAA receptor antagonist, resulted in SCS-mediated excitation (Duggan and Foong 1985), and intrathecal application of CGP 35348 (CGP), a GABAB receptor antagonist, reversed SCS-mediated pain relief in an animal model of neuropathic pain (Stiller et al. 1996). The progression of neuropathic pain involves the a variety of processes not depicted by the Gate Control Theory, including cell death (Moore et al. 2002), synaptic reorganization (Polgár et al. 2004), and injury-induced changes in glial cell function (Ji et al. 2013). These mechanisms result in a loss of GABAergic inhibition or disinhibition of excitatory responses (Braz et al. 2014) and may contribute to the diminishing efficacy of SCS over time (Kumar et al. 2007). However, antagonism of GABAergic inhibition from both segmental and supraspinal sources may additionally modulate the activity of interneurons (Takazawa and MacDermott 2010a) and increase spontaneous and evoked activity in spinal projection neurons (Lin et al. 1996), behaviors that are beyond what are predicted by the Gate Control circuit. As a result, the relationship between GABAergic inhibition and neuronal responses to SCS remains unclear.
We hypothesized that projection neuron responses to SCS would be dependent on stimulation frequency in a manner consistent with the Gate Control Theory and that blockade of GABAergic receptors would reduce inhibition produced by SCS. We found that spinal sensory projection neuron responses to SCS did depend on stimulation frequency in both healthy animals and neuropathic animals following chronic constriction injury (CCI). Although a subset of neurons exhibited frequency-dependent responses to SCS consistent with the Gate Control Theory (Zhang et al. 2014b), responses were substantially more heterogeneous than anticipated. We postulated that spinal microcircuits (Prescott et al. 2014) may underlie the observed heterogeneity and computational models of the spinal microcircuits that included tonic inhibitory neurons in addition to the Gate Control circuit reproduced the spectrum of experimental responses to SCS. In addition, antagonism of GABAA receptors reduced inhibitory responses produced by SCS but also enhanced excitatory responses and increased spontaneous baseline activity in sensory projection neurons. Our results suggest that spinal circuits in addition to the Gate Control circuit underlie the effects of SCS and refine our understanding of the Gate Control Theory within the context of contemporary knowledge of dorsal horn circuitry. In addition, GABAergic mechanisms modulate the activity of sensory neuron projection neurons and neuronal responses to SCS via mechanisms within and beyond the framework of the Gate Control Theory.
METHODS
Animal preparation.
All animal care and experimental procedures were approved by the Institutional Animal Care and Use Committee of Duke University and were in accordance with the Guide for the Care and Use of Laboratory Animals (8th ed.). Male Sprague-Dawley rats (300–500 g) were initially anesthetized with 3.0% isoflurane (inhaled; Abbott Laboratories) and urethane (1.2 g/kg sc; Sigma-Aldrich). Following the subcutaneous injection of urethane, isoflurane was stopped, and supplemental doses of urethane (0.4 g·kg−1·h−1 ip) were administered if a withdrawal reflex occurred in response to pinching of the hindpaw. A tracheotomy was performed, and rats were connected to a pressure-controlled ventilator (Kent Scientific MouseVent G500) via the tracheal tube for artificial respiration. Oxygen saturation and heart rate were monitored using a pulse oximeter (Kent Scientific MouseVent G500 module) on the hindpaw and maintained within physiological levels (SpO2 > 90%, heart rate < 450). Body temperature was monitored using a rectal thermometer and maintained at 35–37°C using a heating blanket (Gaymar T/Pump). The head was mounted in a stereotaxic frame (Kopf Instruments), and the vertebral column was suspended from vertebral clamps (Kopf Instruments) attached to T9 and L2. The sciatic nerve was exposed, and a 1.5-mm diameter bipolar cuff electrode with an electrode spacing of 1 mm was wrapped around the nerve just proximal to the popliteal fossa. Laminectomies were performed to expose the cervical (C1) and thoracic-lumbar (T10-L2) spinal cord. The dura over these regions was resected, and warm (37°C) saline or mineral oil was applied to the exposed cord. At the completion of the surgery, but before neuron search and recording, rats were paralyzed using gallamine triethiodide (20 mg/h; Sigma-Aldrich) delivered through an intraperitoneal catheter, and a 1.5 mm wide bipolar paddle electrode with 1.0-mm-long platinum electrodes spaced 2.0 mm apart was inserted into the epidural space at T10 to deliver SCS (Fig. 1A).
Fig. 1.

Recording the responses of single lumbar dorsal horn projection neurons to epidural spinal cord stimulation (SCS) in urethane-anesthesized rats. A: rats were implanted with sciatic nerve cuffs for peripheral stimulation and bipolar paddles for SCS. Peripheral stimuli [brush, press, pinch, crush (BPPC)] were applied to the hindpaw ipsilateral to the recording electrode for neuron characterization. A Pt-Ir microelectrode was lowered into the ventral aspect of the contralateral cervical spinal cord for antidromic identification of ascending projections from lumbar dorsal horn neurons. DAQ: data acquisition. B: timeline of experimental recordings: antidromic identification and neuron characterization preceded randomized blocks of SCS.
Cord dorsum potentials.
The amplitude of SCS in each experiment was determined by recording the cord dorsum potential (CDP) evoked by single cathodic-first biphasic SCS pulses (pulse width = 100 μs) ∼1 cm caudal from the caudal SCS contact following paralysis but before neuron search. The CDP was validated by ensuring that the response did not switch polarities during polarity-inverted SCS (Fig. 2A) and observing that the magnitude and timing of the peaks of the CDP varied with proximity to the distal contact of the SCS electrode, i.e., responses propagated (Fig. 2B). The amplitude of SCS was set such that a prominent CDP (>50 μVp-p) was recorded from the surface of the dorsal columns both before and after paralysis; CDPs >50 μVp-p could typically be evoked postparalysis at stimulation amplitudes corresponding to 80–90% of preparalysis visible ipsilateral motor threshold, consistent with amplitudes reported in prior studies of SCS (Guan et al. 2010). However, if the postparalysis CDP amplitude was ≤50 μVp-p at 90% of the preparalysis motor threshold, then the SCS amplitude was increased until the CDP was >50 μVp-p. In some experiments, SCS at amplitudes greater than required to evoke a 50-μVp-p CDP and/or up to and past motor threshold were verified not to alter substantially neuronal responses to SCS (Fig. 2C).
Fig. 2.

Cord dorsum potentials (CDP) were used to determine SCS amplitudes. A: inverting the polarity of biphasic SCS (100-μs pulse width) inverted the artifact (red) but not the CDP signal (blue) recorded 1 cm caudal to the site of SCS and over the approximate neuron search region. B: Moving the electrode 3 and 6 mm caudal to the original recording site (A) resulted in a dispersed CDP with later peak latencies consistent with compound action potential propagation at ∼10 m/s. C: representative examples of peristimulus responses to SCS at 50 Hz applied at different amplitudes corresponding to different evoked CDP amplitudes. Increasing the amplitude of SCS beyond the threshold required to evoke a ∼50 μV CDP (e.g., to motor threshold) did not significantly affect peristimulus responses to SCS.
Neuron recording and identification.
A stimulation microelectrode (platinum-iridium, 3- to 4-μm tip diameter, 0.1-MΩ impedance) was lowered into the cervical spinal cord contralateral to the sciatic incision (Fig. 1A). A recording microelectrode (stainless steel, 1- to 2-μm tip diameter, 1.5-MΩ impedance; Microprobes) was lowered into lumbar spinal cord ipsilateral to the sciatic incision (Fig. 1A). Following electrode placement and paralysis, a search stimulus (maximum: 500 μA, 0.2 ms) was applied to the C1 stimulation electrode at no greater than 5 Hz while the recording electrode was advanced and retracted through the dorsal horn until a neuron was observed to follow the search stimulus, i.e., a candidate projection neuron was found. Neuronal signals were amplified and filtered using an X-Cell amplifier (FHC; gain = 10,000, passband = 500-2,000 Hz), recorded using a Powerlab data acquisition system (ADInstruments), and sorted post hoc using manual principal component-based clustering in Offline Sorter (Plexon).
Once a neuron was isolated, three criteria were used to verify that the neuron was indeed a projection neuron activated by the C1 stimulus: the neuron followed single pulse C1 stimulation with a constant latency that varied less than the pulse width of the search stimulus (< 200 μs; Fig. 3A), the neuron followed triplets of C1 stimulation delivered at ≥200 Hz (Fig. 3B), and collisions between orthodromic and antidromic spikes occurred during spontaneous activity or activity evoked by mechanical stimulation of the neuron's receptive field (Fig. 3C) (Lipski 1981).
Fig. 3.

Characterization of recorded neurons. A–C: all included neurons met 3 criteria required for antidromic identification (Lipski 1981): neurons followed single pulse stimulation of the contralateral ventral cervical spinal cord (A), neurons followed trains of 3 pulses of stimulation at 200–333 Hz (B), and neurons exhibited orthodromic-antidromic collisions during activation of the neuron's peripheral receptive field (C). Axes in B and C are the same as in A. D: neurons fell into established low-threshold (LT), wide dynamic range (WDR), and nociceptive-specific (NS) categories as determined based on firing rates during brush-press-pinch-crush (BPPC) stimulation of peripheral receptive fields. E: neuron locations (right) based on 22 recovered Prussian Blue lesions (example left).
Characterization of peripheral response.
Responses of each neuron to mechanical stimulation of the hindlimb were recorded during successive brushing of the receptive field using a camel hair paintbrush (brush), mild pressure using a loose arterial clip (press), moderate pressure using a tight arterial clamp (pinch), and heavy manual pressure using forceps (crush) with 10-s pauses between stimuli. Neurons were classified as “low threshold” (LT) if they responded to innocuous stimuli and/or were inhibited by more noxious stimuli, “wide dynamic range” (WDR) if they responded to all stimuli in a graded manner, and “nociceptive specific” (NS) if they only responded to “pinch” and/or “crush” stimulation (Fig. 3D). At least 30 s after the brush, press, pinch, crush (BPPC) test, sciatic stimulation at C-fiber threshold (50–100 times the motor threshold with a limit of 8 mA) was applied at 1 Hz for 60 s to verify that sciatic nerve stimulation could alter activity of the recorded neuron.
SCS protocol.
After antidromic identification and sensory characterization of an isolated neuron, we applied SCS at 1, 10, 30, 50, 100, and 150 Hz, both with and without accompanying 1-Hz sciatic stimulation during randomized experimental blocks (Fig. 1B). The order of SCS frequencies was randomized between experiments as well, and, among responders to SCS, neither raw nor normalized neuronal responses to SCS were significantly associated with the trial number, i.e., the time course of the experiment [raw firing rate: P = 0.396, repeated-measures (rm)ANOVA; normalized response: P = 0.580, rmANOVA]. To account for possible potentiation or adaption of a neuron during SCS and sciatic stimulation, each individual application of SCS was preceded and followed by 20-s periods with no stimulation or 1-Hz sciatic stimulation at C-fiber threshold (Fig. 1B) (Guan et al. 2010; Herrero et al. 2000; You et al. 2003), and individual trials were separated by 60 s. The stability of the isolated neuron was verified through antidromic C1 stimulation at the beginning and end of each block.
GABAergic modulation.
In two subsets of rats, we administered intrathecally the GABAA receptor antagonist BIC methiodide (Tocris Bioscience) or the GABAB receptor antagonist CGP (Tocris Bioscience) locally to the recording site immediately following control recordings. Either 10 μl of 0.3 μg/μl (6 nmol total) BIC in 0.9% saline solution (n = 12) or 10 μg/μl of CGP 35348 (443 nmol total) in 0.9% saline solution (n = 8) were applied using a 250-μl microsyringe (model 1725; Hamilton) positioned within 1 mm of the recording site. Dosages were subconvulsive but sufficient to produce behaviors associated with hyperalgesia and allodynia in awake rats (Hao et al. 1994; Sivilotti and Woolf 1994). Ten minutes elapsed before BPPC characterization and sciatic stimulation tests were repeated to assess changes in the response profile of the neuron due to GABAergic antagonism, and the SCS experiment was then repeated. In a few cases, both blockers were administered together, after the effects of one blocker in isolation were recorded (BIC + CGP, n = 1; CGP + BIC, n = 2).
Chronic constriction injury.
Neuropathic pain was produced in eight animals using CCI of the sciatic nerve (Bennett and Xie 1988), and the effects of CCI were validated with quantitative behavioral measurements. Briefly, adult Sprague-Dawley rats (male, 250 ± 20 g) were anesthetized with isoflurane, and the site of the incision was disinfected with povidone-iodine and 70% ethanol. The left sciatic nerve was exposed, three ligatures (4-0 Prolene) were placed around the nerve proximal to the popliteal fossa with a distance of 1 mm between each ligature, and the ligatures were loosely tied until a short flick of the ipsilateral hindlimb was observed. For 7–10 days following the surgical procedure, rats were doubly housed under a 12-h light-dark cycle and provided with food and water ad libitum; no supplemental antibiotics were necessary or provided. At the end of this period, animals were subjected to single unit recordings under urethane anesthesia during SCS in a terminal experiment as described above.
Behavioral analysis.
Animals were habituated to the testing environment daily for at least 2 days before baseline testing. Before being tested for mechanical sensitivity, animals were put in boxes on an elevated metal mesh floor and allowed to habituate for 30 min. A series of von Frey hairs with logarithmically increasing stiffness (0.4–26 g; Stoelting) was then presented to the plantar surface of the ipsilateral hindpaws of each animal several times for 3–5 s per hair. Once paw withdrawal behavior was observed, six von Frey hair tests centered around the minimum hair stiffness that resulted in 50% paw-withdrawal incidence were recorded per animal, and the final paw-withdrawal threshold was determined from the composite response using Dixon's up-down method (Chaplan et al. 1994; Dixon 1980).
Single unit activity analysis.
To determine if neurons were responsive to SCS, we constructed poststimulus time histograms (PSTHs) of each neuron's activity and accounted for stimulus artifacts by applying a 0.5- to 2-ms width blanking mask corresponding to the duration of the averaged stimulus artifact; PSTH bin widths were set according to the number of stimulation trials per SCS On period (i.e., by SCS frequency) (Shimazaki and Shinomoto 2007). For comparison of activity between SCS On and SCS Off periods, stimulus times and artifact blanking periods from the On periods were superimposed onto the Off periods, and PSTHs of activity in the Off periods were generated using this virtual stimulus train. We then normalized each neuron's PSTH by expressing the number of spikes per bin in the SCS On condition as Z-scores based on the mean and SD of the neuron's virtual PSTH during a time-matched pre-SCS interval (Montgomery 2006). A neuron's response to SCS at a specific frequency was defined as significant if the Z-scores from at least three contiguous PSTH bins met or exceeded a magnitude of 1.96, with Z = 1.96 corresponding to an individual response magnitude whose likelihood of occurrence was <5% by random chance. Some neurons, in particular nociceptive-specific neurons, exhibited relatively low firing rates (Craig et al. 2001; Kawamata et al. 2005) and these neurons' PSTHs during both the SCS Off and SCS On conditions were occasionally insufficient to calculate Z-scores (<4 spikes/bin) (Dayan 2001). In those cases, if the total number of spikes exceeded 30 in both the SCS On and SCS Off intervals, the Kolmogorov-Smirnoff (K-S) test (cutoff P < 0.05) was used to determine if SCS altered the distribution of poststimulus responses between the SCS Off and SCS On cases (Xu et al. 2008). Neurons were classified as being nonresponsive if they did not meet both the Z-score and the K-S test criteria or if they exhibited insufficient activity [<30 spikes, <4 spikes/bin, <1.5 spikes/s (Dayan 2001)] in pre-SCS and SCS On conditions to conduct statistical analyses. Unless otherwise noted, only responders were included in our data analysis.
Mean firing rates were calculated by dividing the total number of spikes by 20 s minus the total amount of time included in artifact blanking periods. Because baseline firing rates fluctuated between neurons, and to a lesser extent within neurons, we quantified the effects of SCS on neuron activity using the change in firing rates between 20 s on periods and the 20-s off period immediately before SCS. For clustering analysis, relationships between firing rate and SCS frequency were normalized such that the magnitude of the largest firing rate change between off and on conditions in a neuron corresponded to +1 (excitatory) or −1 (inhibitory); this scaling prevented large responses from dominating the field in clustering analysis (Lemay and Grill 2004). A principal component analysis was performed on the entire field of normalized responses, and the resulting principal component loadings were used to classify neurons with k-means clustering and hierarchal clustering algorithms in MATLAB (MathWorks, Natick, MA).
Histology.
At the end of each experiment, an electrolytic lesion (15 μA, 20 s) was made at the recording site in the lumbar spinal cord. Rats were euthanized with an intraperitoneal injection of 0.5 ml Euthasol (Virbac) and perfused with 200 ml 0.9% NaCl followed by 100 ml of 10% formalin + 1% ferrocyanide solution to produce a Prussian Blue reaction at the site of the electrolytic lesion. The lumbar spinal cord was removed, fixed in 10% formalin + 1% ferrocyanide for several days, and then blocked and sectioned into 50-μm sections using a microtome. The tissue was counterstained with nuclear red dye (NovaUltra; IHC World) to facilitate visualization of the electrolytic lesion.
Computational modeling of spinal microcircuits.
We implemented computational network models of dorsal horn circuits representing each of four observed SCS response classes by connecting biophysically based compartmental models of dorsal horn neurons via representations of excitatory and inhibitory synapses. Membrane dynamics of the individual neurons, synaptic properties, and the strengths and types of synaptic connections were based largely on a prior model of the dorsal horn network (Zhang et al. 2014b) and were tuned to match experimental observations.
Networks architectures were based on spinal microcircuits hypothesized to underlie inhibitory (“opponency”), excitatory (“coloring”), and mixed (“mixing”) responses (Prescott and Ratté 2012) to increasing frequencies of SCS. The connections of 15 Aδ- and C-fiber afferents onto the projection neuron and the C-fiber connections onto an intervening interneuron were unaltered from the original model (Zhang et al. 2014b). However, to account for the full range of interactions between SCS and peripherally evoked activity, a template motif based on the Gate Control circuit was developed that included monosynaptic excitatory connections between 15 Aβ afferents and the projection neuron (Duan et al. 2014; Foreman et al. 1976) as well as a disynaptic inhibitory connection between an inhibitory interneuron that received inputs from 15 Aβ afferents and the projection neuron (Duan et al. 2014; Zhang et al. 2014b). In addition, we included GABAergic and glycinergic inputs onto the inhibitory interneuron (gGABAa = 19.9 nS, gGlycine = 5.32 nS) and projection neuron (gGABAa = 15.0 nS; gGlycine = 4.0 nS) from spontaneously active tonic neurons whose activity was decoupled from SCS, as tonic inhibition from segmental or supraspinal sources has been hypothesized as a regulatory influence on excitatory or inhibitory inputs to projection neurons (Peng et al. 1996; Takazawa and MacDermott 2010a). We applied Ornstein-Uhlenbeck noisy currents (Lánský and Sacerdote 2001) with a mean amplitude of 15 pA, a variance of 5 pA, and a relaxation time of 100 ms, to induce firing rates in the inhibitory neurons (40–50 spikes/s) that represented the combined effect of inhibition from segmental (Takazawa and MacDermott 2010a) and supraspinal inhibitory inputs (Carlson et al. 2007; Li et al. 1998).
Conversion of the template circuit to the mixing, opponency, and coloring microcircuits was accomplished by weakening the excitatory or inhibitory paths of the template circuit and/or modulation of the degree of tonic inhibition of the inhibitory interneuron and projection neuron. The mixing microcircuit was generated by setting total excitatory and inhibitory synaptic weights from SCS to 75% of their values in the original model (Zhang et al. 2014b); tonic inhibition of the projection neuron and the inhibitory interneuron was excluded to be consistent with the original model and because excitatory and inhibitory influences on the projection neuron were balanced in this circuit. To generate coloring (excitatory) and opponency (inhibitory) circuits, the synaptic weight onto the projection neuron of the opposing connection was weakened to 20% of its strength in the mixing circuit, while the synaptic weight of the dominant connection was kept at its default strength. To modulate the dominant pathway, external tonic inhibition onto the projection neuron (coloring) or the inhibitory interneuron (opponency) via GABAergic and glycinergic synapses was preserved from the template circuit while tonic inhibition onto the weaker pathway was removed.
To model the adaptation to SCS observed in some projection neurons, the model projection neuron was replaced with a previously published model of an adapting neuron (Melnick et al. 2004). In addition to already existing adaptation mechanisms in the model neuron, a first order, noninactivating slow potassium current (IKm) was added to the soma and dendrites to account for transient adaptation (McCormick et al. 1993; Prescott and De Koninck 2005). The activating gating variable for IKm (n) and its respective rate constants (αn, βn) described as functions of temperature (T) in degrees Celsius, membrane potential Vm, and the potassium reversal potential Ek of −84 mV, are shown in Eqs. 1–3.
| (1) |
| (2) |
| (3) |
Additionally, to account for slow adaptation (Benda and Herz 2003; Powers et al. 1999), a slow inactivating gate, s, was introduced to the Hodgkin-Huxley sodium conductance in the adapting firing neuron with rate constants (αs, βs) defined in Eqs. 4–6; other variables governing the sodium current (m, h) were modeled as previously described (Melnick et al. 2004).
| (4) |
| (5) |
| (6) |
Tonic inhibition was also implemented onto the adapting projection neuron, but the synaptic conductance of tonic inhibition onto the adapting projection neuron was reduced to 50% of the strength of the connection between the tonic inhibition and projection neuron in the coloring microcircuit.
Simulations and analysis.
All simulations of microcircuit responses to SCS were conducted with background activation of the model to generate baseline activity required to visualize inhibitory responses. Sciatic stimulation, as delivered in the experiments, was simulated by applying a 1-Hz train to the local Aβ-, Aδ-, and C-fiber afferent inputs with arrival latencies determined according to published conduction velocities (Aβ: 14–30 m/s; Aδ: 2.2–8 m/s; C: 0.6–1.5 m/s) and an assumed conduction distance of 100 mm (Harper and Lawson 1985). Additionally, spike trains with individual interspike intervals drawn from a homogeneous Poisson process (mean = 0.75 spikes/s) were delivered through C fibers to simulate stochastic activity after surgical sciatic nerve exposure (Michaelis et al. 1995). SCS was simulated by applying a constant frequency train through separate inputs representing dorsal column collaterals of both the local and surround Aβ fibers. Simulations were conducted in the NEURON v. 7.2 simulation environment (Hines and Carnevale 1997) using a time step of 0.0125 ms and second order implicit Crank-Nicholson integration.
The output of the computational model before and during 20 s of simulated SCS was quantified and normalized in the same manner as the experimental data. PSTHs were generated from model and responder neuron spike trains for 10-, 50-, and 150-Hz SCS using a 10-Hz virtual SCS train; generation of smoothed PSTHs using a “virtual” 10-Hz SCS train enabled visualization of the interactions between transient and longer duration features at higher frequencies of SCS. Model and experimental PSTHs were smoothed by convolving raw peristimulus times with a bin-width optimized Gaussian kernel (σ = 0.870 ms) in Chronux (Bokil et al. 2010) and then normalized by setting the maximum increase or decrease vs. the corresponding virtual PSTH during the SCS Off periods across SCS frequencies for a given neuron to 1 and scaling all responses accordingly. This scaling prevented large responses from dominating analysis of groups of simulations and allowed analysis of small but significant responses.
RESULTS
Population of recorded neurons.
We recorded the responses of 50 antidromically identified sensory neurons to at least one repetition of all frequencies of epidural SCS. Eleven of these neurons were classified as LT neurons, 18 neurons were classified as WDR neurons, and 19 neurons were classified as NS neurons (Fig. 3D). The response types of two neurons could not be determined, but they exhibited strong A-fiber and C-fiber-evoked responses during sciatic stimulation. The widespread locations of 22 neurons, confirmed by electrolytic lesions, were consistent with previous reports of locations of sensory projection neurons (Burstein et al. 1990) (Fig. 3E).
Analysis of PSTHs using Z-scores and K-S testing identified 33 neurons that were responsive to SCS and 17 neurons as nonresponsive to SCS at SCS amplitudes sufficient to evoke a >50-μV CDP. (Fig. 4). Responders and nonresponders were located throughout the dorsal horn; there was no significant difference in the dorsal-ventral depths of the responders vs. nonresponders for which location data were recovered: responder n = 24, median = 565 μm, nonresponder n = 13, median = 750 μm, P = 0.12, Kruskal-Wallis, and the distribution of responder and nonresponder depths was also not different (P = 0.31, K-S test). The amplitude of SCS applied to the population of responders (125 ± 9 μA, means ± SE, n = 33) vs. nonresponders (121 ± 9 μA, means ± SE, n = 17) was also not different (P = 0.75, Welch's t-test). Although the proportion of responders that were LT neurons (10/33) was higher than the proportion of nonresponders that were LT neurons (1/17), and the proportion of responders that were NS neurons (10/33) was lower than the proportion of nonresponders that were NS neurons (9/17), the overall distribution of LT, WDR, and NS neurons was not different between responders and nonresponders (P = 0.12, Pearson's χ2). Taken together, these data suggest that responsive and nonresponsive sensory spinal projection neurons are intermingled throughout the superficial and deep dorsal horn and that these neurons do not receive a homogeneous set of inputs from Aβ/dorsal column fibers.
Fig. 4.

Analysis of poststimulus time histograms (PSTHs) was used to differentiate responders to SCS from nonresponders. A: representative peristimulus responses during SCS Off (left) and SCS On (middle) during 10-, 30-, 50-, 100-, and 150-Hz SCS. Normalization of the SCS On response to the SCS Off response using Z-scores (right) was used to determine if a response to SCS was significant. B, left: responder/nonresponder, sorted by incidences of significant peristimulus responses, with individual significant responses highlighted in gray. A neuron was classified as a responder if the Z-score result from 2 or more contiguous frequencies in either the SCS only or SCS + sciatic condition was significant. Neurons for which Z-score analysis could not be conducted due to low firing rates but for which Kolmogorov-Smirnoff tests yielded significant responses were included. B, right: map of neurons that were included in firing rate (Fig. 5), principal component, and clustering analyses (Fig. 6). Only individual frequency-response relationships that met the contiguous frequency criterion were included in principal component analysis (PCA) and clustering. C: distribution of LT, WDR, and NS neurons among nonresponders (top) and responders (bottom).
Responses to SCS are dependent on stimulation frequency and are heterogeneous.
Neither the mean firing rate during SCS nor the mean change in firing rates between SCS On and SCS Off were significantly different across the frequencies of SCS tested (P = 0.43, rmANOVA; Fig. 5A). Mean firing rates and mean firing rate differences were also not significantly different across SCS frequencies when 1-Hz sciatic stimulation was applied concurrently with SCS (P = 0.92, rmANOVA; Fig. 5C). Responses to SCS were heterogeneous both across neurons and within individual neurons: neurons could be inhibited or excited at any of the applied frequencies of SCS, and some frequency-response relationships were nonmonotonic with increasing SCS frequency. Inhibitory and excitatory responses were present during both SCS only and SCS + sciatic conditions, and the averaging of excitation and inhibition across neurons resulted in the lack of significant change in mean firing rates across SCS frequencies (Fig. 5, B and D). However, individual neurons exhibited stereotyped responses to SCS: some neurons showed progressive inhibition with increasing SCS frequency, some neurons showed progressive excitation with increasing SCS frequency, and some neurons exhibited a nonmonotonic relationship between changes in firing rate and SCS frequency consistent with prior computational modeling predictions (Fig. 6A) (Zhang et al. 2014b). The heterogeneous but stereotyped responses suggested that quantitative classification would provide insight into the origins of the responses to SCS.
Fig. 5.

Effect of SCS frequency on firing rate of projection neurons. A and C: median and 25-75th percentiles (boxes) of responder neuron firing rates between SCS Off and SCS On conditions when only SCS was applied (A) or when SCS was applied with sciatic stimulation (C). Outliers beyond 1.5 interquartile ranges (whiskers) are denoted with the “+” symbol. B and D: changes in firing rates of individual responder neurons between SCS On and SCS Off conditions of individual neurons when only SCS was applied (B) or when SCS was applied with sciatic stimulation (D). Each column represents the continuum of responses across neurons for a given SCS frequency. All columns in the color maps are sorted according to response during SCS independently of the neuron from which the response originated.
Fig. 6.

Classification of individual neuron responses to different frequencies of SCS. A: representative examples of increasing (red), nonmonotonic (violet), and decreasing (blue) responses relative to “Off” baselines (dotted line) with increasing SCS frequency. Although the magnitudes of deviations vs. “Off” baselines were heterogeneous, all examples exhibited significant on vs. off PSTH responses to SCS, and the shapes of the responses were stereotyped, motivating classification of normalized responses. B: principal component based k-means clustering of normalized responses. C: aggregate normalized responses (means ± SE of normalized responses shown) formed by averaging individual responses from each cluster in B. D: hierarchal analysis using squared Euclidean distance between points corresponding to the 1st 2 PCA loadings as the proximity measure. *Frequency-response relationship of the neuron (numbering same as in Fig. 4B) during the SCS + Sciatic condition. Discrepancies between k-means and hierarchal clustering are identified in B and D with highlighting using the color corresponding to classification by the other method. E: distribution of response type classification transitions between SCS only and SCS + sciatic stimulation conditions. Groups of slices represent response classifications during SCS only, and individual slices represent response classifications during SCS + sciatic. The number of neurons exhibiting a particular transition is denoted by the white numeral in each inner slice. Neurons that were nonresponsive in either the SCS only or SCS + sciatic condition were grouped according to their response classification in the other condition. F: distribution of response type classifications by physiological response class (LT, WDR, and NS) in the SCS only (left) and SCS + sciatic (right) conditions. Only the 31 neurons that could be classified as LT, WDR, or NS were included. The number of neurons exhibiting a particular response is denoted by the white numeral in each inner slice. Neurons that were nonresponsive in either the SCS only or SCS + sciatic condition were grouped according to their response classification in the other condition.
We quantitatively classified neurons by clustering the results of a principal component analysis on the normalized SCS frequency-response curves, with the normalized response to SCS as the dependent variable and SCS frequency as the independent variable (Fig. 6B). We pooled normalized SCS only and SCS + sciatic response curves together because changes in baseline activity due to sciatic stimulation occasionally unmasked responses that were not evident in the SCS only case. k-Means clustering of the resulting first two principal component scores revealed four different groupings of frequency-dependent responses to SCS (Fig. 6B). These groupings represented the minimal total squared Euclidean centroid distance out of 100 repetitions of k-means clustering with centroids and accounted for 81% of the total variance of the distribution. Increasing the number of centroids in the k-means analysis to five accounted for only 6% more of the total variance, and increasing the number of centroids beyond five did not increase the total variance accounted for by >2.9% per additional centroid, suggesting that four clusters were sufficient to classify the responses.
Hierarchal clustering using squared Euclidean distance between cluster centroids as the linkage distance measure (Distance Cluster Combine) between clusters and visualized through a dendrogram confirmed the results of the k-means clustering (Fig. 6D). Although five clusters are evident in the dendrogram, the fifth cluster was small, comprising responses that were classified by the k-means analysis with other inhibitory responses (i.e., cluster 1), and the small cluster merged with the branch corresponding to the other inhibitory responses on the dendrogram. In addition, only 4 out of 59 responses, on the boundaries of their respective clusters, were inconsistent between k-means and hierarchal analysis clustering (Fig. 6, B and D), indicating that the observed clusters are independent of the clustering algorithm.
Averaging the normalized frequency-response curves corresponding to each cluster revealed distinct SCS frequency-dependent neuronal responses (Fig. 6C). Cluster 2 responses (“gate”) were nonmonotonic and exhibited significant inhibition to 30- and 50-Hz SCS (n = 10, rmANOVA P = 0.001) consistent with predictions of neuronal responses to SCS by a computational model of the Gate Control circuit (Zhang et al. 2014b). However, clusters with behaviors that could not be described by the Gate Control circuit model alone were also present: cluster 1 (“inhibitory”) responses were inhibited with increasing SCS frequency (n = 17, rmANOVA P < 0.0001), cluster 3 (“excite”) responses exhibited increased excitation with increasing SCS frequency (n = 18, rmANOVA P < 0.0001), and cluster 4 (“plateau”) responses exhibited excitation up to 50-Hz SCS followed by accommodation of their responses at higher SCS frequencies (n = 14, rmANOVA P = 0.0006). All clusters, including the gate cluster, contained LT, WDR, and NS neurons.
Fluctuations in baseline activity resulted in the frequency-response curves of 13/33 responders being classified differently between SCS only and SCS + sciatic conditions, excluding seven neurons that were “nonresponsive” in one or the other baseline condition. Changes in responder status occurred when the baseline evoked by sciatic stimulation allowed visualization of inhibition characteristic of inhibitory (n = 5) responses or when high levels of activity evoked by sciatic stimulation sometimes saturated excite or plateau neurons (n = 2). Of the remaining 13 neurons, 3 neurons changed from gate in the SCS only case to inhibitory in the SCS + sciatic case due to a higher baseline in the latter required to visualize inhibition in the gate response. Three neurons changed from excite or plateau in the SCS only condition to gate in the SCS + sciatic condition due to a higher baseline in the latter. Three neurons changed from excite in the SCS only case to plateau in the SCS + sciatic case, possibly due to adaptation to sciatic stimulation. The four remaining neurons exhibited unique response class transitions between the SCS only and SCS + sciatic conditions (Fig. 6E). However, in 7/13 neurons, the features of the SCS-related PSTH, in particular sharp transient excitation at 2–3 ms, diffuse excitation, and/or inhibition immediately following excitation if present, were preserved across SCS only and SCS + sciatic conditions. Six out of 13 neurons exhibited significant differences in PSTH features between SCS only and SCS + sciatic conditions. In addition, no relationships among physiological response type (LT, WDR, and NS), cluster classification, and baseline condition were apparent (Fig. 6F). Taken together, these observations indicate that changes in classification between conditions were generally due to changes in baseline activity resulting from coincident sciatic stimulation rather than changes in the effects of SCS.
Stereotyped responses also evident in CCI rats.
In a subset of eight rats, we induced a CCI (Bennett and Xie 1991) and conducted terminal electrophysiology 7–10 days following the injury. CCI animals (n = 8) exhibited lower paw withdrawal thresholds 7 days postinjury (3.97 ± 0.31 g, means ± SE) than at baseline (9.96 ± 0.65 g, P = 9.5 × 10−7, Student's t-test; Fig. 7A); all experiments on CCI animals were conducted 7–10 days postinjury, as the injury response peaks and remains steady within that time interval (Bennett and Xie 1988; Kim et al. 1997). Seven antidromically identified projection neurons were recorded, and all recorded neurons exhibited either WDR or NS physiological response type during BPPC testing of the ipsilateral hindpaw, confirming that the transmission of sensory information through the sciatic nerve was preserved following CCI (Fig. 7B). Six of the seven neurons were classified as responders based on Z-score comparisons of SCS On vs. SCS Off PSTHs. In addition, these six neurons were classified into the “excite” (n = 2), “inhibit” (n = 1), and “adapt” (n = 3) groups with least squares regression using a generalized linear model with the principal components used to classify the responses shown in Fig. 6 as predictors (Fig. 7C), suggesting that spinal microcircuits underlying response heterogeneity are also present in an animal model of chronic pain.
Fig. 7.

Heterogeneous responses of dorsal horn projection neurons to SCS were also present in animals following constriction injury (CCI). A: CCI produced a significant reduction in paw withdrawal thresholds (n = 8; P < 9.5 × 10−7, Student's t-test). B: representative brush-sensitive WDR neuron, brush-insensitive WDR neuron, and NS neuron indicating that neurons are responsive to stimulation of the peripheral receptive field following CCI. C: normalized responses from 6 responsive projection neurons out of 7 neurons found from CCI animals. C, insets: results of least squares regression classification of the frequency-response curves plotted against the original classification scheme (Fig. 6). Data from one nonresponsive neuron are not shown.
Dorsal horn microcircuits are sufficient to explain heterogeneous responses to SCS.
We implemented computational models of coloring, mixing, and opponency spinal microcircuits (Prescott et al. 2014; Prescott and Ratté 2012) to determine whether different spinal microcircuits could account for the different classes of responses to SCS by weakening or removing specific elements in an overall template circuit (Fig. 8A). Single parameterizations of the computational microcircuits reproduced three of the four normalized SCS frequency-response relationships observed experimentally (Fig. 8, B–D), and each microcircuit model reproduced features of PSTHs from experimental neurons classified into the corresponding cluster, supporting the connectivity delineated by the specific microcircuit as the driver for the corresponding frequency-response relationship. The coloring microcircuit corresponding to cluster 3, with weak SCS-mediated inhibition and strong excitation modulated by SCS-independent tonic inhibition of the projection neuron, reproduced progressive excitation at SCS frequencies exceeding 50 Hz as well as recurring excitatory peaks in neuron activity that occurred 100 ms (10-Hz SCS), 20 ms (50-Hz SCS), and 6.7 ms (150 Hz) apart with latencies similar to those observed experimentally (Fig. 8B). The mixing microcircuit corresponding to cluster 2, featuring strong excitatory and inhibitory connections from SCS-affected inputs but no tonic inhibition, reproduced the nonmonotonic frequency-response relationship, the pattern of strong transient excitation followed by prolonged inhibition following 10- and 50-Hz SCS, and recurring excitatory events observed during 150-Hz SCS (Fig. 8C). Model-predicted suppression of transient excitation by prolonged inhibition at 50-Hz SCS was also observed in experimental neurons classified to cluster 2. Finally, the opponency microcircuit corresponding to cluster 1, which included weak SCS-mediated excitation and strong inhibition modulated by SCS-independent tonic inhibition of the interneuron, reproduced experimentally observed progressively inhibited responses with increasing SCS frequency and total suppression by 150-Hz SCS, as well as PSTH peaks corresponding to weak SCS-mediated excitation during 10- and 50-Hz SCS (Fig. 8D).
Fig. 8.

Computational models of spinal microcircuits reproduced frequency-response curves and features of PSTHs. A: template circuit that describes all features that contributed to the individual microcircuits. B–D: individual spinal microcircuits, normalized frequency-response relationships, and normalized smoothed PSTHs constructed using a “virtual” 10-Hz SCS train time-aligned onto real 10-, 50-, and 150-Hz SCS comparing model and experimental responses. In PSTH comparisons, the model response (bold) was overlaid onto all experimental PSTH responses (faint) from neurons whose frequency-response relationships were classified into the cluster corresponding to the microcircuit. The colors of the graph lines indicate the cluster to which each microcircuit corresponds (Fig. 6).
Spike-frequency adaptation mechanisms reproduce the plateau response.
The nonmonotonic relationship between neuron firing rate and SCS frequency observed in cluster 4 could not be reproduced by any parameterization of the microcircuits described above. Specifically, the decline in firing rate from 50- to 100-Hz SCS, coupled with the absence of an increase in the recurring excitatory component in the PSTHs from 50- to 150-Hz SCS, suggested that adaptation mechanisms might underlie the responses observed in cluster 4. Therefore, we replaced the tonically firing projection neuron in the coloring microcircuit with a model of an adapting dorsal horn neuron. As few data exist regarding the specific mechanisms of spike frequency adaptation in dorsal horn neurons, we conducted a sensitivity analysis wherein we varied the parameters underlying three possible drivers of adaptation-Na conductance (gNa), fast K+ conductance (gK), and slow K+ conductance (gKm) within ranges centered around previously published values (Melnick et al. 2004; Prescott and De Koninck 2005) (Fig. 9A).
Fig. 9.

Spike frequency adaptation mechanisms allowed reproduction of the aggregate response representing neurons in cluster 4 (Fig. 6). A: normalized model projection neuron responses to different frequencies of SCS across different values for gNa, gK, and gKm. In all cases, the Na+ conductance slowly inactivates according to Eqs. 4–6. gNa = 100% refers to a hillock Na+ conductance of 2.19 S/cm2. gK = 100% refers to a hillock K+ conductance of 0.076 S/cm2, a soma K+ conductance of 0.0043 S/cm2, and a dendritic K+ conductance of 0.034 S/cm2. gKm = 100% refers to a soma and dendritic Km conductance of 0.0005 S/cm2. B: normalized individual and average experimental responses (left) juxtaposed with the average of the normalized model responses shown in A (right). C: experimental (left) and all model (right) smoothed normalized PSTHs generated using a virtual 10-Hz train for real 10-, 50-, and 150-Hz SCS responses.
Inclusion of these adaptation mechanisms in the projection neuron was sufficient to reproduce individual features of but not the overall normalized SCS response curve representing cluster 4 (Fig. 9A). Increasing fast or slow K+ conductances and reducing Na+ conductances generated the convex increase in responses to SCS up to 50 Hz and the reduction from 50- to 100-Hz SCS but could not reproduce the plateau at 150 Hz. Reducing K+ conductances and increasing Na+ conductances reproduced the downturn at 100 Hz and the plateau at 150 Hz but could not reproduce the convex increase in response from 1- to 50-Hz SCS. However, consistent with the experimental data, averaging the model responses across parameterizations reproduced the overall cluster 4 response (Fig. 9B), and the range of individual model PSTH responses spanned the range of responses observed experimentally, including the excitatory peaks at similar latencies and the absence of progressive growth of the recurring excitatory component (Fig. 9C). Increasing the strength of tonic inhibition onto the projection neuron to its original value from the coloring microcircuit did not prevent adaptation but resulted in a peristimulus response delay in A-fiber-mediated excitation in the model that exceeded the delays observed experimentally, suggesting that adapting neurons may be subject to less tonic inhibition than nonadapting neurons. Taken together, these results suggest that biophysical rather than network mechanisms account for the differences between the responses observed between experimental clusters 3 and 4 and that heterogeneity in the conductances responsible for projection neuron adaptation could underlie the diversity within cluster 4.
Disruption of GABAA mechanisms alters responses to SCS.
In a subset of 20 neurons, we intrathecally applied BIC, a GABAA receptor antagonist, and/or CGP, a GABAB receptor antagonist, and repeated measurements of the response of the neuron to different frequencies of SCS. BIC was applied in 14 instances, and CGP was applied in 9 instances; these instances include 2 neurons to which BIC was applied after an experimental block with CGP and 1 neuron to which CGP was applied after an experimental block with BIC. BIC administration increased baseline firing in 6/14 neurons and unmasked responses during BPPC testing in 9/14 neurons (Fig. 10A). In contrast, CGP administration increased baseline firing in one out of nine neurons and only enhanced the response to crushing of the peripheral receptive field in one out of nine neurons, suggesting that tonic GABAA mechanisms had a greater modulatory effect than GABAB mechanisms on the behavior of the recorded neurons.
Fig. 10.
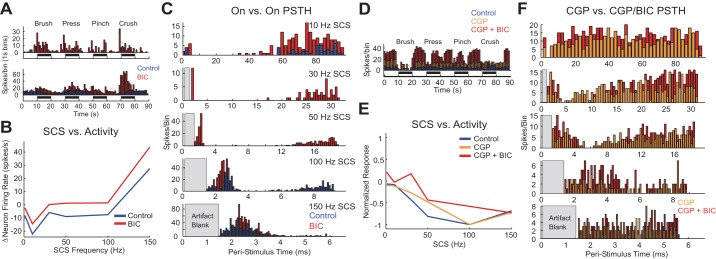
Administration of bicuculline (BIC) unmasked or disinhibited responses to peripheral stimulation and SCS. A: BPPC response profiles of 2 example neurons during control conditions (blue) and after application of BIC (red). BIC unmasked responses to BPPC in the top neuron and increased baseline firing/disinhibited evoked responses in the bottom neuron. B: raw frequency-response relationship of neuronal activity vs. SCS frequency before and after the application of BIC in a representative example. C: peristimulus responses to SCS before (blue) and after (red) intrathecal application of BIC for the representative example shown in B. D: BPPC response of neuron to which CGP 35348 (CGP), then CGP + BIC was applied. E: normalized relationship between SCS frequency and neuron response to SCS before and after the application of CGP, then CGP + BIC in 1 neuron. F: PSTHs during SCS after application of CGP (orange) and after application of CGP + BIC (red) in a representative example. Raw neuron firing rate vs. SCS frequency curves are not shown because the application of CGP and CGP + BIC resulted in increased baseline activity (D) that would have distorted the plot axes.
Application of BIC but not CGP had substantial effects on SCS-evoked responses in sensory projection neurons. Based on Z-score comparisons of PSTH responses during SCS between control and BIC/CGP conditions, BIC significantly disinhibited neuronal responses to SCS in either the SCS only or SCS + sciatic conditions in 7 of the 14 neurons (e.g., Fig. 10B), while CGP only altered significantly poststimulus responses to SCS in 1 of 9 neurons. Five out of seven neurons in which BIC had a disinhibitory effect also exhibited increased baseline activity and enhanced responses to BPPC stimulation. In the seven neurons for which BIC had an effect on the SCS response, the primary effects of BIC application were either the accentuation of transient excitation, suggesting disinhibition of excitatory inputs, or a reduction in the period of transient inhibition following the SCS pulse, suggesting a shift to other inhibitory mechanisms, such as glycinergic mechanisms, that are unaffected by BIC (Fig. 10C) (Braz et al. 2014). Disinhibition was evident even in neurons classified into excitatory clusters: five out of seven neurons were classified into either the excite or plateau clusters in SCS only or SCS + Sciatic conditions, and one out of seven neurons was classified into the gate cluster during SCS only and the excite cluster during SCS + sciatic due to a reduction in baseline activity during sciatic stimulation. Included in these counts is one neuron that was classified into the inhibit cluster during both SCS only and SCS + sciatic conditions and for which BIC was applied after CGP; BIC administration disinhibited this neuron's response to BPPC and SCS (Fig. 10D), particularly at SCS frequencies <50 Hz, whereas CGP administration had only a modest effect on the neuron's normalized response to SCS (Fig. 10, E and F). In addition, two neurons that did not respond to SCS during the control block responded to SCS following BIC administration, whereas CGP did not unmask responses in any neuron that was nonresponsive in the control case.
Finally, three neurons exhibited adaptation during SCS + sciatic stimulation following BIC, and one of these three neurons exhibited an enhanced response during SCS when sciatic stimulation was off but exhibited adaptation to SCS when sciatic stimulation was on. For all three neurons that adapted following BIC, BIC administration resulted in substantially increased baseline firing rates when SCS was off, suggesting that removal of GABAA-mediated inhibition resulted in excessive excitatory drive onto these neurons sufficient to cause adaptation or an increase in SCS-independent modulation, such as from supraspinal nuclei, as has been previously reported (Sorkin et al. 1998). CGP did not induce adaptation during the SCS response in any of the tested neurons, including the neuron for which CGP application raised the baseline activity level.
DISCUSSION
We hypothesized that SCS-mediated inhibition of spinal sensory projection neurons would be dependent on stimulation frequency and follow a nonmonotonic trend predicted by a computational model of the Gate Control circuit (Zhang et al. 2014b). The responses of antidromically identified spinal sensory projection neurons were dependent on SCS frequency in both healthy and CCI animals, and a subset of neurons exhibited SCS frequency dependence consistent with predictions based on the Gate Control Theory. However, responses were unexpectedly heterogeneous, and computational models of excitatory, mixed, and inhibitory spinal microcircuits (Prescott and Ratté 2012) along with the Gate Control circuit were required to reproduce the range of observed responses. This finding suggested that spinal microcircuits in addition to the Gate Control circuit underlie the experimentally observed heterogeneity in responses to SCS (Prescott et al. 2014). Consistent with the Gate Control Theory, the administration of BIC, a GABAA receptor antagonist, reduced the amount of inhibition produced by SCS in some neurons or unmasked responses to SCS in others. However, BIC also altered baseline neural activity and enhanced excitatory responses to SCS, suggesting that tonic GABAergic inhibition beyond that depicted by the Gate Control Theory plays an important role in modulating responses of sensory neurons to peripheral stimuli and SCS (Takazawa and MacDermott 2010b). Our in vivo characterization of sensory projection neuron responses to SCS combined with our computational assessments of spinal microcircuits refines our understanding of the neural circuits underlying SCS both within and beyond the context of the Gate Control Theory. The insights gained from our study add to our knowledge of the mechanisms underlying SCS and may ultimately contribute to the improvement of the efficacy of SCS.
Projection neuron responses and spinal microcircuits.
This is the first study to assess the effects of different frequencies of SCS on antidromically verified projection neurons. Quantitative analysis of projection neuron activity during SCS directly tests the Gate Control-based hypothesis that SCS acts to inhibit the output neurons of the dorsal horn circuit (Guan 2012; Shealy et al. 1972). As recorded neurons were verified as sensory projection neurons, the results of our study more accurately reflect the effects of SCS on the output of the spinal circuit and possibly the transmission of nociceptive information to the brain than did prior studies that did not identify neurons as projection neurons (Guan et al. 2010; Yakhnitsa et al. 1999). In addition, that 8 of the 33 responsive neurons, including 2 WDR and 3 NS neurons, exhibited SCS frequency-response relationships consistent with the Gate Control Theory (Zhang et al. 2014b) reinforces the hypothesis that the Gate Control circuit is sufficient to explain some aspects of the SCS response.
Projection neuron responses to SCS were unexpectedly heterogeneous in both healthy and CCI animals, suggesting that networks in addition to the Gate Control circuit may mediate different aspects of the population response to SCS. Spinal microcircuits that describe inhibitory and excitatory interactions between peripheral inputs and SCS beyond those proposed by the Gate Control Theory may explain the heterogeneous responses (Prescott et al. 2014; Prescott and Ratté 2012). Our experiments included coactivation of nociceptive and nonnociceptive afferents necessary to test the proposed interactions, and both frequency-response curves and PSTHs were reproduced by computational models of the Gate circuit and proposed microcircuits. Furthermore, the dominant response of the excitatory coloring and inhibitory opponency microcircuits could be reproduced with attenuation rather than deletion of the opposing pathway, and PSTHs during SCS were not affected by 1-Hz sciatic stimulation at C-fiber threshold in 14 of 20 neurons for which the change in baseline activity resulted in a dual classification. These observations suggests that microcircuits may share common features or represent points along a continuum of a population of dorsal horn circuits with architectures similar to the Gate Control circuit (Fig. 7A) responsible for processing nociception and mediating the analgesic effects of SCS.
In addition, heterogeneous membrane properties of the model adapting neuron were required to reproduce the overall adaptation response, suggesting that heterogeneity among individual neurons may also contribute to the population code underlying nociception. Diverse biophysics affect neuronal synchrony in response to simulated synchronized synaptic inputs, and as one possibility, adaptation mechanisms may act as a safeguard against noise from primary afferent inputs (Ratté et al. 2013). These findings motivate the future consideration of pain as the result of a population response in addition to the output of a specific neural circuit or the result of direct information transfer from a specific group of neurons (e.g., a “labeled line”) (Prescott et al. 2014).
Sensory projection neuron activity is modulated by GABAA receptor pathways.
Our finding that SCS frequency dependence is modulated by activation of GABAA receptors builds on a prior study showing that SCS-mediated inhibition in healthy animals may be removed by BIC (Duggan and Foong 1985) and supports the Gate Control Theory prediction that as GABAergic inhibition is lost, SCS becomes less effective at inhibiting neuronal activity (Zhang et al. 2014b). As the loss of GABAergic inhibition is associated with the development of hyperalgesia and allodynia (Sivilotti and Woolf 1994), and as the progression of neuropathic pain is accompanied by the loss of GABAergic inhibition in the dorsal horn (Braz et al. 2014), the altered SCS-frequency dependence following the loss of GABAergic inhibition may underlie the loss of SCS efficacy with the progression of neuropathic pain (Kumar et al. 2007). However, we did not observe loss of inhibitory responses to SCS following administration of CGP 35348, a GABAB receptor antagonist. This result is inconsistent with the observation of reversals of SCS-mediated increases in paw withdrawal thresholds by CGP 35348 in animals with sciatic nerve mononeuropathy (Cui et al. 1998; Meyerson et al. 1995). This inconsistency may reflect a conversion of inhibitory mechanisms to GABAB receptor-mediated pathways following the loss of GABAA receptor-mediated pathways during the transition to neuropathic pain. In addition, inhibitory responses to SCS were preserved following CCI in one neuron, suggesting that inhibitory processes beyond GABAergic mechanisms may contribute to SCS-mediated inhibition of sensory neurons and resultant pain relief.
Tonically active neurons from segmental (Takazawa and MacDermott 2010a; Torsney and MacDermott 2006) and supraspinal (Carlson et al. 2007; Lin et al. 1996) sources beyond the Gate Control Theory may also mediate GABAergic inhibition of dorsal horn inhibitory interneurons and projection neurons that modulate neuronal responses to pain and SCS. Altering the magnitude of tonic GABAergic inhibition in the models enabled tuning of the frequencies at which spinal microcircuits exhibited SCS frequency-dependent inhibition and excitation. Furthermore, that BIC had a disinhibitory effect even on six neurons that exhibited excite or plateau excitatory responses during SCS and that excitatory responses to SCS were unmasked in two nonresponders by BIC support tonic inhibition as a general modulator of sensory neuron activity regardless of peripheral afferent inputs. However, we could not determine the extent to which BIC administration affected direct SCS-mediated inhibition vs. indirect inhibition from SCS-independent tonic inhibitory interneurons or to what extent the inhibition from these neurons may have been due to other inhibitory neurotransmitters, such as glycine (Braz et al. 2014; Takazawa and MacDermott 2010a).
General limitations.
We conducted acute experiments on healthy animals using 1- to 150-Hz SCS without behavioral assessments, whereas clinically SCS is used to treat chronic neuropathic pain. Changes involved in the transition from the healthy state to neuropathic pain include but are not limited to altered network connections (Todd 2010), glial cell activation (Ji et al. 2013; Scholz and Woolf 2007), modifications in synaptic dynamics due to KCC2 malfunction (Coull et al. 2005), and loss of GABAergic inhibition in the dorsal horn (Braz et al. 2014), not all of which could be assessed in our experiments. Although the firing rate of sensory projection neurons correlate with the magnitude of perceived pain (Simone et al. 1991), our study did not directly assess how SCS at different frequencies affects pain-related behaviors. Importantly, we addressed some of the possible differences in responses between healthy and neuropathic pain states by demonstrating that block of GABAA mechanisms reduced inhibitory responses to SCS in some neurons and that the same microcircuit responses to SCS were observed in CCI animals that exhibited decreased paw withdrawal thresholds after injury. In addition, we applied SCS concurrently with sciatic stimulation at amplitudes sufficient to produce purportedly painful C-fiber-evoked responses. That four neurons, among them one WDR and three NS neurons, were classified as exhibiting an inhibitory response to SCS during on-going sciatic stimulation when they were inactive during SCS with no other accompanying stimulation (Fig. 6F) further suggests that suppression of sensory neuron activity by SCS underlies SCS-mediated pain relief. However, behavioral studies are needed to link the observed neuronal responses to analgesic effects of SCS, especially as targeted modulation of only a subset of sensory projection neurons may be sufficient to reverse hyperalgesia in neuropathic animals (Lavertu et al. 2013).
The presence of nonresponders despite a robust SCS-evoked CDP over their resident spinal segment and a lack of correlation between microcircuit classification and peripheral sensory type (LT, WDR, and NS) among responders suggests that primary afferents may modulate dorsal horn neuron responses to SCS through more complex, polysynaptic circuits beyond those considered in this study (Braz et al. 2014; Zheng et al. 2010). The existence of nonresponders to SCS has not been previously explicitly reported, suggests that not all projection neurons receive inputs affected by SCS, and may explain why SCS does not inhibit acute pain or sensation from the affected dermatome (Lindblom and Meyerson 1975). That the microcircuit classification of a neuron was independent of its physiological response class (LT, WDR, and NS) supports the hypothesis that SCS-evoked effects may override baseline activity resulting from low-frequency (<2.3 Hz) Aδ/C-fiber afferent drive (Zhang et al. 2014b). However, both the SCS frequency response and the peristimulus response to SCS were significantly different between SCS only and SCS + sciatic conditions in 6/33 responder neurons, suggesting that interactions more complex than those depicted by the spinal microcircuits may play a role in mediating responses to SCS. In addition, inhibition due to activation of dorsal column collaterals of afferent inputs from surround receptive fields by SCS (Hillman and Wall 1969) or excitatory interactions between projection neurons corresponding to distinct receptive fields (Luz et al. 2010) may also be independent of projection neuron physiological response class and may have contributed to the observed response heterogeneity. However, we could not target specific fiber tracts in the dorsal columns with our electrode, as CDPs do not provide sufficient spatial information to discern activation of specific fibers, and the relationship between the somatotopic organization of the dorsal columns and the somatotopic organization of dorsal horn sensory neurons is unclear.
Our study sought to determine if the Gate Control Theory circuit was sufficient to describe responses to SCS in sensory projection neurons regardless of laminar location, but recently proposed schemes for the dorsal horn sensory circuit suggest that sensory processing by superficial and deep projection neurons may be significantly different (Braz et al. 2014; Todd 2010). “Transmission Cells” in the Gate were theorized as being responsive to both nonnociceptive myelinated and nociceptive unmyelinated afferent inputs, but unlike the inhibitory interneurons, which were thought to reside specifically in the substantial gelatinosa, the morphology and laminar locations of the projection neurons described as the Transmission Cells were never explicitly defined (Melzack and Wall 1965). Although superficial nociceptive-specific neurons may be inhibited by SCS (Foreman et al. 1976) and have been more recently implicated as a potential labeled line for pain (Craig 2003; Todd 2010), projection neurons from lamina IV–VI are also sensitive to nociceptive and nonnociceptive sensory inputs (Chung et al. 1979). These deeper neurons project to the same nuclei in the sensory thalamus as do superficial neurons (Burstein et al. 1990), they are excited and inhibited by SCS (Foreman et al. 1976; Handwerker et al. 1975), and their firing rates correlate with perceived pain (Simone et al. 1991) and applied pressure to the hindpaw in neuropathic rats (Lavertu et al. 2013). Recent data show that glycinergic inhibitory interneurons critical to normal sensory processing are distributed across superficial and deep dorsal horn, and both superficial and deeper neurons may synapse onto PKCγ+ interneurons in lamina IIi that are important to the gating of pain transmission (Foster et al. 2015; Lu et al. 2013). These observations contribute to ambiguity regarding whether the Transmission Cells described by the Gate Control Theory refer to a generalized functional class of neurons or a specific anatomically localized population of neurons and motivated our strategy of recording from projection neurons at all depths. However, studies focusing on the responses of sensory neurons from specific laminae to SCS are needed to ascertain whether sensory circuit architectures, and therefore sensory processing, differ between the superficial and deep dorsal horn.
Other mechanisms of segmental inhibition beyond the GABAergic mechanisms assessed in our study, including glycinergic inhibition and presynaptic inhibition, also play an important role in sensory processing. Blockade of glycinergic inhibition by intrathecal administration of strychnine (Lu et al. 2013; Sivilotti and Woolf 1994; Yaksh 1989) or selective knockdown of spinal glycinergic interneurons (Foster et al. 2015) is sufficient to produce hyperalgesia and allodynia characteristic of neuropathic pain in rats. In addition, disinhibition of PKCγ-positive interneurons that receive primarily glycinergic inhibitory inputs and that modulate signal transmission between nonnociceptive and nociceptive pathways in the superficial dorsal horn is implicated as a factor in the development and progression of neuropathic pain (Braz et al. 2014; Lu et al. 2013; Miraucourt et al. 2007). Importantly, we implemented glycinergic inhibition in the model at the inhibitory connections from the inhibitory interneurons affected by SCS (Zhang et al. 2014b) and at the inhibitory connections from the tonic inhibitory interneurons unaffected by SCS (Lu et al. 2013; Takazawa and MacDermott 2010b), but the effects of the loss of glycinergic inhibition on neuropathic pain and the response to SCS remain to be clarified. As well, the original Gate Control circuit depicted presynaptic inhibition as the primary means by which inhibitory interneurons gate the transmission of nociceptive information by projection neurons (Melzack and Wall 1965). However, the original Gate Control Theory was published before establishing many mechanisms underlying postsynaptic inhibition, and the mechanism of inhibition was depicted as being presynaptic to be consistent with knowledge at the time. Subsequent experiments provided strong evidence for postsynaptic inhibition of sensory neurons (Hongo et al. 1968) and led to a revision of the Gate Control Theory to include postsynaptic inhibition (Wall 1978). Furthermore, the absence of anatomical evidence of presynaptic inhibition among sensory primary afferent terminals in the dorsal horn (Coull et al. 2003; Rudomin and Schmidt 1999) motivated some to depict postsynaptic inhibition as the exclusive inhibitory mechanism in Gate Control (Basbaum and Jessell 2000). Finally, recent studies suggest that mechanisms underlying the loss of inhibition that accompanies the progression of neuropathic pain primarily affect postsynaptic inhibition (Braz et al. 2014; Coull et al. 2003; Moore et al. 2002). As such, we focused exclusively on postsynaptic inhibition in our computational studies. However, recent work identified axo-axonic GABAergic synapses among a substantial portion of primary afferents in the superficial dorsal horn (Paul et al. 2012), suggesting that presynaptic inhibition may play a role in the mechanisms underlying SCS.
Finally, we did not account for the effects of SCS on supraspinal circuits (Heinricher et al. 2009) or on descending pathways, such as the dorsolateral funiculus, that could have been activated by SCS. Although segmental mechanisms have been shown to be sufficient in describing inhibitory and excitatory responses to SCS (Foreman et al. 1976; Smits et al. 2012), supraspinal loops may also modulate analgesia by SCS (Barchini et al. 2012). For example, neurons in supraspinal nuclei exhibited more activity during SCS in neuropathic rats that showed increased paw withdrawal thresholds than in nonresponding rats, suggesting that effective SCS for chronic pain requires supraspinal modulation (Song et al. 2013), but knowledge regarding the extent of supraspinal contributions to SCS is sparse.
DISCLOSURES
Funding for this work was provided by the Stryker Corporation. J. J. Janik is a Stryker Corporation employee and owns Stryker Corporation stock.
AUTHOR CONTRIBUTIONS
Author contributions: T.C.Z., J.J.J., and W.M.G. conception and design of research; T.C.Z., R.V.P., and G.C. performed experiments; T.C.Z. and R.V.P. analyzed data; T.C.Z., J.J.J., R.V.P., R.-R.J., and W.M.G. interpreted results of experiments; T.C.Z. prepared figures; T.C.Z. drafted manuscript; T.C.Z., J.J.J., R.-R.J., and W.M.G. edited and revised manuscript; T.C.Z., J.J.J., R.V.P., G.C., R.-R.J., and W.M.G. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Gilda I. Mills for assistance with the animal experiments and the Duke Computing Cluster for assistance with computational experiments.
REFERENCES
- Barchini J, Tchachaghian S, Shamaa F, Jabbur S, Meyerson B, Song Z, Linderoth B, Saade N. Spinal segmental and supraspinal mechanisms underlying the pain-relieving effects of spinal cord stimulation: an experimental study in a rat model of neuropathy. Neuroscience 215: 196–208, 2012. [DOI] [PubMed] [Google Scholar]
- Basbaum AI, Jessell TM. The perception of pain. In: Principal of Neural Science, edited by Kandel ER. New York: McGraw-Hill, 2000, chapt. 24. [Google Scholar]
- Benda J, Herz AV. A universal model for spike-frequency adaptation. Neural Comput 15: 2523–2564, 2003. [DOI] [PubMed] [Google Scholar]
- Bennett GJ, Xie YK. A peripheral mononeuropathy in rat that produces disorders of pain sensation like those seen in man. Pain 33: 87–107, 1988. [DOI] [PubMed] [Google Scholar]
- Bokil H, Andrews P, Kulkarni JE, Mehta S, Mitra PP. Chronux: a platform for analyzing neural signals. J Neurosci Methods 192: 146–151, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braz J, Solorzano C, Wang X, Basbaum AI. Transmitting pain and itch messages: a contemporary view of the spinal cord circuits that generate gate control. Neuron 82: 522–536, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burstein R, Dado RJ, Giesler GJ Jr. The cells of origin of the spinothalamic tract of the rat: a quantitative reexamination. Brain Res 511: 329–337, 1990. [DOI] [PubMed] [Google Scholar]
- Carlson JD, Maire JJ, Martenson ME, Heinricher MM. Sensitization of pain-modulating neurons in the rostral ventromedial medulla after peripheral nerve injury. J Neurosci 27: 13222–13231, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods 53: 55–63, 1994. [DOI] [PubMed] [Google Scholar]
- Chung JM, Kenshalo DR, Gerhart KD, Willis WD. Excitation of primate spinothalamic neurons by cutaneous C-fiber volleys. J Neurophysiol 42: 1354–1369, 1979. [DOI] [PubMed] [Google Scholar]
- Coull JA, Beggs S, Boudreau D, Boivin D, Tsuda M, Inoue K, Gravel C, Salter MW, De Koninck Y. BDNF from microglia causes the shift in neuronal anion gradient underlying neuropathic pain. Nature 438: 1017–1021, 2005. [DOI] [PubMed] [Google Scholar]
- Coull JAM, Boudreau D, Bachand K, Prescott SA, Nault F, Sík A, De Koninck P, De Koninck Y. Trans-synaptic shift in anion gradient in spinal lamina I neurons as a mechanism of neuropathic pain. Nature 424: 938–942, 2003. [DOI] [PubMed] [Google Scholar]
- Craig A. Pain mechanisms: labeled lines vs. convergence in central processing. Annu Rev Neurosci 26: 1–30, 2003. [DOI] [PubMed] [Google Scholar]
- Craig AD, Krout K, Andrew D. Quantitative response characteristics of thermoreceptive and nociceptive lamina i spinothalamic neurons in the cat. J Neurophysiol 86: 1459–1480, 2001. [DOI] [PubMed] [Google Scholar]
- Cui J, Meyerson B, Sollevi A, Linderoth B. Effect of spinal cord stimulation on tactile hypersensitivity in mononeuropathic rats is potentiated by simultaneous GABAB and adenosine receptor activation. Neurosci Lett 247: 183–186, 1998. [DOI] [PubMed] [Google Scholar]
- Dayan PA. Theoretical Neuroscience: Computational and Mathematical Modeling of Neural Systems. Cambridge, MA: MIT Press, 2001. [Google Scholar]
- Dixon WJ. Efficient analysis of experimental observations. Annu Rev Pharmacol Toxicol 20: 441–462, 1980. [DOI] [PubMed] [Google Scholar]
- Duan B, Cheng L, Bourane S, Britz O, Padilla C, Garcia-Campmany L, Krashes M, Knowlton W, Velasquez T, Ren X. Identification of spinal circuits transmitting and gating mechanical pain. Cell 159: 1417–1432, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duggan AW, Foong FW. Bicuculline and spinal inhibition produced by dorsal column stimulation in the cat. Pain 22: 249–259, 1985. [DOI] [PubMed] [Google Scholar]
- Foreman RD, Beall JE, Coulter JD, Willis WD. Effects of dorsal column stimulation on primate spinothalamic tract neurons. J Neurophysiol 39: 534–546, 1976. [DOI] [PubMed] [Google Scholar]
- Foster E, Wildner H, Tudeau L, Haueter S, Ralvenius WT, Jegen M, Johannssen H, Hösli L, Haenraets K, Ghanem A. Targeted ablation, silencing, and activation establish glycinergic dorsal horn neurons as key components of a spinal gate for pain and itch. Neuron 85: 1289–1304, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan Y. Spinal cord stimulation: neurophysiological and neurochemical mechanisms of action. Curr Pain Headache Rep 16: 217–225, 2012. [DOI] [PubMed] [Google Scholar]
- Guan Y, Wacnik PW, Yang F, Carteret AF, Chung CY, Meyer RA, Raja SN. Spinal cord stimulation-induced analgesia: electrical stimulation of dorsal column and dorsal roots attenuates dorsal horn neuronal excitability in neuropathic rats. Anesthesiology 113: 1392–1405, 2010. [DOI] [PubMed] [Google Scholar]
- Handwerker H, Iggo A, Zimmermann M. Segmental and supraspinal actions on dorsal horn neurons responding to noxious and non-noxious skin stimuli. Pain 1: 147–165, 1975. [DOI] [PubMed] [Google Scholar]
- Hao JX, Xu XJ, Wiesenfeld-Hallin Z. Intrathecal γ-aminobutyric acidB(GABAB) receptor antagonist CGP 35348 induces hypersensitivity to mechanical stimuli in the rat. Neurosci Lett 182: 299–302, 1994. [DOI] [PubMed] [Google Scholar]
- Harper AA, Lawson SN. Conduction velocity is related to morphological cell type in rat dorsal root ganglion neurones. J Physiol 359: 31–46, 1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinricher MM, Tavares I, Leith JL, Lumb BM. Descending control of nociception: specificity, recruitment and plasticity. Brain Res Rev 60: 214–225, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrero JF, Laird J, Lopez-Garcia JA. Wind-up of spinal cord neurones and pain sensation: much ado about something? Prog Neurobiol 61: 169–203, 2000. [DOI] [PubMed] [Google Scholar]
- Hillman P, Wall PD. Inhibitory and excitatory factors influencing the receptive fields of lamina 5 spinal cord cells. Exp Brain Res 9: 284–306, 1969. [DOI] [PubMed] [Google Scholar]
- Hines M, Carnevale N. The NEURON simulation environment. Neural Comput 9: 1179–1209, 1997. [DOI] [PubMed] [Google Scholar]
- Hongo T, Jankowska E, Lundberg A. Post-synaptic excitation and inhibition from primary afferents in neurones of the spinocervical tract. J Physiol 199: 569–592, 1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji RR, Berta T, Nedergaard M. Glia and pain: is chronic pain a gliopathy? Pain 154: S10–S28, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamata M, Koshizaki M, Shimada SG, Narimatsu E, Kozuka Y, Takahashi T, Namiki A, Collins JG. Changes in response properties and receptive fields of spinal dorsal horn neurons in rats after surgical incision in hairy skin. Anesthesiology 102: 141–151, 2005. [DOI] [PubMed] [Google Scholar]
- Kim KJ, Yoon YW, Chung JM. Comparison of three rodent neuropathic pain models. Exp Brain Res 113: 200–206, 1997. [DOI] [PubMed] [Google Scholar]
- Kumar K, Taylor RS, Jacques L, Eldabe S, Meglio M, Molet J, Thomson S, O'Callaghan J, Eisenberg E, Milbouw G, Buchser E, Fortini G, Richardson J, North RB. Spinal cord stimulation versus conventional medical management for neuropathic pain: a multicentre randomised controlled trial in patients with failed back surgery syndrome. Pain 132: 179–188, 2007. [DOI] [PubMed] [Google Scholar]
- Lánský P, Sacerdote L. The Ornstein-Uhlenbeck neuronal model with signal-dependent noise. Physics Lett A 285: 132–140, 2001. [DOI] [PubMed] [Google Scholar]
- Lavertu G, Côté SL, De Koninck Y. Enhancing K-Cl co-transport restores normal spinothalamic sensory coding in a neuropathic pain model. Brain 137: 724–738, 2013. [DOI] [PubMed] [Google Scholar]
- Lemay MA, Grill WM. Modularity of motor output evoked by intraspinal microstimulation in cats. J Neurophysiol 91: 502–514, 2004. [DOI] [PubMed] [Google Scholar]
- Li H, Monhemius R, Simpson B, Roberts M. Supraspinal inhibition of nociceptive dorsal horn neurones in the anaesthetized rat: tonic or dynamic? J Physiol 506: 459–469, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Q, Peng YB, Willis WD. Role of GABA receptor subtypes in inhibition of primate spinothalamic tract neurons: difference between spinal and periaqueductal gray inhibition. J Neurophysiol 75: 109–123, 1996. [DOI] [PubMed] [Google Scholar]
- Lindblom U, Meyerson BA. Influence on touch, vibration and cutaneous pain of dorsal column stimulation in man. Pain 1: 257–270, 1975. [DOI] [PubMed] [Google Scholar]
- Lipski J. Antidromic activation of neurones as an analytic tool in the study of the central nervous system. J Neurosci Methods 4: 1–32, 1981. [DOI] [PubMed] [Google Scholar]
- Lu Y, Dong H, Gao Y, Gong Y, Ren Y, Gu N, Zhou S, Xia N, Sun YY, Ji RR, Xiong L. A feed-forward spinal cord glycinergic neural circuit gates mechanical allodynia. J Clin Invest 123: 4050–4062, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luz LL, Szucs P, Pinho R, Safronov BV. Monosynaptic excitatory inputs to spinal lamina I anterolateral-tract-projecting neurons from neighbouring lamina I neurons. J Physiol 588: 4489–4505, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormick DA, Wang Z, Huguenard J. Neurotransmitter control of neocortical neuronal activity and excitability. Cereb Cortex 3: 387–398, 1993. [DOI] [PubMed] [Google Scholar]
- Melnick IV, Santos SF, Safronov BV. Mechanism of spike frequency adaptation in substantia gelatinosa neurones of rat. J Physiol 559: 383–395, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melzack R, Wall PD. Pain mechanisms: a new theory. Science 150: 971–979, 1965. [DOI] [PubMed] [Google Scholar]
- Meyerson BA, Ren B, Herregodts P, Linderoth B. Spinal cord stimulation in animal models of mononeuropathy: effects on the withdrawal response and the flexor reflex. Pain 61: 229–243, 1995. [DOI] [PubMed] [Google Scholar]
- Michaelis M, Blenk K, Janig W, Vogel C. Development of spontaneous activity and mechanosensitivity in axotomized afferent nerve fibers during the first hours after nerve transection in rats. J Neurophysiol 74: 1020–1027, 1995. [DOI] [PubMed] [Google Scholar]
- Miraucourt LS, Dallel R, Voisin DL. Glycine inhibitory dysfunction turns touch into pain through PKCgamma interneurons. PLoS One 2: e1116, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montgomery EB. Effects of GPi stimulation on human thalamic neuronal activity. Clin Neurophysiol 117: 2691–2702, 2006. [DOI] [PubMed] [Google Scholar]
- Moore K, Kohno T, Karchewski L, Scholz J, Baba H, Woolf C. Partial peripheral nerve injury promotes a selective loss of GABAergic inhibition in the superficial dorsal horn of the spinal cord. J Neurosci 22: 6724–6731, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakley JC, Prager JP. Spinal cord stimulation: mechanisms of action. Spine 27: 2574–2583, 2002. [DOI] [PubMed] [Google Scholar]
- Paul J, Zeilhofer HU, Fritschy JM. Selective distribution of GABAAreceptor subtypes in mouse spinal dorsal horn neurons and primary afferents. J Comp Neurol 520: 3895–3911, 2012. [DOI] [PubMed] [Google Scholar]
- Peng YB, Lin Q, Willis WD. Effects of GABA and glycine receptor antagonists on the activity and PAG-induced inhibition of rat dorsal horn neurons. Brain Res 736: 189–201, 1996. [DOI] [PubMed] [Google Scholar]
- Polgár E, Gray S, Riddell JS, Todd AJ. Lack of evidence for significant neuronal loss in laminae I–III of the spinal dorsal horn of the rat in the chronic constriction injury model. Pain 111: 144–150, 2004. [DOI] [PubMed] [Google Scholar]
- Powers RK, Sawczuk A, Musick JR, Binder MD. Multiple mechanisms of spike-frequency adaptation in motoneurones. J Physiol (Paris) 93: 101–114, 1999. [DOI] [PubMed] [Google Scholar]
- Prescott SA, De Koninck Y. Integration time in a subset of spinal lamina I neurons is lengthened by sodium and calcium currents acting synergistically to prolong subthreshold depolarization. J Neurosci 25: 4743–4754, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prescott SA, Ma Q, De Koninck Y. Normal and abnormal coding of somatosensory stimuli causing pain. Nat Neurosci 17: 183–191, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prescott SA, Ratté S. Pain processing by spinal microcircuits: afferent combinatorics. Curr Opin Neurobiol 22: 631–639, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratté S, Hong S, De Schutter E, Prescott SA. Impact of neuronal properties on network coding: roles of spike initiation dynamics and robust synchrony transfer. Neuron 78: 758–772, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudomin P, Schmidt RF. Presynaptic inhibition in the vertebrate spinal cord revisited. Exp Brain Res 129: 1–37, 1999. [DOI] [PubMed] [Google Scholar]
- Scholz J, Woolf CJ. The neuropathic pain triad: neurons, immune cells and glia. Nat Neurosci 10: 1361–1368, 2007. [DOI] [PubMed] [Google Scholar]
- Shealy CN, Mortimer J, Hagfors NR. Dorsal column electroanalgesia. Survey Anesthesiol 16: 15–16, 1972. [DOI] [PubMed] [Google Scholar]
- Shimazaki H, Shinomoto S. A method for selecting the bin size of a time histogram. Neural Comput 19: 1503–1527, 2007. [DOI] [PubMed] [Google Scholar]
- Simone DA, Sorkin LS, Oh U, Chung JM, Owens C, LaMotte RH, Willis WD. Neurogenic hyperalgesia: central neural correlates in responses of spinothalamic tract neurons. J Neurophysiol 66: 228–246, 1991. [DOI] [PubMed] [Google Scholar]
- Sivilotti L, Woolf CJ. The contribution of GABAA and glycine receptors to central sensitization: disinhibition and touch-evoked allodynia in the spinal cord. J Neurophysiol 72: 169–179, 1994. [DOI] [PubMed] [Google Scholar]
- Smits H, van Kleef M, Joosten EA. Spinal cord stimulation of dorsal columns in a rat model of neuropathic pain: Evidence for a segmental spinal mechanism of pain relief. Pain 153: 177–183, 2012. [DOI] [PubMed] [Google Scholar]
- Song Z, Ansah O, Meyerson B, Pertovaara A, Linderoth B. Exploration of supraspinal mechanisms in effects of spinal cord stimulation: role of the locus coeruleus. Neuroscience 253: 426–434, 2013. [DOI] [PubMed] [Google Scholar]
- Sorkin LS, Puig S, Jones DL. Spinal bicuculline produces hypersensitivity of dorsal horn neurons: effects of excitatory amino acid antagonists. Pain 77: 181–190, 1998. [DOI] [PubMed] [Google Scholar]
- Stiller CO, Cui JG, O'Connor WT, Brodin E, Meyerson BA, Linderoth B. Release of gamma-aminobutyric acid in the dorsal horn and suppression of tactile allodynia by spinal cord stimulation in mononeuropathic rats. Neurosurgery 39: 367–367, 1996. [DOI] [PubMed] [Google Scholar]
- Takazawa T, MacDermott AB. Glycinergic and GABAergic tonic inhibition fine tune inhibitory control in regionally distinct subpopulations of dorsal horn neurons. J Physiol 588: 2571–2587, 2010a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takazawa T, MacDermott AB. Synaptic pathways and inhibitory gates in the spinal cord dorsal horn. Ann NY Acad Sci 1198: 153–158, 2010b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor RS, Desai MJ, Rigoard P, Taylor RJ. Predictors of pain relief following spinal cord stimulation in chronic back and leg pain and failed back surgery syndrome: a systematic review and meta-regression analysis. Pain Pract 14: 489–505, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todd AJ. Neuronal circuitry for pain processing in the dorsal horn. Nat Rev Neurosci 11: 823–836, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torsney C, MacDermott AB. Disinhibition opens the gate to pathological pain signaling in superficial neurokinin 1 receptor-expressing neurons in rat spinal cord. J Neurosci 26: 1833–1843, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wall PD. The Gate Control Theory of pain mechanisms: a re-examination and re-statement. Brain 101: 1–18, 1978. [DOI] [PubMed] [Google Scholar]
- Xu W, Russo GS, Hashimoto T, Zhang J, Vitek JL. Subthalamic nucleus stimulation modulates thalamic neuronal activity. J Neurosci 28: 11916–11924, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yakhnitsa V, Linderoth B, Meyerson BA. Spinal cord stimulation attenuates dorsal horn neuronal hyperexcitability in a rat model of mononeuropathy. Pain 79: 223–233, 1999. [DOI] [PubMed] [Google Scholar]
- Yaksh TL. Behavioral and autonomic correlates of the tactile evoked allodynia produced by spinal glycine inhibition: effects of modulatory receptor systems and excitatory amino acid antagonists. Pain 37: 111–123, 1989. [DOI] [PubMed] [Google Scholar]
- You HJ, Dahl Morch C, Chen J, Arendt-Nielsen L. Simultaneous recordings of wind-up of paired spinal dorsal horn nociceptive neuron and nociceptive flexion reflex in rats. Brain Res 960: 235–245, 2003. [DOI] [PubMed] [Google Scholar]
- Zhang TC, Janik JJ, Grill WM. Mechanisms and models of spinal cord stimulation for the treatment of neuropathic pain. Brain Res 1569: 19–31, 2014a. [DOI] [PubMed] [Google Scholar]
- Zhang TC, Janik JJ, Grill WM. Modeling the effects of spinal cord stimulation on wide dynamic range dorsal horn neurons: influence of stimulation frequency and GABAergic inhibition. J Neurophysiol 112: 552–567, 2014b. [DOI] [PubMed] [Google Scholar]
- Zheng J, Lu Y, Perl ER. Inhibitory neurones of the spinal substantia gelatinosa mediate interaction of signals from primary afferents. J Physiol 588: 2065–2075, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]