

Abstract
Genetic lesions and other regulatory events lead to silencing of the 13q14 locus in a majority of chronic lymphocytic leukemia (CLL) patients. This locus encodes a pair of critical pro-apoptotic microRNAs, miR-15a/16-1. Decreased levels of miR-15a/16-1 are critical for the increased survival exhibited by CLL cells. Similarly, in a de novo murine model of CLL, the NZB strain, germline-encoded regulation of the syntenic region resulted in decreased miR-15a/16-1. In this paper we have identified additional molecular mechanisms regulating miR-15a/16-1 levels and shown that the transcription factor BSAP (B cell Specific Activator Protein) directly interacts with Dleu2, the host gene containing the mir-15a/16-1 loci and via negative regulation of the Dleu2 promoter results in repression of mir-15a/16 expression. CLL patient B cell expression levels of BSAP were increased compared to control sources of B cells. With the use of siRNA mediated repression, the levels of BSAP were decreased in vitro in the NZB derived malignant B1 cell line, LNC, and in ex vivo CLL patient PBMC. BSAP knockdown led to an increase in the expression of miR-15a/16-1 and an increase in apoptosis and a cell cycle arrest in both the cell line and patient PBMC. Moreover, using Dleu2 promoter analysis by chromatin immunoprecipitation (ChIP) assay we have shown that BSAP directly interacts with the Dleu2 promoter. Derepression of the Dleu2 promoter via inhibition of histone deacetylation combined with BSAP knockdown increased miR-15a/16 expression and increased malignant B cell death. In summary, therapy targeting enhanced host gene Dleu2 transcription may augment CLL therapy.
Keywords: miR-15a/16-1, BSAP, HDAC inhibitor, CLL, NZB
Introduction
CLL is an age-associated B cell malignancy characterized by the accumulation of hyper-diploid B-1 cells in the bone marrow, spleen and blood [1]. It is the most common lymphoid malignancy in the Western hemisphere. Since its first documented diagnosis more than 150 years ago, the etiology of CLL is largely unknown and it remains incurable with current therapy [2]. FCR therapy - combination of Fludarabine, Cyclophosphamide, and Rituximab - is the new gold standard for CLL therapy [3]. Although the response rate to therapy is higher as compared to other cancers, almost all patients relapse due to persistence of minimal residual disease [4, 5]. Hence, novel treatment strategies need to be developed. Modulating microRNA expression is one such promising but under-explored therapeutic area [6]. We have previously shown that miR-15a/16-1 upregulation is one such promising therapeutic strategy [7].
13q14 (region that encodes miR-15a/16-1 in humans) deletion is the most common chromosomal abnormality in CLL, occurring in 50–60% of patients [8]. It is believed to encode critical tumor suppressor genes since it is frequently deleted or silenced in various other malignancies like prostate cancer, mantle cell lymphoma, and multiple myeloma [9–11]. Detailed cytogenetic analysis has revealed the presence of a 130kb Minimal Deleted Region (MDR) centromeric to the marker D13S272 that contains several candidate tumor suppressor genes like Dleu1, Dleu2, Dleu5 and Dleu7 [12, 13]. However, currently only Dleu2 (host gene of miR-15a/16-1) and Dleu7 have been demonstrated to have tumor suppressive functions in CLL [14, 15].
MicroRNAs are often located in intronic regions within host genes which can be both coding and non-coding host genes [16]. mir-15a/16-1 is encoded within an intronic region of the non-coding Dleu2 gene in both human and mouse and is transcribed off the Dleu2 promoter. A point mutation (in several CLL patients and NZB mice – de novo mouse model of CLL) and a point deletion (in NZB mice) in the 3′ flanking region of mir-16-1 was discovered and was associated with 50% reduction in expression of mature miR-15a/16-1 in patients as well as NZB mice and LNC cell line (NZB derived mouse B-CLL line) [17–19]. Correcting the reduced miR-15a/16-1 level gives rise to growth inhibitory effect [20]. In order to develop strategies to modulate miR-15a/16-1 levels, it is imperative to understand the molecular mechanisms that control its expression.
BSAP, encoded by the pax5 gene, acts as a transcription factor and contains a DNA binding domain and recently it has been shown that BSAP negatively regulates Dleu2, the host gene of mir-15a/16-1 in mouse lymphoma cells [21]. BSAP is expressed at the pro-B cell stage and is maintained until the plasma cell stage is reached [22]. BSAP can function either as an oncogene or a tumor suppressor depending on the cell type [23]. BSAP can result in increased or decreased gene expression and this can be regulated by additional proteins that BSAP is capable of interacting with via its protein-binding domain. BSAP overexpression generally confers proliferative phenotype in lymphoid malignancies especially B-ALL [24, 25]. In light of this background, we explored the BSAP-Dleu2 regulation in mouse and human CLL cells. It has become increasingly clear that combination therapies are much more effective at fighting cancer {Reviewed in [26]}. Hence we also report herein the combined effect of BSAP knockdown and HDAC inhibition (HDAC activity is increased in CLL) on miR-15a/16-1 levels and malignant cell death.
Results
1) BSAP levels are increased and inversely correlate with miR-15a/16 levels in B-1 malignant cells from CLL patient PBMC
PBMC from untreated CLL or age-matched normal controls were stained for surface expression of CD19 and CD5 and intracellular levels of BSAP. Cells were gated on CD19+ (B cell gate) and the mean fluorescence intensity (MFI) of BSAP determined (Fig 1A). The BSAP levels in a non-B cell source, patient T cells (CD3+, CD19−), is shown for comparison. The CLL B cells demonstrated increased expression of BSAP when compared to non-CLL sources. However, all sources of B cells had increased expression of BSAP relative to their T cell population. In addition, the CLL B cells were sorted into two different B-1 populations, CD19+CD5+ BSAPhi and CD19+CD5+BSAPlo (Fig. 1B). RNA was obtained from the sorted populations and analyzed by PCR for the expression of miR-15a. The B-1 cells with high BSAP had reduced levels of miR-15a relative to the expression in B-1 cells with low expression of BSAP (Fig. 1B). Similarly, since miR-15a/16-1 targets Bcl-2, the expression of Bcl-2 was low in the BSAP low expressing CLL cells suggesting that the resultant high levels of miR-15a/16 permits these cells to readily undergo apoptosis. Indeed the BSAP low cells are the minor population of B-1 cells in the CLL patient. Hence, we hypothesized that by knocking down BSAP via siBSAP, miR15a/16-1 levels will be increased, and their target, Bcl-2 decreased which would lead to the induction of apoptosis.
Fig. 1. BSAP Levels are Elevated in CLL B cells and Inversely Correlate with miR-15a Levels.
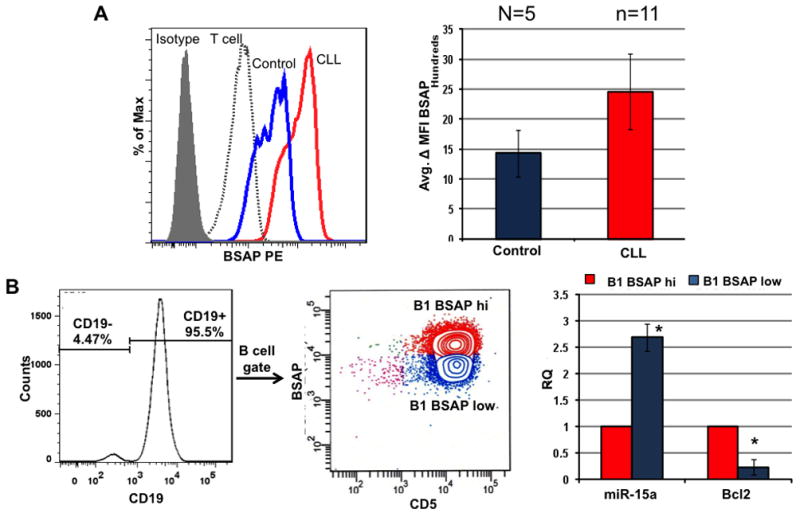
A) Representative histogram overlay of intracellular BSAP levels in PBMC from normal controls (n=5) or CLL (n=11). Control CD19+ (B cells) in blue are decreased relative to CLL B cells (red). For comparison, CLL patient T cells (CD3+ dashed histogram) and the isotype (grey shaded histogram) are shown. The average delta MFI ± SD (change in MFI relative to isotype) is shown in the graph to the right. B) CLL B cells were gated on CD19+ and sorted into CD5+BSAPhi and CD5+ BSAPlow populations. The mean expression levels of miR-15a and Bcl2 in the sorted populations, measured using 100 cell PCR, is shown in the graph.* p<0.05, n=11, two tailed paired Student’s t-test.
2) Negative regulation of miR-15a/16-1 by BSAP can be exploited for CLL therapy
CLL is a very heterogeneous disease but downregulation of miR-15a/16-1 remains the single most commonly occurring pathological event in more than 70% of CLL patients [27]. Hence, we hypothesized that by attenuating the BSAP mediated negative regulation of the promoter of the host gene for miR-15a/16-1; we can increase miR-15a/16-1 levels in ex vivo treated patient PBMC. Total PBMC isolated from patient blood were used for the BSAP knockdown studies instead of purified B1 cells since more than 90% of the PBMC were malignant CD19+CD5+ B1 cells (Fig. 2A). BSAP expression was knocked down by about 25% using siRNA-BSAP as compared to the Neg.CTRL siRNA (Fig. 2D). The reduced expression of BSAP translated into a significant increase in the expression of miR-15a/16-1 (Fig. 2B). Different patients exhibited different kinetics of BSAP knockdown and hence the peak reduction in BSAP is shown. BSAP positively regulates CD19 expression and post siRNA-BSAP treatment, CD19 expression was reduced, further validating the success of the BSAP knockdown (Fig. 2E). Since, the CLL patient PBMC are not actively cycling, increase in the percentage of apoptotic cells was used as a read-out for biologically significant increase in miR-15a/16-1 expression, instead of cell cycle analysis. The percentage of Annexin V positive cells (apoptotic cells) was significantly higher in the siRNA-BSAP treated group as compared to the Neg.CTRL treated group (Fig. 1F), indicating that reduction in BSAP induced by siRNA-BSAP increased both the level of miR-15a/16-1 and the amount of apoptosis.
Fig. 2. BSAP Knockdown in ex vivo CLL Patient PBMC Increases miR-15a/16-1 and Apoptosis.
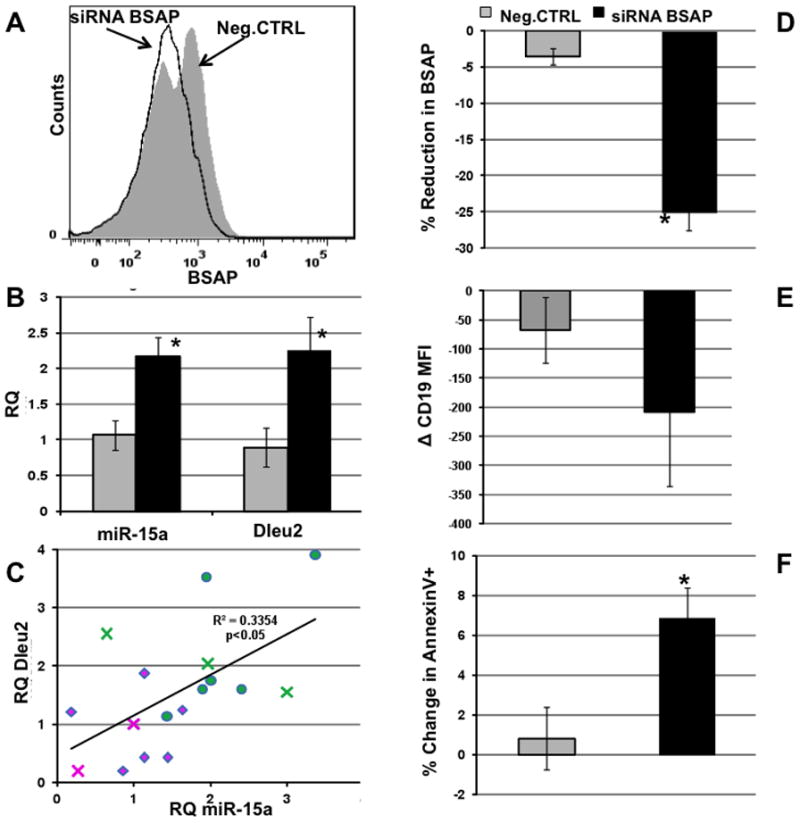
A) Representative histogram overlay of BSAP expression in CD19+CD5+ cells from PBMC of CLL patients. Neg.CTRL (grey filled) and siRNA BSAP (black open) treated cells 24h post transfection. B) Mean peak RQ PCR ± SD of miR-15a and Dleu2 post siRNA BSAP or Negative control treatment. C) Regression analysis of levels of Dleu2 mRNA levels versus mR15a. diamond = neg siRNA, circle + siBSAP, cross = sorted CD5+ B cells D) Average reduction in BSAP expression following treatment with Neg.CTRL or siRNA BSAP transfection. %Reduction in BSAP = (MFI treated-MFI mock)*100/MFI Mock. BSAP, 24hr post transfection.. E) Mean delta CD19 MFI as compared to mock treated cells in Neg.CTRL (grey) and siRNA BSAP (black). F) Mean % change in apoptotic cells in Neg.CTRL (grey) and siRNA BSAP (black) % change = (%AnnexinV+ treated − %AnnexinV+ Mock). *p<0.05, n=7.
3) BSAP regulates miR-15a/16-1 expression at the level of host gene transcription
Many microRNAs are encoded within the introns of other bigger coding or non-coding genes, with or without their own promoters. In the latter case, the expression of the encoded miR would depend on the promoter status of the host gene. To confirm that BSAP regulates Dleu2 transcription (host gene for mir-15a/16-1) the Dleu2 mRNA level was measured in the CLL patient PBMC following siRNA-BSAP. A significant increase was observed in the expression of Dleu2 following BSAP knockdown (Fig. 2B). The increase in Dleu2 was found to be positively correlated to the level of miR-15a/16-1 (Fig. 2C). Hence, we conclude that BSAP inhibits mir-15a/16-1 expression by inhibiting the transcription of its host gene, Dleu2. This was further verified by in situ FISH analysis at a single cell level to simultaneously quantitate the amount of Pax5 mRNA (which encodes the BSAP protein) and the amount of Dleu2 mRNA. In non-CLL B cells, the amount of PAX5 (BSAP) was decreased relative to the CLL B cells (Fig 3). In addition, the amount of PAX5 expression was inversely related to the amount of Dleu2 on a single cell level (Fig 3), with CLL B cells having low levels of miR15a/16 expression and high levels of BSAP and non-CLL B cells expressing lower levels of BSAP but higher levels of miR15a.
Fig. 3. In situ RNA FISH Analysis of Dleu2 and Pax5 mRNA levels.

Cell were grown on poly L-lysine coated coverslips and hybridized with single molecule FISH probes. Dleu2 probes labeled were with TMR and Pax5 probes were labeled with Alexa 594. The z-stacks obtained from each fluorescence channel were merged and coded to represent Pax5 as red and Dleu2 as green. Each spot represent single mRNA molecule [65]. The right panel is the merge of both channels and the chart represents the mean values from multiple analysis ± SEM.
4) BSAP interacts with Dleu2 promoter
It is currently unknown whether BSAP regulates Dleu2 promoter directly or indirectly. Promoter analysis using CONSITE failed to show any canonical BSAP binding sites in the human Dleu2 promoter. However, we cannot rule out the possibility that BSAP might be present in a complex with some of its binding partners at the Dleu2 promoter. Binding sites for some of the proteins that BSAP has been known to interact with were found in the human Dleu2 promoter (c-Myb, AML1, E2F). We performed a ChIP assay on Daudi cells to assess BSAP-Dleu2 promoter interactions. Daudi is a B lineage cell line (Burkitt’s Lymphoma) and has BSAP expression [28]. Similar to the positive control CD19 promoter, the Dleu2 promoter was enriched in the BSAP pulldown. No enrichment of the negative control Kras promoter was observed (Fig. 4A, B). The ChIP assay was performed employing 4 CLL PBMC samples in 4 independent pulldown experiments (Fig 4C). The Dleu2 promoter region was also enriched in the CLL samples and was similar to a known promoter, which encodes a BSAP binding site, the CD19 promoter. In contrast, the Kras promoter, which does not encode a BSAP binding site, was not pulldowned in CLL samples (Fig 4C). This data suggests that a BSAP binding site is present at the Dleu2 promoter in human B cells. However, a canonical BSAP binding site was found at +676 position in the mouse Dleu2 gene. A ChIP assay was performed on murine B cells, the LNC cells, to assess BSAP-Dleu2 promoter interactions. Since, we knew the position of the putative BSAP binding site in mouse Dleu2; we were able to design a ChIP assay having an internal control in addition to the GAPDH control as shown in Fig. 4D. Indeed, the Dleu2 promoter fragments were enriched in the BSAP pulldown and the amount of enrichment was inversely proportional to the distance from the BSAP binding site (Fig. 4E). The upstream sequences (U) were not as enriched by the BSAP pulldown as were the downstream sequences (D) which contained the BSAP binding site. The presence in the BSAP pulldown of the Dleu2 U region may be due to DNA fragments which contain both the D and U region. The human data indicating BSAP interaction with the Dleu2 promoter were validated in the mouse system which has a canonical BSAP binding site in its promoter.
Fig. 4. Dleu2 Promoter Occupancy by BSAP in Human and Mouse.
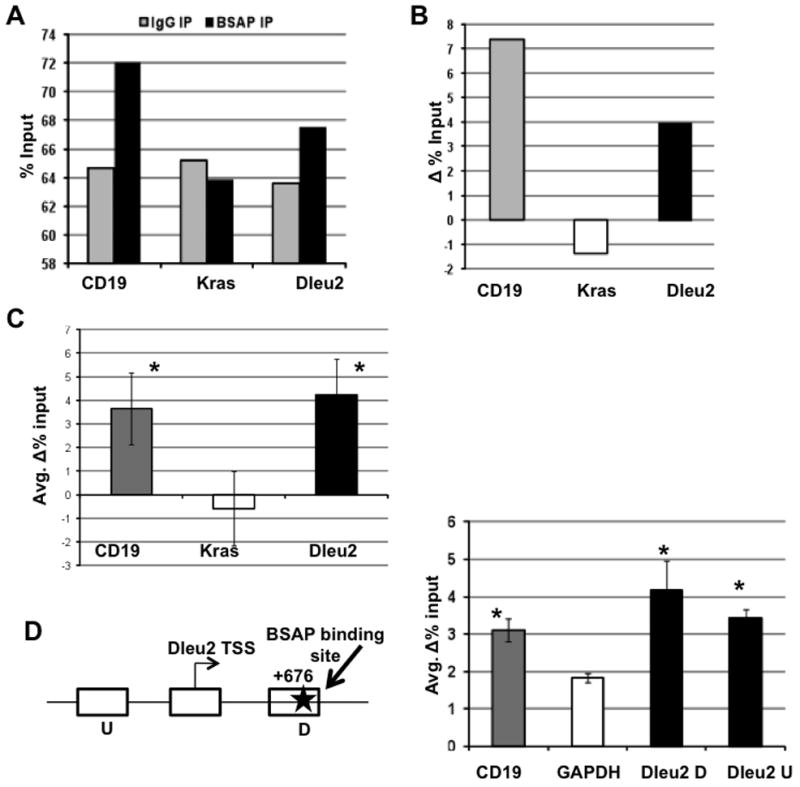
A) Representative data of promoter occupancy in human B cell line (Daudi) following immunoprecipitation with IgG (grey) or BSAP (black) antibody. B) Delta % Input = (% Input BSAP IP − % Input IgG), indicates promoter enrichment over pull down with non-specific IgG antibody in Daudi cells. C) Cumulative data of promoter enrichment ChIP experiments in CLL PBMC, n=4, D) Promoter analysis in mouse, schematic of the Dleu2 promoter in mouse, the transcription start site (Dleu2 TSS), the flanking upstream (U) and downstream (D) regions are indicated and the total region is approximately 1kb. The BSAP binding site is shown as a star. Chart is enrichment of CD19, GAPDH, Dleu2 D and Dleu2 U fragments in LNC, the NZB mouse CLL cell line (mean values of 3 independent experiments). *p<0.05, Student’s t-test. Error bars indicate ± SEM.
5) BSAP knockdown leads to cell cycle arrest and apoptosis in NZB
We and others have previously shown that miR-15a/16-1 expression is reduced by almost 50% in CLL (in both patients and NZB mice) [20]. Using microarray based expression analysis, negative correlation was shown between BSAP and Dleu2 (host gene of miR-15a/16-1) levels in Myc5 cells [21]. Myc5 is a B-lymphoma cell line derived by transducing p53 null bone marrow cells with c-Myc retrovirus. These cells can oscillate between B cell and macrophage lineages based on the culture conditions and BSAP levels in vitro [29]. On the contrary, LNC is a natural malignant B-1 cell line derived from the spontaneously occurring murine model of human CLL and has constitutively very high level of BSAP expression and low miR-15a/16-1 expression (Fig. S1). In light of the above disparities in the two systems, we wanted to confirm whether BSAP could negatively regulate miR-15a/16-1 levels in our system.
The NZB derived CLL cell line LNC was transfected with either 3ug non-targeting negative control siRNA (Neg.CTRL) or with 3ug siRNA BSAP (siRNA-BSAP) using Amaxa Nucleofection, in order to transiently knockdown BSAP. The expression of BSAP was reduced as early as 12hr post transfection and the suppression began to diminish after 48hr. Flow cytometric analysis via histogram overlays of BSAP levels on a per cell basis showed a decrease in BSAP in siRNA treated cells compared to Neg.CTRL treated cells as early as 12hr post-transfection (Fig. 5A). The average BSAP MFI at different time-points post-transfection is shown in Fig. 5B and was found to be lowest at 24hr post siRNA-BSAP treatment when compared to the Neg.CTRL at that time-point (p<0.05). BSAP protein was reduced by 53% at 24hr as compared to the Neg.CTRL. The miR-15a levels were measured in cells with successful knocked down BSAP to determine the potential causal relationship between BSAP and miR-15a/16-1 expression in our system. miR-15a level was measured using TaqMan MicroRNA Assays and an approximately two-fold increase in miR-15a levels was observed in the siRNA-BSAP treated cells at 24hr (p<0.05) (Fig. 5C). Interestingly, the increase in miR-15a expression corresponded with the peak reduction in BSAP suggesting a strong influence of BSAP on the expression of the microRNA. LNC cells mimic aggressive CLL and are very rapidly dividing. We and others have shown that an increase in miR-15a/16-1 leads to cell cycle arrest and reduced proliferation [30–32]. Initially, we have shown that reducing BSAP levels results in an increase in miR-15a/16-1 expression. To test the biological significance of this, the increased miR-15a/16-1 should result in decreased downstream targets like cyclin D leading to cell cycle arrest. As compared to the Neg.CTRL treated cells which have baseline level of BSAP and miR-15a/16-1, the siRNA-BSAP treated cells exhibited a significant increase in percentage of cells in G1 and a reduction in the percentage of cells in S phase (Fig. 5D and 5E, p<0.05). Moreover, we also observed a decrease in the expression of Cyclin D1 in the siRNA-BSAP treated cells as compared to the Neg.CTRL treated cells (Fig. 5F) indicating that the cell cycle arrest is a direct consequence of increased miR-15a/16-1 since it has been shown to be a key regulator of Cyclin D1.
Fig. 5. BSAP Knockdown Leads to Increased miR-15a/16-1 and Cell Cycle Arrest in vitro.
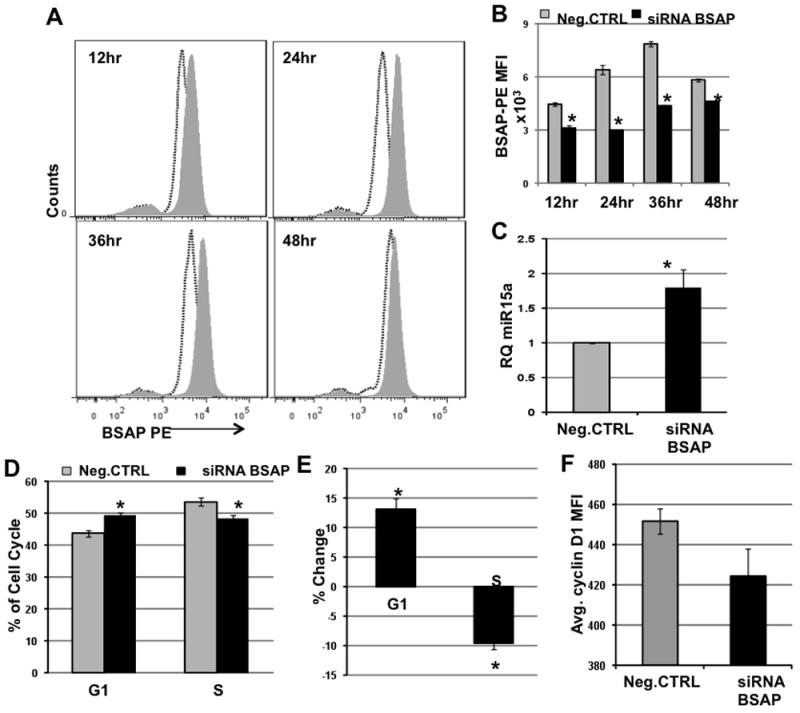
3ug siRNA BSAP or non-targeting siRNA (Neg.CTRL) was transiently transfected into the murine NZB CLL cell line LNC and analyzed at the indicated time-points. A) Representative histogram overlay of cells transfected with either siRNA BSAP (black dotted line) or with Neg.CTRL (Grey filled) and stained with BSAP-PE antibody. B) The BSAP MFI ± SEM at indicated time-points post transfection with siRNA BSAP (Black bar) or with Neg.CTRL (Grey Bar). n=3, *p<0.05, C) Average miR-15a levels 24hr post-transfection, n=6, *p<0.05 D) Percentage of cells in the G1 and S phase of the cell cycle at 36hr post-transfection. n=7, *p<0.01. E) Percent change in cells in G1 phase and S phase, 36hr post-transfection. Percent Change = [(%G1 or %S in siRNA BSAP group − %G1 or %S in Neg.CTRL group)/(%G1 or %S in Neg.CTRL group)]*100. n=7, *p<0.01, large effect size. F) Decrease in the CyclinD1 MFI in the siRNA BSAP group in relation to the Neg.CTRL group, 24hr post transfection. n=3. Statistics employed: Two-tailed paired Student’s t test.
6) Combination treatment with HDAC Inhibitor
Next we wanted to assess whether the anti-proliferative effect of BSAP knockdown can be enhanced by using a HDAC inhibitor. LNC cells were treated with siRNA-BSAP and the HDAC inhibitor SAHA (clinically known as Vorinostat). Dose response curves were calculated for SAHA (data not shown) and a sub-optimal dose of 0.8uM was selected for the combination treatment. An additive effect was observed on the increase in the %G1 (Fig. 6A) and decrease in %S (Fig. 6B) as compared to the untreated cells. However, a synergistic effect was observed on the percentage of cells undergoing apoptosis when treated with the siRNA BSAP and SAHA together (Fig. 6C). Additionally we observed a significant increase in miR-15a/16-1 expression following treatment (Fig. 6D). In order to confirm that the observed cell cycle and apoptosis effects are mediated by increased miR-15a/16-1, LNC cells were treated with SAHA + siRNA BSAP + antagomiR to miR-15a and miR-16-1. The antagomiR lead to a stable reduction in miR-15a/16-1 up to 48hrs post tranfection (Fig. 6E). This gave rise to a partial rescue from increased malignant cell death (Fig. 6F). In conclusion, these findings indicate that combination of HDAC inhibition and BSAP knockdown leads to a significantly higher increase in malignant cell death, in part by increasing the level of miR-15a/16-1.
Fig. 6. HDAC Inhibitor Enhances the Apoptotic Effect of BSAP Knockdown.
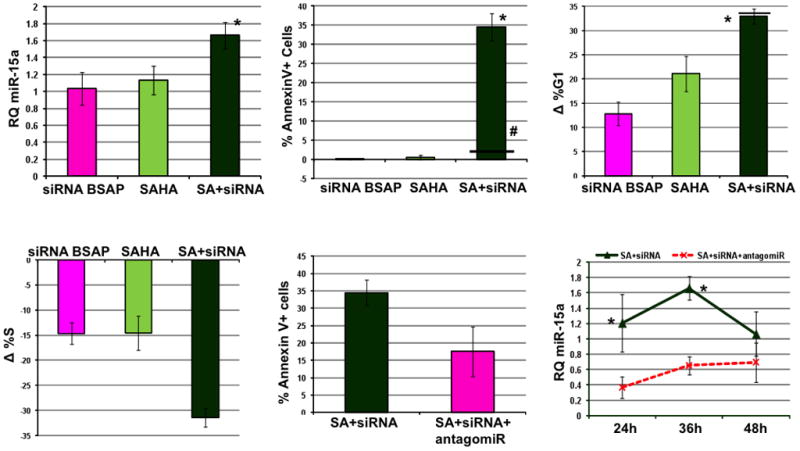
LNC cells were treated with 0.8uM SAHA alone or in combination with 2.5uM siRNA-BSAP. The delta values were calculated in relation to untreated cells. A) RQ miR-15a 36hrs post treatment with siRNA BSAP, SAHA and SAHA+siRNA BSAP (SA+siRNA). B) Percentage of AnnexinV+ cells after indicated treatments. *p<0.05, combination versus single agent treatment, horizontal line (−) indicates predicted value if effect of siRNA BSAP and SAHA were additive, #p<0.05, observed increase in apoptosis significantly different from the predicted additive value for the combination treatment. C) Change in percentage of cells in G1 phase after indicated treatments. D) Change in percentage of cells in S phase after indicated treatments. E) Bar graph showing average % Annexin V+ cells in SAHA+siRNA BSAP (SA+siRNA) and SAHA+siRNA BSAP+ antgomiR to miR-15a/16-1 (SA+siRNA+antagomiR) groups at 24hr. F) Line graph showing the level of miR-15a in SA+siRNA (solid line) and SA+siRNA+antagomiR (dashed line) at 24h, 36h and 48h post treatment. *Significant difference in the miR-15a levels between the two groups at that time-point, paired Student’s t test, n>3, error bars indicate ± SEM.
DISCUSSION
In this study we found that in CLL patients, B cells constitutively express higher levels of BSAP than do control sources of B cells. However, within a CLL malignant clone, a minority of the B-1 cells had relatively low levels of BSAP. Analysis of the two types of B-1 cells demonstrated that increased expression of the microRNAs miR-15a/16-1 was found in the patient B-1 subpopulation with the higher expression of BSAP (minor population). To further dissect the role of BSAP in the regulation of the levels of miR-15a/16-1, addition of siRNA pools with antisense regions to the PAX5 gene (which encodes BSAP) resulted in an increase in miR-15a/16-1 levels. Together these studies indicate that BSAP is a negative regulator of miR-15a/16-1. The BSAP mediated regulation of miR-15a/16-1 levels is occurring at the level of the host gene transcription as seen by the significant positive correlation between Dleu2 and miR-15a/16-1 levels that is dependent on the BSAP levels and independent of downstream mutations in that loci. This was also observed at the level of single cell RNA FISH analysis. We have also shown that BSAP interacts with the Dleu2 promoter (direct interaction in mice and indirect interaction in humans). This finding is very interesting given that the human Dleu2 promoter lacks a canonical BSAP binding site. We speculate that, BSAP interacts with another protein that binds to the Dleu2 promoter directly and this complex is responsible for the repression of the Dleu2 promoter. The present report concentrated on Dleu2 promoter repression and other studies have also indicated that the Dleu2 promoter regulates miR15a/16 levels [33]. However, a potential promoter region located near the mir15a/16-1 loci within Dleu2 which has myb binding sites has been reported and myb interacts with BSAP [34]. Further studies need to be performed to identify binding partners of BSAP and additional promoter regulation of miR15a/16.
Earlier studies focused mainly on the role of BSAP in the B lineage development program [35]. This study highlights the role of BSAP in cancer cell survival via its regulation of an important pro-apoptotic microRNA cluster, mir-15a/16-1. BSAP appears to act as an oncogene in CLL and play an important role in down-regulating mir-15a/16-1, a known tumor suppressor in CLL. Other groups have also demonstrated the role of BSAP in regulating apoptosis [36]. Previously, we demonstrated that inhibition of BSAP lead to decreased proliferation and apoptosis in malignant B-1 cells from the NZB murine model of CLL [37] [38] [39]. In the present study we have shown that knocking down BSAP leads to malignant cell death not only in our murine CLL cell line but also in ex vivo patient PBMC via upregulation of miR15a/16 levels. We propose that targeting BSAP using siRNA or small molecule inhibitors could serve as a novel therapeutic strategy for CLL. The role of BSAP in tumorigenesis is highly cell type specific, in that it can act both as an oncogene (non-Hodgkins lymphoma, neuroblastoma) or as tumor suppressor (multiple myeloma) [40–43]. Targeting BSAP has already been shown to have therapeutic effects in other types of cancers like small cell lung cancer [44]. BSAP has been shown to be a positive regulator of CD19 and indirectly to control c-myc levels via regulation of CD19 [45]. In the present studies, BSAP binding to the promoter region of CD19 in the ChIP analysis was verified and the additional binding to the promoter of Dleu2 (and miR15a/16-1) determined. The elevated levels of BSAP found in CLL malignant B cell clones may have multiple effects in addition to negative regulation of miR15a/16 via repression of Dleu2 including elevation of c-myc via upregulation of CD19.
Because the bicistronic miR-15a/16-1 is a tumor suppressor miRNA [46], a wide variety of malignancies involve the repression of this microRNA cluster. There are different mechanisms by which this repression occurs. Several other studies have also found that the Dleu2 host gene promoter is repressed. Recently, c-Myc has been shown to repress the Dleu2 promoter via recruitment of histone deacetylase 3 (HDAC3) [47]. HDAC activity is increased in a variety of cancers including CLL [48–50]. Histone deacetylases have emerged as attractive targets in the treatment of both solid and hematological malignancies [51–53]. Several groups have reported the efficacy of HDAC inhibitors in inducing CLL cell death in vitro and in clinical trials [54, 55]. In addition to inducing direct cell death, HDAC inhibition also increases the immunogenicity of CLL cells, thereby facilitating anti-tumor immune response [56, 57]. HDAC has also been shown to repress the Dleu2 promoter in CLL malignant B-1 [58]. Our results support these previous findings. We have shown that similar to BSAP mediated Dleu2 repression, HDAC mediated repression is also reversible and it opens up avenues for therapeutic intervention. Based on the data presented in this paper we propose that BSAP and histone deacetylases co-operate to bring about repression of the Dleu2 promoter in CLL. This study is critical because for the first time it simultaneously addresses the interaction between three key players (13q14 locus, BSAP and HDAC) involved in CLL pathogenesis. Out of the many genetic alterations found in CLL, the frequency of only 13q14 deletions is significantly elevated in MBL (Monoclonal B-cell Lymphocytosis), the precursor stage of CLL [59]. In addition, epigenetic silencing of 13q14 has been found in CLL [60] This finding indicates that 13q14 silencing via its deletion or epigenetic modification is one of the first events leading to CD19+CD5+ B cell expansion and understanding the regulation of this locus will be instrumental in delineating CLL etiology. A recent paper by Laurie C et al that analyzed clonal mosaicism in 50,000 subjects further underscores the importance of the 13q14 region in CLL [61]. In this study the authors discovered that 13q deletions were over-represented in normal subjects with age and its presence increased the chances of a future CLL diagnosis.
In summary we have shown that in CLL, promoter regulation by a transcriptional repression via, BSAP and HDAC co-coordinately repress Dleu2, the host gene of miR-15a/16-1 and targeting this loop leads to malignant cell death via increase in mature miR-15a/16-1 expression (Fig. S2).
Materials and Methods
Patient Samples and Cell Lines
4–5ml of blood was collected in EDTA tubes from untreated CLL patients after obtaining informed consent in accordance with UMDNJ human subjects IRB. Peripheral blood mononuclear cells (PBMC) were isolated from patient blood using Ficoll-Hypaque solution according to manufacturer’s instructions (StemCell Technologies, Inc; Vancouver, Canada). Human Burkitt’s lymphoma cell line Daudi (ATCC No. CCL-213) was employed. Additionally the NZB (murine model of human CLL) derived malignant B-1 cell line LNC was also employed [62]. Both cell lines were maintained in RPMI 1640 supplemented with 10% FBS, 1% Sodium pyruvate, 1% Penicillin-Streptomycin at 37°C and 5% CO2.
siRNA Nucleofection
2.5×106 PBMC were nucleofected with 3ug of ON-TARGETplus SMARTpool - siRNA targeting the pax5 gene that encodes the BSAP protein (referred to as siRNA-BSAP from here on) or ON-TARGETplus Non-targeting Pool siRNA (neg CTRL) as a control (Dharmacon, Lafayette, CO) or nothing (mock) using human B cell nucleofector kit program with the AMAXA instrument (Lonza, Switzerland). Similarly 2.5×106 LNC cells were nucleofected with 3ug of ON-TARGETplus SMARTpool – Mouse siRNA BSAP or ON-TARGETplus Non-targeting Pool siRNA as a control (Dharmacon) or nothing (mock) using Cell Line Nucleofector Kit T, program G-016 (Lonza, Switzerland). % Reduction in BSAP protein was calculated as [(siRNA BSAP MFI – Neg.CTRL MFI)/Neg.CTRL MFI]*100. The cells were harvested at different time points (24, 36, 48, 72 and 96hrs) for further analysis. Wherever indicated, 1uM each of antagomiRs to miR-15a and miR-16-1 (Dharmacon) were also nucleofected.
Surface and Intracellular Flow Cytometry
Approximately 0.5×106 cells were stained with indicated surface antibodies for 25min at 4°C. Cells were then fixed and permeablized using BD Cytofix/Cytoperm kit (BD Biosciences, Franklin Lakes, NJ) according to manufacturer’s instructions and stained with the indicated intracellular antibodies for 30min. Antibodies for surface markers were anti-human CD19-FITC (BD Biosciences), anti-human CD5-PE.Cy7 (BD Biosciences), and for intracellular markers were anti-human/mouse Pax5-PE (eBioscience, San Diego, CA) and anti-mouse Cyclin D1-AF647 (Santa Cruz Biotechnology Inc, Santa Cruz CA). The stained cells were acquired on BD LSR II (BD Biosciences) and analyzed using the FloJo Software (TreeStar Inc, Ashland, OR).
Sorting of B-1 Cells and 100 Cell PCR
CLL patients PBMC were stained with CD19-FITC, CD5-PE.Cy7 and BSAP-PE antibodies. The CD19+ population in the lymphoid gate was further sorted into CD5+BSAPhi (B1 BSAP hi) and CD5+BSAPlow (B1 BSAP low) using the BD FACS Aria II cell sorter. Due to the low number of cells obtained after sorting, we performed 100 cell PCR in order to measure the miR-15a/16-1 and Bcl2 levels in the sorted sub-populations. Briefly, for the microRNA measurement, 100 cells in 4.84ul 1X PBS were heat disrupted at 95°C for 10min to release RNA and immediately kept on ice. This extract was then used to prepare cDNA using the TaqMan microRNA RT kit (Applied Biosystems, Carlsbad, CA), followed by real time PCR. Bcl2 levels were measured using the Power SYBR green cells-to-ct kit (Ambion Inc, Austin, TX)
RNA Isolation and Quantitation
Total RNA was extracted using Trizol reagent (Invitrogen, Carlsbad, CA) according to manufacturer’s instructions. microRNA specific cDNA was prepared using the TaqMan MicroRNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA) according to manufacturer’s instructions. The following pre-made TaqMan Assays (Applied Biosystems) were used for real time quantitation – mmu-miR-15a (Assay ID 000389), U6 (Assay ID 001973). Dleu2 transcripts were quantified from random hexamer primed cDNA using TaqMan hsa-Dleu2 assay (Assay ID Hs00863925 m1). microRNA and Dleu2 levels were normalized to U6 (mouse and human) and 18s rRNA (human) respectively.
Cell cycle and Apoptosis Assay
For cell cycle analysis 0.5×106 were washed with 1X PBS and resuspended in 300ul hypotonic PI solution, acquired on a BD Calibur cytometer and analyzed using the ModFit LT Software (Verity Software House, Topsham, ME). For apoptosis quantitation, cells were stained with Annexin V-PE (BD Bioscience) according to manufacturer’s protocol, acquired on BD Calibur IV.
Chromatin Immunoprecipitation (ChIP)
5×106 cells were crosslinked with 0.37% formaldehyde and sonicated to obtain 1kb or 400bp fragments using the S220 sonicator (Covaris Inc, Woburn, MA). The ChIP-ready chromatin was pulled down with either goat anti-human Pax5 (Santa Cruz Biotechnology) or normal goat IgG (Santa Cruz Biotechnology). EpiTect One Day ChIP kit (Qiagen, Valencia, CA) was used. The pulled down DNA was then used for real time SYBR green PCR of Dleu2 promoter using primers described in [63]. CD19 promoter was used as a positive control and Kras promoter was used as the negative control. Similar protocol was followed for the mouse cell line. Dleu2 was primed at two separate sites (with and without the BSAP binding site as an internal control). CD19 was used as a positive control and GAPDH was used as a negative control. Refer supplementary materials (Table S1) for mouse primer sequences.
HDAC Inhibitor Treatment
Suberoylanilide hydroxamic acid (SAHA) (clinically known as Vorinostat [57, 64]) (Selleckchem, Houston, TX) was dissolved in 100% ethanol to obtain a 10mM stock. Final working concentration used was 0.8uM.
RNA Fluorescence In Situ Hybridization (FISH)
Dleu2 and Pax5 RNAs were imaged using single molecule FISH probes as described previously [65]. Briefly, a set of 35 probes was designed to hybridize to each target RNA and was synthesized with a 3′ amino modification from Biosearch Technologies, CA. The individual probes for a given target were pooled in equimolar amounts and then coupled with succinimidyl ester of either TMR (for Dleu2) or Alexa 594 (for Pax5). The coupled fraction was purified using HPLC and the concentration was determined using nanodrop. The coverslips were washed with 1XPBS, fixed in 4% formaldehyde, permeabilized with 70% ethanol and hybridized with the Dleu2 TMR and Pax5 Alexa 594 probes. Hybridization was done overnight at 37C. The cover slips were washed (with 10% formamide in 2X SSC) to remove unbound probes and imaged using Zeiss wide field fluorescence microscope. For each image, z stacks were obtained and merged to get the final image. The image acquisition was done by openLab software and numbers of mRNAs were counted using custom written alogrithms in MATLAB [66].
Statistics
Data was analyzed with a paired student’s T test, unless otherwise specified. p<0.05 was considered significant.
Supplementary Material
Primers used to amplify promoter regions in the mouse BSAP-ChIP assay.
non-NZB = A20, a Balb/c B cell line and NZB = LNC, mouse B-CLL cell line. A) Representative single color histogram overlay of BSAP expression in unstained (solid black, open), non-NZB (grey filled) and NZB (dashed, open). B) Average BSAP MFI in non-NZB (grey bar) and NZB (black bar) cell line. C) RQ miR-15a in non-NZB (grey bar) and NZB (black bar) cell line measured using real time TaqMan microRNA expression assays. * p<0.05, Unpaired student’s t test, n=3.
Left) Dleu2 transcription start site (Dleu2 TSS) is not easily accessible to the transcription machinery because increased HDAC activity in CLL cells leads to reduced acetylation and tightly wound nucleosome conformation; and steric hindrance by promoter bound BSAP. Net effect of the above events is reduced mature miR-15a/16-1 leading to increased anti-apoptotic factors including cyclin D1 and Bcl2. Right) Addition of HDAC inhibitor increases acetylation giving rise to open nucleosome conformation while BSAP inhibitor frees the steric hindrance of Dleu2 promoter. This gives rise to increased transcription of miR-15a/16-1. Malignant cell number decreases due to cell cycle arrest and apoptosis.
Acknowledgments
This work was supported by NSF/FDA/SIR #1238375 and NIH R01CA12926 (ESR). We would like to thank the UMDNJ-NJMS Flow Cytometry Core.
Footnotes
Conflict of Interest Declaration: The authors declare that there is no conflict of interest.
References
- 1.Swerdlow SH. WHO classification of tumours of haematopoietic and lymphoid tissues. Vol. 2. World Health Organization; 2008. [Google Scholar]
- 2.Kalyana-Sundaram S, et al. Expressed pseudogenes in the transcriptional landscape of human cancers. Cell. 2012;149(7):1622–34. doi: 10.1016/j.cell.2012.04.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Montserrat E. Further progress in CLL therapy. Blood. 2008;112(4):924–5. doi: 10.1182/blood-2008-05-155721. [DOI] [PubMed] [Google Scholar]
- 4.Robertson LE, et al. Response assessment in chronic lymphocytic leukemia after fludarabine plus prednisone: clinical, pathologic, immunophenotypic, and molecular analysis. Blood. 1992;80(1):29–36. [PubMed] [Google Scholar]
- 5.Bottcher S, et al. Minimal residual disease quantification is an independent predictor of progression-free and overall survival in chronic lymphocytic leukemia: a multivariate analysis from the randomized GCLLSG CLL8 trial. J Clin Oncol. 2012;30(9):980–8. doi: 10.1200/JCO.2011.36.9348. [DOI] [PubMed] [Google Scholar]
- 6.Kasinski AL, Slack FJ. Epigenetics and genetics. MicroRNAs en route to the clinic: progress in validating and targeting microRNAs for cancer therapy. Nature reviews Cancer. 2011;11(12):849–64. doi: 10.1038/nrc3166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kasar S, et al. Systemic in vivo lentiviral delivery of miR-15a/16 reduces malignancy in the NZB de novo mouse model of chronic lymphocytic leukemia. Genes and immunity. 2012;13(2):109–19. doi: 10.1038/gene.2011.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dohner H, et al. Genomic aberrations and survival in chronic lymphocytic leukemia. N Engl J Med. 2000;343(26):1910–6. doi: 10.1056/NEJM200012283432602. [DOI] [PubMed] [Google Scholar]
- 9.Chang H, et al. Detection of chromosome 13q deletions and IgH translocations in patients with multiple myeloma by FISH: comparison with karyotype analysis. Leuk Lymphoma. 2004;45(5):965–9. doi: 10.1080/10428190310001638832. [DOI] [PubMed] [Google Scholar]
- 10.Chen L, et al. Molecular cytogenetic aberrations in patients with multiple myeloma studied by interphase fluorescence in situ hybridization. Exp Oncol. 2007;29(2):116–20. [PubMed] [Google Scholar]
- 11.Flordal Thelander E, et al. Detailed assessment of copy number alterations revealing homozygous deletions in 1p and 13q in mantle cell lymphoma. Leuk Res. 2007;31(9):1219–30. doi: 10.1016/j.leukres.2006.10.022. [DOI] [PubMed] [Google Scholar]
- 12.Corcoran MM, et al. Detailed molecular delineation of 13q14.3 loss in B-cell chronic lymphocytic leukemia. Blood. 1998;91(4):1382–90. [PubMed] [Google Scholar]
- 13.Migliazza A, et al. Nucleotide sequence, transcription map, and mutation analysis of the 13q14 chromosomal region deleted in B-cell chronic lymphocytic leukemia. Blood. 2001;97(7):2098–104. doi: 10.1182/blood.v97.7.2098. [DOI] [PubMed] [Google Scholar]
- 14.Klein U, et al. The DLEU2/miR-15a/16-1 cluster controls B cell proliferation and its deletion leads to chronic lymphocytic leukemia. Cancer cell. 2010;17(1):28–40. doi: 10.1016/j.ccr.2009.11.019. [DOI] [PubMed] [Google Scholar]
- 15.Palamarchuk A, et al. 13q14 deletions in CLL involve cooperating tumor suppressors. Blood. 2010;115(19):3916–22. doi: 10.1182/blood-2009-10-249367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rodriguez A, et al. Identification of mammalian microRNA host genes and transcription units. Genome Res. 2004;14(10A):1902–10. doi: 10.1101/gr.2722704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Salerno E, et al. The New Zealand black mouse as a model for the development and progression of chronic lymphocytic leukemia. Cytometry Part B, Clinical cytometry. 2010;78(Suppl 1):S98–109. doi: 10.1002/cyto.b.20544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Raveche ES, et al. Abnormal microRNA-16 locus with synteny to human 13q14 linked to CLL in NZB mice. Blood. 2007;109(12):5079–86. doi: 10.1182/blood-2007-02-071225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Calin GA, et al. A MicroRNA signature associated with prognosis and progression in chronic lymphocytic leukemia. N Engl J Med. 2005;353(17):1793–801. doi: 10.1056/NEJMoa050995. [DOI] [PubMed] [Google Scholar]
- 20.Salerno E, et al. Correcting miR-15a/16 genetic defect in New Zealand Black mouse model of CLL enhances drug sensitivity. Molecular cancer therapeutics. 2009;8(9):2684–92. doi: 10.1158/1535-7163.MCT-09-0127. [DOI] [PubMed] [Google Scholar]
- 21.Chung EY, et al. c-Myb oncoprotein is an essential target of the dleu2 tumor suppressor microRNA cluster. Cancer biology & therapy. 2008;7(11):1758–64. doi: 10.4161/cbt.7.11.6722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Medvedovic J, et al. Pax5: a master regulator of B cell development and leukemogenesis. Advances in immunology. 2011;111:179–206. doi: 10.1016/B978-0-12-385991-4.00005-2. [DOI] [PubMed] [Google Scholar]
- 23.O’Brien P, et al. The Pax-5 gene: a pluripotent regulator of B-cell differentiation and cancer disease. Cancer research. 2011;71(24):7345–50. doi: 10.1158/0008-5472.CAN-11-1874. [DOI] [PubMed] [Google Scholar]
- 24.Firtina S, et al. Evaluation of PAX5 gene in the early stages of leukemic B cells in the childhood B cell acute lymphoblastic leukemia. Leukemia research. 2012;36(1):87–92. doi: 10.1016/j.leukres.2011.07.017. [DOI] [PubMed] [Google Scholar]
- 25.Robichaud GA, Perreault JP, Ouellette RJ. Development of an isoform-specific gene suppression system: the study of the human Pax-5B transcriptional element. Nucleic acids research. 2008;36(14):4609–20. doi: 10.1093/nar/gkn432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Humphrey RW, et al. Opportunities and Challenges in the Development of Experimental Drug Combinations for Cancer. Journal of the National Cancer Institute. 2011 doi: 10.1093/jnci/djr246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hanlon K, Rudin CE, Harries LW. Investigating the Targets of MIR-15a and MIR-16-1 in Patients with Chronic Lymphocytic Leukemia (CLL) PLoS ONE. 2009;4(9):e7169. doi: 10.1371/journal.pone.0007169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hamada T, et al. Expression of the PAX5/BSAP transcription factor in haematological tumour cells and further molecular characterization of the t(9;14)(p13;q32) translocation in B-cell non-Hodgkin’s lymphoma. British journal of haematology. 1998;102(3):691–700. doi: 10.1046/j.1365-2141.1998.00842.x. [DOI] [PubMed] [Google Scholar]
- 29.Yu D, et al. Oscillation between B-lymphoid and myeloid lineages in Myc-induced hematopoietic tumors following spontaneous silencing/reactivation of the EBF/Pax5 pathway. Blood. 2003;101(5):1950–5. doi: 10.1182/blood-2002-06-1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Salerno E, et al. Correcting miR-15a/16 genetic defect in New Zealand Black mouse model of CLL enhances drug sensitivity. Molecular Cancer Therapeutics. 2009;8(9):2684–2692. doi: 10.1158/1535-7163.MCT-09-0127. [DOI] [PubMed] [Google Scholar]
- 31.Cimmino A, et al. miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc Natl Acad Sci U S A. 2005;102(39):13944–13949. doi: 10.1073/pnas.0506654102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Calin GA, et al. MiR-15a and miR-16-1 cluster functions in human leukemia. Proceedings of the National Academy of Sciences. 2008;105(13):5166–5171. doi: 10.1073/pnas.0800121105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chung EY, et al. c-Myb oncoprotein is an essential target of the dleu2 tumor suppressor microRNA cluster. Cancer Biol Ther. 2008;7(11) doi: 10.4161/cbt.7.11.6722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhao H, et al. The c-myb proto-oncogene and microRNA-15a comprise an active autoregulatory feedback loop in human hematopoietic cells. Blood. 2009;113(3):505–16. doi: 10.1182/blood-2008-01-136218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cobaleda C, et al. Pax5: the guardian of B cell identity and function. Nature immunology. 2007;8(5):463–70. doi: 10.1038/ni1454. [DOI] [PubMed] [Google Scholar]
- 36.Robichaud GA, Perreault JP, Ouellette RJ. Development of an isoform-specific gene suppression system: the study of the human Pax-5B transcriptional element. Nucleic acids research. 2008;36(14):4609–4620. doi: 10.1093/nar/gkn432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang M, Raveche ES. Apoptosis induction in fludarabine resistant malignant B-1 cells by G2-M cell cycle arrest. Oncol Rep. 1998;5(1):23–30. doi: 10.3892/or.5.1.23. [DOI] [PubMed] [Google Scholar]
- 38.Chong SY, et al. The growth-regulatory role of B-cell-specific activator protein in NZB malignant B-1 cells. Cancer Immunol Immunother. 2001;50(1):41–50. doi: 10.1007/s002620000165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang M, Chong SY, Raveche ES. The role of B-cell-specific activator protein in the response of malignant B-1 cells to LPS. Exp Cell Res. 2001;264(2):233–43. doi: 10.1006/excr.2000.5122. [DOI] [PubMed] [Google Scholar]
- 40.Baumann Kubetzko FB, et al. The PAX5 oncogene is expressed in N-type neuroblastoma cells and increases tumorigenicity of a S-type cell line. Carcinogenesis. 2004;25(10):1839–1846. doi: 10.1093/carcin/bgh190. [DOI] [PubMed] [Google Scholar]
- 41.Proulx M, et al. Overexpression of PAX5 induces apoptosis in multiple myeloma cells. International Journal of Hematology. 2010;92(3):451–462. doi: 10.1007/s12185-010-0691-9. [DOI] [PubMed] [Google Scholar]
- 42.Krenacs L, et al. Transcription Factor B-Cell–Specific Activator Protein (BSAP) Is Differentially Expressed in B Cells and in Subsets of B-Cell Lymphomas. Blood. 1998;92(4):1308–1316. [PubMed] [Google Scholar]
- 43.Poppe B, et al. PAX5/IGH rearrangement is a recurrent finding in a subset of aggressive B-NHL with complex chromosomal rearrangements. Genes, Chromosomes and Cancer. 2005;44(2):218–223. doi: 10.1002/gcc.20214. [DOI] [PubMed] [Google Scholar]
- 44.Kanteti R, et al. PAX5 is expressed in small-cell lung cancer and positively regulates c-Met transcription. Lab Invest. 2009;89(3):301–14. doi: 10.1038/labinvest.2008.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chung EY, et al. CD19 is a major B cell receptor-independent activator of MYC-driven B-lymphomagenesis. The Journal of clinical investigation. 2012;122(6):2257–66. doi: 10.1172/JCI45851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Calin GA, et al. Frequent deletions and down-regulation of micro- RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(24):15524–9. doi: 10.1073/pnas.242606799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang X, et al. Myc represses miR-15a/miR-16-1 expression through recruitment of HDAC3 in mantle cell and other non-Hodgkin B-cell lymphomas. Oncogene. 2012;31(24):3002–8. doi: 10.1038/onc.2011.470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang JC, et al. Histone deacetylase in chronic lymphocytic leukemia. Oncology. 2011;81(5–6):325–9. doi: 10.1159/000334577. [DOI] [PubMed] [Google Scholar]
- 49.Van Damme M, et al. HDAC isoenzyme expression is deregulated in chronic lymphocytic leukemia B-cells and has a complex prognostic significance. Epigenetics. 2012;7(12) doi: 10.4161/epi.22674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Barneda-Zahonero B, Parra M. Histone deacetylases and cancer. Molecular oncology. 2012;6(6):579–589. doi: 10.1016/j.molonc.2012.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Khan O, La Thangue NB. HDAC inhibitors in cancer biology: emerging mechanisms and clinical applications. Immunol Cell Biol. 2012;90(1):85–94. doi: 10.1038/icb.2011.100. [DOI] [PubMed] [Google Scholar]
- 52.Ramalingam SS, et al. Phase II study of belinostat (PXD101), a histone deacetylase inhibitor, for second line therapy of advanced malignant pleural mesothelioma. J Thorac Oncol. 2009;4(1):97–101. doi: 10.1097/JTO.0b013e318191520c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stathis A, et al. Phase I Study of Decitabine in Combination with Vorinostat in Patients with Advanced Solid Tumors and Non-Hodgkin’s Lymphomas. Clinical Cancer Research. 2011;17(6):1582–1590. doi: 10.1158/1078-0432.CCR-10-1893. [DOI] [PubMed] [Google Scholar]
- 54.Lucas DM, et al. The novel deacetylase inhibitor AR-42 demonstrates pre-clinical activity in B-cell malignancies in vitro and in vivo. PLoS One. 2010;5(6):e10941. doi: 10.1371/journal.pone.0010941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.El-Khoury V, et al. The Histone Deacetylase Inhibitor MGCD0103 Induces Apoptosis in B-Cell Chronic Lymphocytic Leukemia Cells through a Mitochondria-Mediated Caspase Activation Cascade. Molecular Cancer Therapeutics. 2010;9(5):1349–1360. doi: 10.1158/1535-7163.MCT-09-1000. [DOI] [PubMed] [Google Scholar]
- 56.Dubovsky JA, et al. Restoring the functional immunogenicity of chronic lymphocytic leukemia using epigenetic modifiers. Leuk Res. 2011;35(3):394–404. doi: 10.1016/j.leukres.2010.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Perez-Perarnau A, et al. Analysis of apoptosis regulatory genes altered by histone deacetylase inhibitors in chronic lymphocytic leukemia cells. Epigenetics. 2011;6(10):1228–35. doi: 10.4161/epi.6.10.17200. [DOI] [PubMed] [Google Scholar]
- 58.Sampath D, et al. Histone deacetylases mediate the silencing of miR-15a, miR-16, and miR-29b in chronic lymphocytic leukemia. Blood. 2012;119(5):1162–72. doi: 10.1182/blood-2011-05-351510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lanasa MC, et al. Immunophenotypic and gene expression analysis of monoclonal B-cell lymphocytosis shows biologic characteristics associated with good prognosis CLL. Leukemia. 2011;25(9):1459–66. doi: 10.1038/leu.2011.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mertens D, et al. Chronic lymphocytic leukemia and 13q14: miRs and more. Leuk Lymphoma. 2009;50(3):502–5. doi: 10.1080/10428190902763509. [DOI] [PubMed] [Google Scholar]
- 61.Laurie CC, et al. Detectable clonal mosaicism from birth to old age and its relationship to cancer. Nat Genet. 2012;44(6):642–50. doi: 10.1038/ng.2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Peng B, et al. A cultured malignant B-1 line serves as a model for Richter’s syndrome. Journal of immunology. 1994;153(4):1869–80. [PubMed] [Google Scholar]
- 63.Bougel S, et al. PAX5 activates the transcription of the human telomerase reverse transcriptase gene in B cells. J Pathol. 2010;220(1):87–96. doi: 10.1002/path.2620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Siegel D, et al. Vorinostat in solid and hematologic malignancies. J Hematol Oncol. 2009;2:31. doi: 10.1186/1756-8722-2-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Batish M, et al. Neuronal mRNAs travel singly into dendrites. Proc Natl Acad Sci U S A. 109(12):4645–50. doi: 10.1073/pnas.1111226109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Batish M, Raj A, Tyagi S. Single molecule imaging of RNA in situ. Methods Mol Biol. 714:3–13. doi: 10.1007/978-1-61779-005-8_1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Primers used to amplify promoter regions in the mouse BSAP-ChIP assay.
non-NZB = A20, a Balb/c B cell line and NZB = LNC, mouse B-CLL cell line. A) Representative single color histogram overlay of BSAP expression in unstained (solid black, open), non-NZB (grey filled) and NZB (dashed, open). B) Average BSAP MFI in non-NZB (grey bar) and NZB (black bar) cell line. C) RQ miR-15a in non-NZB (grey bar) and NZB (black bar) cell line measured using real time TaqMan microRNA expression assays. * p<0.05, Unpaired student’s t test, n=3.
Left) Dleu2 transcription start site (Dleu2 TSS) is not easily accessible to the transcription machinery because increased HDAC activity in CLL cells leads to reduced acetylation and tightly wound nucleosome conformation; and steric hindrance by promoter bound BSAP. Net effect of the above events is reduced mature miR-15a/16-1 leading to increased anti-apoptotic factors including cyclin D1 and Bcl2. Right) Addition of HDAC inhibitor increases acetylation giving rise to open nucleosome conformation while BSAP inhibitor frees the steric hindrance of Dleu2 promoter. This gives rise to increased transcription of miR-15a/16-1. Malignant cell number decreases due to cell cycle arrest and apoptosis.