

Abstract
Sphingosine-1-phosphate receptor 2 (S1PR2) plays an essential role in regulating blood-brain barrier (BBB) function during demyelinating central nervous system (CNS) disease. Increased expression of S1PR2 occurs in disease-susceptible CNS regions of female versus male SJL mice and in female multiple sclerosis (MS) patients. Here we reported a novel sensitive and noninvasive method to quantitatively assess S1PR2 expression using a C-1l labeled positron emission tomography (PET) radioligand [11C]5a for in vivo imaging of S1PR2. Compounds 5a exhibited promising binding potency with IC50 value of 9.52 ± 0.70 nM for S1PR2 and high selectivity over S1PR1 and S1PR3 (both IC50 > 1000 nM). [11C]5a was synthesized in ~40 min withradiochemistry yield of 20 ± 5% (decayed to the end of bombardment (EOB), n > 10), specific activity of 6 – 10 Ci/μmol (decayed to EOB). The biodistribution study in female SJL mice showed the cerebellar uptake of radioactivity at 30 min of post-injection of [11C]5a was increased by Cyclosporin A (CsA) pretreatment (from 0.84 ± 0.04 ID%/g to 2.21 ± 0.21 ID%/g, n = 4, p < 0.01). MicroPET data revealed that naive female SJL mice exhibited higher cerebellar uptake compared with males following CsA pretreatment (standardized uptake values (SUV) 0.58 ± 0.16 vs 0.48 ± 0.12 at 30 min of post-injection, n = 4, p < 0.05), which was consistent with the autoradiographic results. These data suggested that [11C]5a has the capability in assessing the sexual dimorphism of S1PR2 expression in the cerebellum of the SJL mice. The development of radioligands for S1PR2 to identify a clinical suitable S1PR2 PET radiotracer, may greatly contribute to investigating sex differences in S1PR2 expression that contribute to MS subtype and disease progression and it will be very useful for detecting MS in early state and differentiating MS with other patients with neuroinflammatory diseases, and monitoring the efficacy of treating diseases using S1PR2 antagonism.
Keywords: Sphingosine-1-phosphate receptor 2, Radioligand, Positron emission tomography, Multiple sclerosis, Sexually dimorphic expression
1. Introduction
Multiple sclerosis (MS) is a neuroinflammatory demyelinating disease of the central nervous system (CNS) which leads to focal plaques of primary demyelination in conjunction with BBB disruption in the gray and white matter of the brain and spinal cord.1, 2 MS affects 2.5 million people worldwide with a prevalence of approximately 0.1% in the Caucasian population.3 The life expectancy of patients with MS is significantly reduced and the quality of life is adversely affected in terms of mobility and social functioning. MS also has a strong sex bias, with the female to male ratio currently approaching 4:1.4-6 Relapsing-remitting MS (RRMS), the most common form of the disease in women, is a condition in which recurrent episodes of new neurological dysfunction (relapses) are separated by periods of clinical stability (remission). Most RRMS patients (>50%) will develop secondary progressive MS (SPMS), with increased and sustained disability within 10 – 20 years after diagnosis.7, 8 The mechanisms of that contribute to progression of MS are incompletely understood.
Sphingolipids, main components of nervous tissue, have been linked to MS several decades ago. Nowadays, especially sphingosine-1-phosphate (S1P) is in the focus of pathophysiological research and therapy development. S1P is a membrane-derived lysophospholipid that binds with five subtype G-protein coupled receptors S1PR1- S1PR5. The activation of S1PRs modulates a spectrum of key biological events including activities in the CNS and immune response.9 Sphingolipid-like immunomodulator fingolimod (FTY-720, Gilenya) is the first oral disease-modifying therapy approved by the US Food and Drug Administration (FDA) for RRMS.10 Phosphorylated Fingolimod sequesters lymphocytes within the lymph nodes and prevents their trafficking to the CNS, via binding to S1PR subtypes S1PR1, S1PR3, S1PR4, and S1PR5, but not S1PR2.11 Encouraged by the success of Fingolimod, much effort has been devoted to developing selective compounds targeting S1PR subtypes, especially S1PR1 or S1PR2.12
In a recent study, we found increased expression of S1PR2 in disease-susceptible CNS regions of female MS patients, and suggest cell intrinsic sex differences in the S1PR2 expression on CNS vasculature contribute to the development of disease cycles in females compared with males.13 Quantification of S1PR2 levels would greatly contribute to studies examining sex differences in MS, and the identification of patients that may benefit from treatment using S1PR2 antagonists, which are less likely to cause lymphopenia.14 Combined with a suitable radiotracer, positron emission tomography (PET), a reliable well established noninvasive imaging method, is able to offer a unique sensitive way to quantitatively assess neurotransmitter receptors, enzymes, and transporters in vivo.15 Currently PET is widely used in clinical diagnosis. Identifying a suitable PET tracer that can specifically assess the expression of S1PR2 in vivo, will ultimately lead to better understanding of the function of S1PR2 in the neuropathogenesis of MS.
The inbred SJL mouse strain has been used as a model of the sexual dimorphism observed in MS, as SJL females are more susceptible to experimental autoimmune encephalomyelitis (EAE) than males; and exhibit a relapsing-remitting disease pattern similar to that observed in MS patients.16, 17 Moreover, sex difference has also been found in the CNS expression of S1PR2 in SJL mice, especially in the cerebellum, 13 which provides a good target for the validation of new-synthesized S1PR2 radioligands.
Herein we report the design and synthesis of a series of S1PR2 ligands containing similar core structures as the well-known S1PR2 selective antagonist JTE-013.18 In vitro competitive cell membrane binding assays are conducted to determine the binding affinities of the newly synthesized analogues towards S1PR1, S1PR2, and S1PR3. Radiosynthesis of a S1PR2 radioligand [11C]5a, in vivo evaluation of [11C]5a via autoradiography, biodistribution, and microPET studies on SJL mice are accomplished. Our studies suggest that [11C]5a demonstrates sexual dimorphism of S1PR2 expression in the cerebellum of SJL mice.
2. Results
2.1 Chemistry
The synthesis of S1PR2 ligands starts with the construction of key hydrazine intermediate 3. Condensation of 5-amino-1,3-dimethylpyrazole with ethyl isobutyrylacetate using acetic acid as the solvent afforded compound 1.19 The reaction yield was low when propionic acid was employed as the solvent. Moreover, it's very challenging to remove the acylating side product from the reaction of 5-amino-1,3-dimethylpyrazole with solvent propionic acid due to its close polarity as the product. However, use of acetic acid as the solvent allowed the separation of the product from the acylated side product. Bromination of 1 afforded compound 2, followed by reaction with hydrazine to produce the key intermediate 3. The 2-chloropyridine moiety was synthesized from commercially available 2-chloro-6-methoxyisonicotinic acid. Treatment of 2-chloro-6-methoxyisonicotinic acid with diphenylphosphoryl azide afforded acyl azide 4. Reflux of compound 4 in toluene in situ produced the isocyanate. A solution of hydrazine 3 in tetrahydrofuran (THF) was subsequently added to the above solution to give the first target compound 5a in moderate yield (Scheme 1).
Scheme 1.
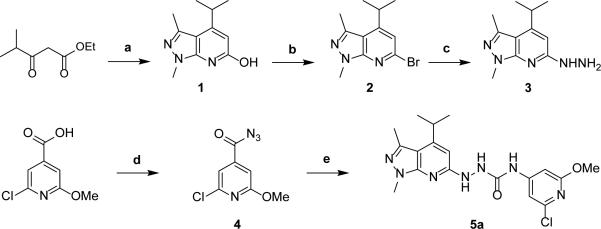
Synthesis of the target compound 5a. Reagents and conditions: (a) 5-amino-1,3-dimethylpyrazole, acetic acid, 140 °C, overnight, 18%; (b) POBr3, anisole, reflux, 54%; (c) NH2NH2, EtOH, reflux, 92%; (d) diphenylphosphoryl azide, Et3N, EtOAc, 57%; (e) (i) toluene, reflux; (ii) THF, 3, 50 °C, 32% over two steps.
Next we focused on the modification of the methoxy group in compound 5a to explore new analogues targeting on S1PR2. The syntheses of the target compounds 5b – 5f are depicted in Scheme 2. Reaction of methyl 2-chloroisonicotinate with silver (II) fluoride, followed a reported procedure 20 to introduce a fluorine atom on the pyridine ring to generate compound 6. Saponification of compound 6 gave carboxylic acid 7 in quantitative yield. Conversion of the acid to azide provided acyl azide 8, which was further reacted with hydrazine 3 to give target compound 5b. By replacing one of the chlorine atoms in 2,6-dichloroisonicotinic acid with methoxyethoxy functional group generated intermediate 9, which was readily converted to target compound 5c using a similar procedure for preparing compound 5a. Alternatively, hydrolysis of 2,6-dichloroisonicotinic acid with aqueous sodium hydroxide solution at 130 °C, followed by precipitation of the product by acidification using 6 M HCl, produced the compound 11 in high yield. Protection of 11 with chloromethyl methyl ether afforded two regioisomers 12 and 13 in a 5/1 ratio, which were readily separated. The esters were saponified to afford acids 14 and 16 in high yields. Both compounds were converted to the acyl azide and reacted with hydrazine 3 to provide target compounds 5d and 5e. Attempts to remove the MOM protecting group in compound 5e using hydrogen chloride/dioxane led to a complex resulted from the poor solubility of compound 5e in the solvent. To resolve the difficulty, deprotection of the MOM group of 5e was accomplished by using trifluoroacetic acid in dichloromethane that smoothly produced the C-11 radiolabeling precursor 18 in moderate yield. One of the chlorine atoms in 2,6-dichloroisonicotinic acid was converted into a fluoroethoxy group to afford fluorine containing intermediate 19. Following the abovementioned procedure, compound 19 reacted with compound 3 to give the target compound 5f.
Scheme 2.

Synthesis of target compounds 5b – 5f. Reagents and conditions: (a) silver(II) fluoride, acetonitrile; (b) LiOH, THF/H2O (1/2, v/v), then 1 M HCl; (c) diphenylphosphoryl azide, Et3N, dioxane; (d) (i) toluene, reflux; (ii) THF, hydrazine 3, 50 °C; (e) 2-methoxyethanol, tBuOK, THF; (f) (i) 2 M aqueous NaOH, 130 °C; (ii) 6 M HCl; (g) MOMCl, DIPEA, CH2Cl2; (h) TFA, CH2Cl2, 0 °C to rt, 2 h; (i) (i) 2-fluoroethanol, tBuOK, THF; (ii) diphenylphosphoryl azide, Et3N, dioxane.
2.2 In vitro competitive binding assay
The in vitro competitive binding assays against [32P]-S1P for the new synthesized target compounds 5a – 5f were conducted following our published protocol.21 Results showed that compounds 5a, 5e, and 5f exhibited promising binding potency with IC50 value of 9.52 ± 0.70 nM, 8.09 ± 0.91 nM, 8.12 ± 0.62 nM, respectively, while compounds 5b (IC50 = 134.9 ± 21.4 nM) and 5c (IC50 = 233.5 ± 34.4 nM) only had moderate binding potency towards S1P2 receptor. No binding potency was observed for compound 5d toward S1PR2. More importantly, compound 5a was seven-fold more potent than the well-known S1PR2 antagonist – JTE-013 (IC50 = 68.47 ± 7.45 nM, Figure 1) and also showed good selectivity towards S1P1 and S1P3 receptors (IC50 > 1000 nM) (Table 1). Compounds 5a, 5e, 5f showed similar calculated LogD7.4 values as JTE-013 except the calculated LogD7.4 for compound 5d was 1.01, which may cause its lose binding potency for S1PR2.
Figure 1.

Competitive binding curves of compound 5a and JTE-013 for S1PR2. A CHO cell membrane containing recombinant human S1PR2 was used in a [32P]S1P competitive binding assay to measure the binding affinity for compound 5a (red line, fitted IC50 = 9.52 ± 0.70 nM) and JTE-013 (blue dashed line, fitted IC50 = 68.47 ± 7.45 nM).
Table 1.
Binding affinities (IC50 values, nM) of new synthesized compounds towards S1P2, S1P2, S1P3 receptors.
| ||||
|---|---|---|---|---|
| Compound | cLogD7.4* | IC50 (nM) |
||
| S1PR2 | S1PR1 | S1PR3 | ||
| JTE-013 | 4.37 | 68.47 ± 7.45 | > 1000 | > 1000 |
| 5a | 4.43 | 9.52 ± 0.70 | > 1000 | > 1000 |
| 5b | 3.36 | 135 ± 21 | > 1000 | > 1000 |
| 5c | 4.10 | 234 ± 34 | > 1000 | > 1000 |
| 5d | 1.01 | > 1000 | > 1000 | > 1000 |
| 5e | 3.83 | 8.09 ± 0.91 | > 1000 | > 1000 |
| 5f | 4.66 | 8.12 ± 0.62 | > 1000 | > 1000 |
Calculated value at pH = 7.4 by ACD/I-Lab ver. 7.0 (Advanced Chemistry Development, Inc., Canada).
2.3 Radiochemistry
With the promising in vitro competitive binding potency for several ligands having IC50 < 10 nM, we elected to radiolabel 5a using [11C]methyl iodide to make [11C]5a for further in vivo validation. Various labeling conditions in terms of 11C-methylating agents, reaction temperature, base, mobile phase were explored, the optimized reaction conditions of this reaction were: 1.0 - 1.3 mg precursor, 200 μL DMF, 3 μL of 5 M aqueous KOH solution, 85 °C, 5 min (Scheme 3). Under optimized semi-preparative HPLC conditions (Agilent Zorbax SB-C18 column, 250 × 9.2 mm, mobile phase 45% acetonitrile in 0.1 M ammonium formate, pH 6.5, flow rate 4.0 mL/min, detection wavelength 254 nm), the retention time was 15 – 17 min for [11C]5a, 4 – 5min for precursor 18, and 5 – 6 min for a radioactive vice-product. It took about 40 min starting from the release of [11C]methyl iodide to formation of the final injection dose of [11C]5a with radiochemistry yield of 20 ± 5% (decayed to the end of bombardment (EOB), n > 10), specific activity of 6 – 10 Ci/μmol (EOB).
Scheme 3.

Radiolabeling of the precursor 18 for [11C]5a and postulated by-product.
2.4 Biodistribution study
Biodistribution studies were conducted to assess the tracer uptake in the brain and peripheral organs/tissues.The uptake of [11C]5a in female SJL mice at 30 min post injection showed that the heart, lung, kidney and liver had the highest uptake, which was consistent with S1PR2 distribution in mice.22 The cerebellum and total brain displayed moderate tracer accumulation with the ID%/gram values of 0.84 ± 0.04 and 0.78 ± 0.04 respectively. Pretreatment of mice with Cyclosporin A (CsA), which improves blood brain barrier permeability via modulation of P-glycoprotein (P-gp) with inhibition of the ATP binding cassette efflux transporters,23, 24 the cerebellar uptake (ID%/g) within female mice was notably increased to 2.21 ± 0.21 (n = 4, p < 0.01) upon CsA treatment. The uptake of [11C]5a in lung, thymus, kidney and liver showed similar trend in the CsA treatment group shown in Table 2.
Table 2.
Biodistribution of [11C]5a at 30min post-injection in control versus CsA pre-treated female SJL mice.
| ID%/g | Female control | Female with CsA |
|---|---|---|
| Blood | 3.52 ± 0.29 | 5.55 ± 0.61 |
| Heart | 3.56 ± 0.56 | 5.89 ± 0.62 |
| Lung | 5.71 ± 0.47 | 13.79 ± 2.56 |
| Muscle | 1.81 ± 0.18 | 2.65 ± 0.30 |
| Fat | 3.30 ± 0.54 | 3.13 ± 1.70 |
| Thymus | 2.81 ± 0.16 | 4.60 ± 0.22 |
| Spleen | 2.95 ± 0.24 | 3.99 ± 0.39 |
| Kidney | 7.96 ± 1.29 | 9.43 ± 0.96 |
| Liver | 13.31 ± 1.34 | 15.91 ± 1.06 |
| Cerebellum | 0.84 ± 0.04 | 2.21 ± 0.21 |
| Total brain | 0.78 ± 0.04 | 1.96 ± 0.18 |
2.5 Ex vivo autoradiography and microPET/computerized tomography (CT) study
To further validate the application of [11C]5a for imaging S1PR2, ex vivo autoradiography studies and microPET/CT scans in male and female SJL mice with CsA pre-treatment were also performed. The results from both autoradiography and PET/CT studies consistently demonstrated higher accumulation of [11C]5a in the cerebellum of female SJL mice than that observed in males (indicated by white dashed circles in Figure 2A). Furthermore, quantitative microPET analysis showed that the average cerebellar uptake of [11C]5a in female mice was statistically higher (n = 5, p = 0.021) than that in male mice (standardized uptake values (SUV) 0.58 ± 0.16 vs 0.48 ± 0.12, Figure 2B). The autoradiography and microPET/CT scans clearly demonstrated a sex difference in the cerebellar tracer uptake of [11C]5a in SJL mice and was consistent with our previous report of S1PR2 expression in the cerebellum of SJL mice, 13 which support the target selectivity and specificity of this newly developed S1PR2 radiotracer.
Figure 2.
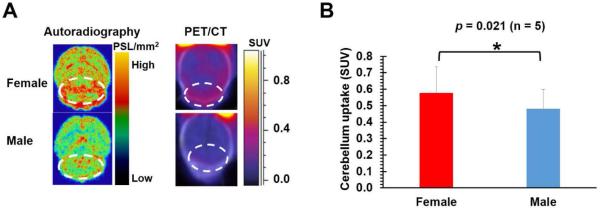
A, Autoradiography and PET/CT images of [11C]5a in the brains of female and male SJL mice ( n = 5) upon CsA pre-treatment; B, Quantification of cerebellar uptake of [11C]5a in female and male SJL mice upon CsA treatment from microPET. All the histograms were average values from 5 independent experiments, error bars were the standard derivations.
3. Discussion
Our previous study revealed that S1PR2 plays an essential role in regulating blood-brain barrier (BBB) function during demyelinating CNS disease.13 Development of a reliable and noninvasive imaging method that detects S1PR2 expression levels will facilitate better understanding of the neuropathologenesis of MS, especially with regard to the role of S1PR2 in neuroinflammation. Here we reported the design and synthesis of six S1PR2 ligands containing similar core structures as the well-known S1PR2 selective antagonist JTE-013. The synthesis took 6 – 9 steps. The synthesized ligands 5a – 5f was tested for binding potency towards S1PR1, S1PR2, and S1PR3 by in vitro competitive cell membrane binding assay. Three compounds showed good binding potency (IC50 < 10 nM) for S1PR2, and high selectivity over S1PR1 and S1PR3. Based on the in vitro data, we conclude that minor optimization of the functional group may led to significant change on the binding affinities, which will be useful for our future exploring the these structures reported in this manuscript.
The radiolabeling for the [11C]5a was challenging due to two competitive O-11C-methylation and N-11C-methylation product formulated in the reaction. To increase the desired O-11C-methylation radiolabeling product, different conditions in terms of reaction temperature, 11C-methylating agents including [11C]methyl iodide and [11C]methyl triflate, base and mobile phase were tested (see Table S1). Although cesium carbonate was reported to be an optimized base for the O-11C-methylation of similar pyridin-2-ol substrates,25, 26 only moderate yield was achieved in our reaction when using cesium carbonate as the base. In our system, we observed that potassium hydroxide combined with [11C]methyl iodide gave the highest O-11C-methylation to N-11C-methylation ratio compared to other condition including [11C]MeOTf/KOH, [11C]MeI/Cs2CO3, [11C]MeI/CsOH. Using pH 6.5 buffered aqueous ammonium formate solution improved resolution and radiochemical purity over pH 4.5 buffered aqueous ammonium formate. Under the optimized semi-preparative HPLC conditions, [11C]5a with retention time at 15 – 17 min was separated well from the precursor 18 (tR = 4 – 5 min) and radiolabeled by-product (tR = 5 – 6 min). [11C]5a was synthesized in about 40 min starting from the release of [11C]methyl iodide to formation of the final injection dose with radiochemistry yield of 20 ± 5% (decayed to EOB, n > 10), specific activity of 6 – 10 Ci/μmol (EOB).
Biodistribution study of [11C]5a indicated a higher cerebellar uptake of the tracer in female SJL mice with CsA pre-treatment than that in control the group. It is notable that CsA increased the cerebellar uptake of [11C]5a in female SJL mice by ~2.6-fold, which suggest the brain uptake towards [11C]5a is modestly modulated by P-gp 27-29 and thus is limited. Although species differences is common in P-gp transport of radioligands 30 and [11C]5a may have relatively high uptake in monkey or human brains. On the other hand, the low brain uptake of [11C]5a might also be partly attributed to the relatively low S1PR2 expression level in the brain, since the localization of S1PR2 in SJL mice brain is mainly on endothelial cells. 13 Structural optimization of 5a in the pyrazole-pyridine moiety to avoid P-gp efflux transport and enhance BBB penetration, while retaining binding potency is ongoing. Among the synthesized ligands, compound 5e with a fluoroethoxy group also displayed high binding potency (IC50 = 8.09 ± 0.91 nM). Here we focused on our first investigation of [11C]5a as a S1PR2 PET ligand. Exploration of 5f and other F-18 labeled ligands is undergoing with our group.
While the pathogenesis of MS involves both genetic susceptibility in terms of immune function and activation,31 and environmental causes such as Epstein-Barr virus infections,32 studies focused on sex differences are needed for new insights into hormonal and genetic sex-related factors that may impact on disease susceptibility and to identify new treatment targets. In addition, defined biomarkers that improve diagnostic evaluation and treatment follow-up in a sex-specific fashion are urgently needed. The identification of sex differences in S1PR2 expression and signaling in the neuropathogenesis of CNS autoimmunity 13 is a critical step towards gender-based medicine for the treatment of MS. Targeting S1PR2 may also treat MS without impacting normal immune function. Detection of S1PR2 via a novel, sensitive and noninvasive imaging PET modality will improve the assessment of the receptor in vivo. The current developed tracer meet the above requirement and is able to detect the sexual dimorphism of S1PR2 expression within autoimmune susceptible SJL mice in terms of biodistribution, autoradiography, microPET/CT scans.
4. Conclusions
We designed and synthesized a spectrum of S1PR2 ligands, chose a ligand that has high binding potency and high selectivity for S1P receptor 1to be labeled with [11C]methyl iodide. Utilizing the radioligand [11C]5a, we successively demonstrated the sexual dimorphism of S1PR2 expression in SJL mice with CsA pre-treatment. Further exploring the lead structures to identify a promising radioligands for S1PR2 with ability to penetrate the blood brain barrier, may greatly contribute to the diagnosis and treatment of MS and other neuroinflammatory diseases.
5. Materials and methods
5.1 Chemistry
General
All reagents and chemicals were purchased from commercial suppliers and used without further purification unless otherwise stated. Melting points were determined on a MEL-TEMP 3.0 apparatus and are uncorrected. 1H NMR and 13C NMR spectra were recorded at 400 or 300 MHz on a Varian Mercury-VX spectrometer with CDCl3, Acetone-d6, CD3OD, DMSO-d6 as solvent. All chemical shift values are reported in parts per million (ppm) (δ). The following abbreviations were used to describe peak patterns wherever appropriate: br = broad, s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet. HRMS analyses were conducted in Washington University Resource for Biomedical and Bio-organic Mass Spectrometry. Preparative chromatography was performed on Chemglass chromatography column using 230 – 400 mesh silica gel purchase from Silicycle. Analytical TLC was carried out on Merck 60 F254 silica gel glass plates, and visualization was aided by UV.
4-Isopropyl-1,3-dimethyl-1H-pyrazolo[3,4-b]pyridin-6-ol (1)33
A mixture of 5-amino-1,3-dimethylpyrazole (5.4 g, 49 mmol) and ethyl isobutyrylacetate (10 g, 63 mmol) in acetic acid (100 mL) was heated at 140 °C in a sealed tube for 20 h, the mixture was concentrated to remove the solvent and subjected to silica gel chromatography using CH2Cl2/MeOH (20/1) as the eluent to afford compound 1 (1.8 g, 18% yield) as a light yellow solid. 1H NMR (400 MHz, CDCl3) δ 6.14 (s, 1H), 3.96 (s, 3H), 3.32 – 3.22 (m, 1H), 2.49 (s, 3H), 1.27 (d, J = 6.4 Hz, 6H).
6-Bromo-4-isopropyl-1,3-dimethyl-1H-pyrazolo[3,4-b]pyridine (2)33
To a solution of 1 (1.0 g, 4.9 mmol) in anisole (10 mL) was added phosphorus oxybromide (2.0 g, 7.0 mmol). The reaction was heated at 130 °C overnight. Water was added to quench the reaction which was then extracted with EtOAc. The combined organic layers were washed with saturated aqueous NaHCO3, and saturated aqueous NaCl. The organic phase was concentrated in vacuo, and the residue was subjected to silica gel chromatography using CH2Cl2/MeOH (30/1) as the eluent to afford compound 2 (0.70 g, 54% yield) as a light yellow solid. 1H NMR (400 MHz, CDCl3) δ 6.99 (s, 1H), 3.95 (s, 3H), 3.53 – 3.43 (m, 1H), 2.59 (s, 3H), 1.28 (d, J = 6.8 Hz, 6H).
6-Hydrazinyl-4-isopropyl-1,3-dimethyl-1H-pyrazolo[3,4-b]pyridine (3)33
To a solution of 2 (0.45 g, 1.7 mmol) in ethanol (1.0 mL) was added anhydrous hydrazine (2.8 g, 88 mmol). The reaction was heated in a sealed tube at 120 °C overnight. The mixture was concentrated in vacuo, and the residue was subjected to silica gel chromatography using CH2Cl2/MeOH (20/1) as the eluent to afford compound 3 (0.34 g, 92% yield) as a yellow solid. 1H NMR (400 MHz, CDCl3) δ 6.14 (s, 1H), 4.00 (br, 2H), 3.84 (s, 3H), 3.40 – 3.30 (m, 1H), 2.50 (s, 3H), 1.21 (d, J = 7.2 Hz, 6H).
2-Chloro-6-methoxyisonicotinoyl azide (4)
To a solution of 2-chloro-6-methoxyisonicotinic acid (0.28 g, 1.5 mmol) in ethyl acetate (3 mL) was added Et3N (0.38 g, 3.8 mmol). The solution was cooled to 0 °C and diphenylphosphoryl azide (0.45 g, 1.7 mmol) was added dropwise. The reaction was warmed to room temperature upon addition and was stirred overnight. The mixture was concentrated in vacuo, and the residue was subjected to silica gel chromatography using hexane/EtOAc (30/1) as the eluent to afford compound 4 (0.18 g, 57% yield) as a white solid. Mp 67 – 69 °C; 1H NMR (400 MHz, Acetone-d6) δ = 7.21 (s, 1H), 6.99 (s, 1H), 3.82 (s, 3H); 13C NMR (100.7 MHz, Acetone-d6) δ 169.8, 164.5, 149.3, 142.9, 114.5, 108.9, 54.2; HRMS (ESI) Calcd for C7H6ClN4O2 213.0179, [M + H]+, found 213.0174.
N-(2-Chloro-6-methoxypyridin-4-yl)-2-(4-isopropyl-1,3-dimethyl-1H-pyrazolo[3,4-b]pyridin-6-yl)hydrazine-1-carboxamide (5a)
Under nitrogen, a solution of 4 (40 mg, 0.19 mmol) in toluene (0.5 mL) was refluxed for 3 hours to in situ produce the isocyanate. The reaction mixture was allowed to cool to 50 °C and a solution of 3 (31 mg, 0.14 mmol) in anhydrous THF (1 mL) was added dropwise. The reaction was then stirred at 50 °C overnight. The mixture was concentrated in vacuo, and the residue was subjected to silica gel chromatography using CH2Cl2/MeOH (20/1) as the eluent to afford compound 5a (18 mg, 32% yield) as a light yellow solid. Mp 243 – 245 °C; 1H NMR (400 MHz, CD3OD) δ 7.09 (s, 1H), 6.82 (s, 1H), 6.40 (s, 1H), 3.74 (s, 3H), 3.73 (s, 3H), 3.42 – 3.37 (m, 1H), 2.45 (s, 3H), 1.22 (d, J = 6.4 Hz, 6H); 13C NMR (100.7 MHz, CD3OD) δ 166.6, 164.7, 160.0, 156.2, 150.6, 148.5, 145.9, 140.0, 110.0, 106.2, 99.1, 96.1, 53.1, 31.8, 29.4, 21.7, 13.4; HRMS (ESI) Calcd for C18H22ClN7NaO2 426.1421, [M + Na]+, found 426.1403.
Methyl 2-chloro-6-fluoroisonicotinate (6)
To a solution of methyl 2-chloroisonicotinate (1.0 g, 6.0 mmol) in anhydrous acetonitrile (50 mL) was added silver(II) fluoride (2.6 g, 18 mmol) under nitrogen. The reaction was stirred in a sealed tube at 40 °C overnight. The suspension was filtered through Celite and the solution was concentrated by rotary evaporation. The residue was subjected to silica gel chromatography using hexane/EtOAc (40/1) as the eluent to afford compound 6 (0.25 g, 0.58 g of the starting material was recovered, 50% yield based on recovered starting material) as a clear liquid. This compound is known compound and commercially available. 1H NMR (400 MHz, Acetone-d6) δ 7.65 – 7.63 (m, 1H), 7.37 – 7.36 (m, 1H), 3.85 (s, 3H).
2-Chloro-6-fluoroisonicotinic acid (7)
To a solution of 6 (0.24 g, 1.3 mmol) in THF (2 mL) was added aqueous lithium hydroxide solution (40 mg in 4 mL water, 1.6 mmol) dropwise at 0 °C. The reaction was stirred at room temperature overnight. 2 M HCl(aq) was added to neutralize the reaction. The mixture was extracted with EtOAc, the combined organic layers were dried over sodium sulfate. The concentrated residue was subjected to silica gel chromatography using CH2Cl2/MeOH (3/1) as the eluent to afford compound 7 (0.22 g, 99% yield) as a white soild. Mp 118 – 120 °C; 1H NMR (400 MHz, CD3OD) δ 7.52 (s, 1H), 7.20 (d, J = 1.6 Hz, 1H); 13C NMR (100.7 MHz, CD3OD) δ 164.7, 162.7 (d, J = 245.3 Hz), 148.8 (d, J = 14.2 Hz), 148.2, 121.3 (d, J = 12.2 Hz), 107.8 (d, J = 44.2 Hz); HRMS (ESI) Calcd for C6H4ClFNO2 175.9915, [M + H]+, found 175.9905.
2-Chloro-6-fluoroisonicotinoyl azide (8)
To a solution of 7 (0.10 g, 0.57 mmol) in anhydrous 1,4-dioxane (2 mL) was added Et3N (0.12 g, 1.1 mmol). The solution was cooled to 0 °C and diphenylphosphoryl azide (0.31 g, 1.1 mmol) was added dropwise. The reaction was warmed to room temperature upon addition and was stirred overnight. The mixture was concentrated in vacuo, and the residue was subjected to silica gel chromatography using hexane/EtOAc (50/1) as the eluent to afford compound 8 (35 mg, 31% yield) as a yellow solid. Mp 39 – 40 °C; 1H NMR (400 MHz, Acetone-d6) δ 7.71 (s, 1H), 7.44 (d, J = 2.0 Hz, 1H); 13C NMR (100.7 MHz, Acetone-d6) δ 169.0, 162.8 (d, J = 244.9 Hz), 149.3, 145.8, 120.9, 107.8 (d, J = 43.6 Hz);
N-(2-Chloro-6-fluoropyridin-4-yl)-2-(4-isopropyl-1,3-dimethyl-1H-pyrazolo[3,4-b]pyridin-6-yl)hydrazine-1-carboxamide (5b)
Under nitrogen, a solution of 8 (30 mg, 0.15 mmol) in toluene (0.8 mL) was refluxed for 3 hours to in situ produce the isocyanate. The reaction mixture was allowed to cool to 50 °C and a solution of 3 (30 mg, 0.12 mmol) in anhydrous THF (0.8 mL) was added dropwise. The reaction was then stirred at 50 °C overnight. The mixture was concentrated in vacuo, and the residue was subjected to silica gel chromatography using CH2Cl2/MeOH (30/1) as the eluent to afford compound 5b (8 mg, 17% yield) as a light yellow solid. Mp 166 – 168 °C; 1H NMR (400 MHz, CDCl3) δ 8.16 (br, 1H), 7.26 (s, 1H), 7.14 (s, 1H), 6.61 (br, 1H), 6.44 (br, 1H), 6.40 (s, 1H), 3.85 (s, 3H), 3.50 – 3.43 (m, 1H), 2.55 (s, 3H), 1.26 (d, J = 6.8 Hz, 6H); 13C NMR (100.7 MHz, CDCl3) δ 164.4, 163.0, 156.3 (d, J = 225.1 Hz), 150.8, 150.6, 149.2, 140.2, 110.5, 110.3, 97.5, 96.8, 96.4, 33.3, 29.8, 22.9, 15.3; HRMS (ESI) Calcd for C17H19ClFN7NaO 414.1221, [M + Na]+, found 414.1207.
2-Chloro-6-(2-methoxyethoxy)isonicotinic acid (9)
To a solution of 2, 6-dichloroisonicotinic acid (1.1 g, 5.7 mmol) in anhydrous THF (5 mL) was added a solution of potassium tert-butoxide (1.6 g, 14.3 mmol) in THF (14 mL) dropwise at 0 °C. 30 min later, 2-methoxyethanol was added dropwise and the reaction was continued at room temperature overnight. 1 N aqueous hydrochloric acid was added to adjust the solution pH to 4. The mixture was extracted with EtOAc, the combined organic layers were dried over sodium sulfate. The concentrated residue was subjected to silica gel chromatography using CH2Cl2/MeOH (4/1) as the eluent to afford compound 9 as a white solid (1.6 g, 98% yield). Mp 138 – 140 °C; 1H NMR (400 MHz, CD3OD) δ 7.36 (s, 1H), 7.17 (s, 1H), 4.41 (t, J = 5.2 Hz, 2H), 3.71 (t, J = 4.4 Hz, 2H), 3.38 (s, 3H); 13C NMR (100.7 MHz, CD3OD) δ 164.9, 163.8, 148.7, 143.8, 115.5, 109.4, 70.3, 65.8, 57.7; HRMS (ESI) Calcd for C9H11ClNO4 232.0377, [M + H]+, found 232.0368.
2-Chloro-6-(2-methoxyethoxy)isonicotinoyl azide (10)
To a solution of 9 (0.15 g, 0.65 mmol) in anhydrous 1,4-dioxane (2 mL) was added Et3N (0.10 g, 0.97 mmol). The solution was cooled to 0 °C and diphenylphosphoryl azide (0.27 g, 0.97 mmol) was added dropwise. The reaction was warmed to room temperature upon completion of the addition and was stirred overnight. The mixture was concentrated in vacuo, and the residue was subjected to silica gel chromatography using hexane/EtOAc (20/1) as the eluent to afford compound 10 (200 mg, 84% yield) as a light yellow liquid. 1H NMR (400 MHz, Acetone-d6) δ 7.25 (s, 1H), 7.05 (s, 1H), 4.33 (t, J = 4.4 Hz, 2H), 3.58 (t, J = 4.8 Hz, 2H), 3.22 (s, 3H); 13C NMR (100.7 MHz, Acetone-d6) δ 169.9, 164.2, 149.1, 143.2, 114.6, 109.1, 70.1, 66.5, 57.9;
N-(2-Chloro-6-(2-methoxyethoxy)pyridin-4-yl)-2-(4-isopropyl-1,3-dimethyl-1H-pyrazolo[3,4-b]pyridin-6-yl)hydrazine-1-carboxamide (5c)
A solution of 10 (126 mg, 0.49 mmol) in toluene (1.2 mL) was refluxed at 120 °C for 3 hours under nitrogen to in situ produce the isocyanate. To the above mixture was added dropwise a solution of 3 (140 mg, 0.64 mmol) in anhydrous THF (1.2 mL) at 50 °C. The reaction continued to stir at 50 °C for overnight. The mixture was concentrated in vacuo, and the residue was subjected to silica gel chromatography using CH2Cl2/MeOH (30/1) as the eluent to afford compound 5c (78 mg, 35% yield) as a light yellow solid. Mp 210 – 212 °C; 1H NMR (400 MHz, CDCl3) δ 8.22 (br, 1H), 7.45 (br, 1H), 7.07 (br, 1H), 6.99 (s, 1H), 6.84 (s, 1H), 6.36 (s, 1H), 4.36 (t, J = 4.0 Hz, 2H), 3.80 (s, 3H), 3.65 (t, J = 4.4 Hz, 2H), 3.41 – 3.38 (m, 1H), 3.35 (s, 3H), 2.50 (s, 3H), 1.21 (d, J = 6.8 Hz, 6H); 13C NMR (100.7 MHz, CDCl3) δ 163.8, 158.4, 156.7, 156.2, 150.5, 149.1, 148.7, 140.0, 109.6, 106.9, 97.7, 97.3, 70.8, 65.6, 58.9, 33.2, 29.6, 22.8, 15.1; HRMS (ESI) Calcd for C20H26ClN7NaO3 470.1683, [M + Na]+, found 470.1674.
6-Chloro-2-oxo-1, 2-dihydropyridine-4-carboxylic acid (11)
2,6-dichloroisonicotinic acid (2.5 g, 13 mmol) was added to 2 N aqueous NaOH solution (32 mL). The solution was refluxed at 130 °C for overnight. 6 N HCl was added to the solution to precipitate the product when the solution was cooled down to room temperature. The suspension was filtered and the white solid was collected, dried in vacuo to afford the product 11 (2.2 g, 99% yield) as a white solid. Mp 186 – 188 °C; 1H NMR (400 MHz, DMSO-d6) δ 12.82 (br, 1H), 7.18 (s, 1H), 7.02 (s, 1H); 13C NMR (100.7 MHz, DMSO-d6) δ 165.2, 164.8, 148.3, 144.7, 114.3, 108.9; HRMS (ESI) Calcd for C6H5ClNO3 173.9958, [M + H]+, found 173.9946.
Methoxymethyl 6-chloro-1-(methoxymethyl)-2-oxo-1,2-dihydropyridine-4-carboxylate (12) /Methoxymethyl 2-chloro-6-(methoxymethoxy)isonicotinate (13)
To a solution of compound 11 (0.87 g, 5 mmol) in CH2Cl2 (100 mL) was added i-Pr2NEt (3.2 g, 25 mmol). The solution was cooled to 0 °C and chloromethyl methyl ether (1.6 g, 20 mmol) was added dropwise. The reaction was warmed to room temperature after completion of the addition and stirred overnight. The solution was quenched with water and extracted with CH2Cl2. The combined organic phase was washed with saturated aqueous NaHCO3, saturated sodium chloride solution, and dried with anhydrous sodium sulfate. The concentrated residue was subjected to silica gel chromatography using CH2Cl2/MeOH (40/1) as the eluent to afford compound 12 (polar, 876 mg, 67% yield) as a clear liquid and 13 (less polar, 180 mg, 14% yield) as a clear liquid.
Compound 12: 1H NMR (400 MHz, CDCl3) δ 7.11 (s, 1H), 6.73 (s, 1H), 5.56 (s, 2H), 5.39 (s, 2H), 3.48 (s, 3H), 3.39 (s, 3H); 13C NMR (100.7 MHz, CDCl3) δ 163.12, 163.05, 140.4, 138.4, 121.3, 105.5, 92.1, 75.8, 58.1, 57.8; HRMS (ESI) Calcd for C10H12ClNNaO5 284.0302, [M + Na]+, found 284.0300.
Compound 13: 1H NMR (400 MHz, CDCl3) δ 7.40 (d, J = 0.8 Hz, 1H), 7.20 (d, J = 0.8 Hz, 1H), 5.43 (s, 2H), 5.40 (s, 2H), 3.47 (s, 3H), 3.44 (s, 3H); 13C NMR (100.7 MHz, CDCl3) δ 163.1, 162.8, 149.2, 142.5, 116.7, 109.8, 92.8, 91.9, 58.0, 57.3; HRMS (ESI) Calcd for C10H12ClNNaO5 284.0302, [M + Na]+, found 284.0289.
6-Chloro-1-(methoxymethyl)-2-oxo-1,2-dihydropyridine-4-carboxylic acid (14)
To a solution of 12 (1.4 g, 5.3 mmol) in THF (5 mL) was added an aqueous solution of lithium hydroxide (0.17 mg in 10 mL water, 7.0 mmol) dropwise at 0 °C. The reaction was stirred at room temperature for overnight. 2 N HCl solution was added to acidify neutralize the reaction. The mixture was extracted with EtOAc, the combined organic phase was dried over sodium sulfate. The concentrated residue was subjected to silica gel chromatography using CH2Cl2/MeOH (4/1) as the eluent to afford compound 14 (0.82 g, 71% yield) as a white solid. Mp 161 – 163 °C; 1H NMR (400 MHz, CD3OD) δ 6.94 (s, 1H), 6.74 (s, 1H), 5.52 (s, 2H), 3.30 (s, 3H); 13C NMR (100.7 MHz, CD3OD) δ 164.5, 163.9, 142.3, 138.3, 119.7, 106.2, 75.7, 56.4; HRMS (ESI) Calcd for C8H8ClNNaO4 284.0040, [M + Na]+, found 284.0034.
6-Chloro-1-(methoxymethyl)-2-oxo-1,2-dihydropyridine-4-carbonyl azide (15)
To a solution of 14 (0.41 g, 1.9 mmol) in anhydrous 1,4-dioxane (16 mL) was added Et3N (0.28 g, 2.8 mmol). The solution was cooled to 0 °C and diphenylphosphoryl azide (0.77 g, 2.8 mmol) was added dropwise. The reaction was warmed to room temperature upon completion of the addition and stirred overnight. The mixture was concentrated in vacuo, and the residue was subjected to silica gel chromatography using hexane/EtOAc (6/1) as the eluent to afford compound 15 (0.28 g, 62% yield) as a yellow solid. Mp 99 – 100 °C; 1H NMR (400 MHz, CDCl3) δ 7.06 (s, 1H), 6.69 (s, 1H), 5.55 (s, 2H), 3.39 (s, 3H); 13C NMR (100.7 MHz, Acetone-d6) δ 170.0, 162.1, 140.7, 138.9, 120.4, 103.5, 75.6, 56.7; HRMS (ESI) Calcd for C16H15Cl2N8O6 485.0492, [2M + H]+, found 485.0490.
N-(6-Chloro-1-(methoxymethyl)-2-oxo-1,2-dihydropyridin-4-yl)-2-(4-isopropyl-1,3-dimethyl-1H-pyrazolo[3,4-b]pyridin-6-yl)hydrazine-1-carboxamide (5d)
A solution of 15 (75 mg, 0.31 mmol) in toluene (2 mL) was refluxed for 3 hours under nitrogen to in situ produce the isocyanate. To the above mixture was added dropwise a solution of 3 (53 mg, 0.24 mmol) in anhydrous THF (2 mL) at 60 °C. The reaction continued to stir at 60 °C overnight. The mixture was concentrated in vacuo, and the residue was subjected to silica gel chromatography using CH2Cl2/MeOH (40/1) as the eluent to afford compound 5d (92 mg, 88% yield) as a light yellow solid. Mp 172 – 174 °C; 1H NMR (400 MHz, CD3OD) δ 6.77 (s, 1H), 6.71 (s, 1H), 6.43 (s, 1H), 5.43 (s, 2H), 3.79 (s, 3H), 3.45 – 3.38 (m, 1H), 3.32 (s, 3H), 2.49 (s, 3H), 1.25 (d, J = 6.8 Hz, 6H); 13C NMR (100.7 MHz, CD3OD) δ 164.6, 159.7, 155.6, 150.8, 150.0, 139.8, 137.6, 107.9, 102.1, 100.2, 98.6, 98.5, 74.9, 56.2, 31.9, 29.4, 21.9, 13.7; HRMS (ESI) Calcd for C19H24ClN7NaO3 456.1527, [M + Na]+, found 456.1514.
2-Chloro-6-(methoxymethoxy)isonicotinic acid (16)
To a solution of 13 (0.48 g, 1.8 mmol) in THF (2.4 mL) was added aqueous lithium hydroxide solution (57 mg in 4.8 mL water, 2.4 mmol) dropwise at 0 °C. The reaction was stirred at room temperature overnight. 2 N HCl(aq) was added to neutralize the reaction at 0 °C. The mixture was extracted with EtOAc, the combined organic phase was dried over sodium sulfate. The concentrated residue was subjected to silica gel chromatography using CH2Cl2/MeOH (4/1) as the eluent to afford compound 16 (0.31 g, 77% yield) as a light yellow solid. Mp 150 – 152 °C; 1H NMR (400 MHz, Acetone-d6) δ 7.33 (s, 1H), 7.12 (s, 1H), 5.36 (s, 2H), 3.35 (s, 3H); 13C NMR (100.7 MHz, Acetone-d6) δ164.0, 163.0, 148.7, 144.1, 116.6, 109.8, 92.7, 56.4; HRMS (ESI) Calcd for C8H8ClNNaO4 240.0040, [M + Na]+, found 240.0036.
2-Chloro-6-(methoxymethoxy)isonicotinoyl azide (17)
To a solution of 16 (0.24 g, 1.1 mmol) in anhydrous 1,4-dioxane (5 mL) was added Et3N (0.14 g, 1.4 mmol). The solution was cooled to 0 °C and diphenylphosphoryl azide (0.39 g, 1.4 mmol) was added dropwise. The reaction was warmed to room temperature and stirred overnight. The mixture was concentrated in vacuo, and the residue was subjected to silica gel chromatography using hexane/EtOAc (50/1) as the eluent to afford compound 17 (0.11 g, 41% yield) as a yellow liquid. 1H NMR (400 MHz, Acetone-d6) δ 7.35 (s, 1H), 7.14 (s, 1H), 5.41 (s, 2H), 3.38 (s, 3H); 13C NMR (100.7 MHz, Acetone-d6) δ 181.1, 169.8, 163.2, 149.0, 115.7, 109.2, 92.9, 56.4; the acyl azide was converted to free amine during mass analysis. HRMS (ESI) Calcd for C7H9ClN2NaO2 211.0250, [M + Na]+, found 211.0243.
N-(2-Chloro-6-(methoxymethoxy)pyridin-4-yl)-2-(4-isopropyl-1,3-dimethyl-1H-pyrazolo[3,4-b]pyridin-6-yl)hydrazine-1-carboxamide (5e)
A solution of 17 (108 mg, 0.45 mmol) in toluene (2.2 mL) was refluxed at 120 °C for 3 hours under nitrogen to in situ produce the isocyanate. To the above mixture was added dropwise a solution of 3 (76 mg, 0.35 mmol) in anhydrous THF (2.5 mL) at 50 °C. The reaction continued to stir at 50 °C for overnight. The mixture was concentrated in vacuo, and the residue was subjected to silica gel chromatography using CH2Cl2/MeOH (40/1) as the eluent to afford compound 5e (130 mg, 86% yield) as a pale white solid. Mp 208 – 209 °C; 1H NMR (400 MHz, CD3OD) δ 7.25 (s, 1H), 7.00 (s, 1H), 6.48 (br, 1H), 5.37 (s, 2H), 3.81 (s, 3H), 3.53 – 3.46 (m, 1H), 3.44 (s, 3H), 2.54 (s, 3H), 1.31 (d, J = 6.8 Hz, 6H); 13C NMR (100.7 MHz, CD3OD) δ 163.0, 159.9, 155.7, 151.1, 150.9, 148.5, 139.9, 107.8, 107.0, 98.6, 98.5, 96.9, 92.0, 55.8, 31.7, 29.3, 21.8, 13.6; HRMS (ESI) Calcd for C19H24ClN7NaO3 456.1527, [M + Na]+, found 456.1515.
N-(2-Chloro-6-hydroxypyridin-4-yl)-2-(4-isopropyl-1,3-dimethyl-1H-pyrazolo[3,4-b]pyridin-6-yl)hydrazine-1-carboxamide (18)
Compound 5e (54 mg, 0.12 mmol) was dissolved in CH2Cl2 (2 mL). Trifluoroacetic acid (1 mL) was added dropwise at 0 °C. The solution was stirred at room temperature for 2 hours. Saturated aqueous NaHCO3 solution was added to quench the reaction. The mixture was extracted with CH2Cl2. The combined organic phase was dried over sodium sulfate and concentrated through rotary evaporation. The residue was subjected to silica gel chromatography using CH2Cl2/MeOH (10/1) as the eluent to afford the precursor 18 (21 mg, 43% yield) for radiolabeling. Mp 226 – 228 °C; 1H NMR (400 MHz, CD3OD) δ 6.90 (s, 1H), 6.71 (s, 1H), 6.40 (s, 1H), 3.74 (s, 3H), 3.46 – 3.37 (m, 1H), 2.46 (s, 3H), 1.24 (d, J = 6.8 Hz, 6H); 13C NMR (100.7 MHz, CD3OD) δ 164.7, 159.9, 158.0, 155.7, 151.4, 151.0, 140.8, 139.9, 107.8, 103.3, 98.6, 98.5, 31.7, 29.4, 21.8, 13.6;
2-Chloro-6-(2-fluoroethoxy)isonicotinoyl azide (19)
To a solution of 2, 6-dichloroisonicotinic acid (0.96 g, 5 mmol) in anhydrous THF (5 mL) was added a solution of potassium tert-butoxide (1.4 g, 12.5 mmol) in THF (12 mL) dropwise at 0 °C. 5 min later, 2-fluoroethanol (480 mg, 7.5 mmol) was added dropwise and the reaction was continued at room temperature for overnight. 1 N aqueous hydrochloric acid was added to adjust the solution pH to 4.0. The mixture was extracted with EtOAc, the combined organic phase was dried with sodium sulfate, and concentrated in vacuo to give the crude ether. The ether was then dissolved in anhydrous 1,4-dioxane (10 mL). Et3N (0.18 mL, 1.3 mmol), diphenylphosphoryl azide (0.36 g, 1.3 mmol) was added successively. The reaction was stirred at room temperature overnight. The mixture was concentrated in vacuo, and the residue was subjected to silica gel chromatography using hexane/EtOAc (20/1) as the eluent to afford compound 19 (103 mg, 8% yield for two steps) as a yellow solid. Mp 47 – 48 °C; 1H NMR (400 MHz, Acetone-d6) δ 7.26 (s, 1H), 7.07 (s, 1H), 4.66 (dt, J = 47.6 Hz, 4.0 Hz, 2H), 4.47 (dt, J = 29.2 Hz, 4.0 Hz, 2H); 13C NMR (100.7 MHz, Acetone-d6) δ 169.8, 163.8, 149.0, 143.4, 115.1, 109.2, 81.4 (d, J = 209.1 Hz), 66.5 (d, J = 24.3 Hz).
N-(2-Chloro-6-(2-fluoroethoxy)pyridin-4-yl)-2-(4-isopropyl-1,3-dimethyl-1H-pyrazolo[3,4-b]pyridin-6-yl)hydrazine-1-carboxamide (5f)
A solution of 19 (64 mg, 0.26 mmol) in toluene (1.5 mL) was refluxed for 3 hours under nitrogen to in situ produce the isocyanate. To the above mixture was added dropwise a solution of 3 (44 mg, 0.2 mmol) in anhydrous THF (2 mL) at 50 °C. The reaction continued to stir at 50 °C for overnight. The mixture was concentrated in vacuo, and the residue was subjected to silica gel chromatography using CH2Cl2/MeOH as the eluent to afford compound 5f (54 mg, 62% yield) as a pale solid. Mp 240 – 242 °C; 1H NMR (400 MHz, CD3OD) δ 7.20 (s, 1H), 6.93 (s, 1H), 6.47 (s, 1H), 4.64 (dt, J = 47.6 Hz, 3.6 Hz, 2H), 4.42 (dt, J = 25.2 Hz, 4.0 Hz, 2H), 3.81 (s, 3H), 3.52 – 3.42 (m, 1H), 2.52 (s, 3H), 1.30 (d, J = 6.8 Hz, 6H); 13C NMR (100.7 MHz, CD3OD) δ 163.7, 159.9, 155.7, 150.9, 150.8, 148.3, 139.9, 119.7, 106.7, 98.6, 98.5, 96.5, 81.4 (d, J = 209.1 Hz), 65.5 (d, J = 24.3 Hz), 31.7, 29.4, 21.8, 13.6; HRMS (ESI) Calcd for C19H23ClFN7NaO2 458.1483, [M + Na]+, found 458.1492.
5.2 Radiochemistry
Production of [11C]CH3I followed the reported method. Briefly, [11C]CH3I was produced on-site using a GE PETtrace MeI Microlab through consecutive reaction starting from [11C]CO2 . Up to 1400 mCi of [11C]CO2 was produced from the JSW BC-16/8 cyclotron by irradiating a gas target of 0.5% O2 in N2 for 15 – 30 min with a 40 μA beam of 16 MeV protons in the Barnard Cyclotron Facility of Washington University School of Medicine. [11C]CH3I was produced by reduction of [11C]CO2 using a nickel catalyst (Shimalite-Ni (reduced), Shimadzu, Japan P.N.221-27719) in the presence of hydrogen gas at 360 °C, followed by iodination of the resulting [11C]CH4 with iodine at 690 °C. Approximately 12.2 min following the end-of-bombardment (EOB), 800 – 1000 mCi of [11C]CH3I were delivered via gas tubing to the reaction vial in the hot cell.
[11C]CH3I was bubbled for a period of 2 – 3 min into a solution of precursor (1.0 – 1.3 mg) in DMF (0.2 mL) containing 3.0 μL of potassium hydroxide (5.0 M) at room temperature. When the trap of radioactivity was completed, the sealed reaction vessel was heated at 85 °C for 5 min, shaking was applied to the vial using a long clamp during the reaction. The oil bath was removed and 1.7 mL of the HPLC mobile phase (45% acetonitrile in 0.1 M ammonium formate, v/v, pH 6.5) was added into to the reaction vessel. The mixture was loaded onto a reversed phase HPLC system to purify the mixture (Agilent Zorbax SB-C18 column, 250 × 9.2 mm, mobile phase 45% acetonitrile in 0.1 M ammonium formate, pH 6.5, flow rate 4.0 mL/min, detection wavelength 254 nm). Under these conditions, the product with retention time at 15 – 17 min was collected into a vial that contained 50 mL Milli-Q water. Then the collected fraction was passed through a C-18 Plus Sep-Pak® cartridge to concentrate the target component in the Sep-Pak cartridge. The Sep-Pak cartridge was rinsed using 20 mL of sterile water. Finally, the tracer trapped on the Sep-Pak® was eluted with 0.3 mL of ethanol, following with 2.7 mL 0.9% sodium chloride solution, passing through a 0.22 μ (Whatman Puradisc 13 mm syringe filter) sterile filter into a sterile pyrogen-free glass vial for delivery. For quality control, an aliquot of sample was assayed by an analytical HPLC system (Agilent Zorbax SB-C18 column, 250 × 4.6 mm, mobile phase 55% acetonitrile in 0.1 M ammonium formate, pH 4.5, flow rate 1.2 mL/min, detection wavelength 254 nm). The sample was authenticated by co-injecting with the corresponding nonradiolabeled standard solution. The retention time was 5.5 min with radiochemical purity was > 99%. The radiosynthesis typically took 40 min starting from the release of [11C]methyl iodide, with yield 20 ± 5% (n > 10, decay corrected to the end of synthesis), specific activity 6 – 10 Ci/μmol (decayed to EOB).
5.3 In vitro competitive binding assay
In vitro competitive binding assay against [32P]S1P assay was conducted according to our published protocol.21
5.4 Biodistribution, autoradiography, PET/CT study
All animal experiments were conducted in compliance with the Guidelines for the Care and Use of Research Animals under protocols approved by Washington University's Animal Studies Committee. For mice biodistribution studies, 64 – 89 μCi of [11C]5a was injected via the tail vein (i.v.) into mature female SJL mice (n = 4 per study group, mice weight 18 – 24 g) under 2 - 3% isoflurane/oxygen anesthesia. Eight mice were divided into two groups: the control group without CsA pretreatment and the CsA pre-treatment group (25 mg/kg of CsA, i.v. 30 min prior to the tracer injection). At 30 min postinjection (p.i.), mice were anesthetized again and euthanized. The whole brain was quickly harvested, the blood was removed by blotting, and the cerebellum was separated, the remainder of the brain was collected to determine the total brain uptake. Peripheral organs and tissues, including blood, heart, lung, muscle, fat, thymus, spleen, kidney, and liver, were also collected; all samples were weighed and counted in an automated well counter with a standard dilution of the injectate. Counts were decay-corrected and the %ID/g was calculated. A two-tailed paired Student's t test was used to calculate the standard derivations. A p value less than 0.05 was considered statistically significant.
For the ex vivo autoradiography study, SJL mice were injected with 400-900 μCi of [11C]5a and euthanized 30 min p.i. as described above. The whole brain was quickly removed and snap-frozen. Horizontal sections (100 μm) were cut with a Microm cryotome and mounted on Superfrost Plus glass slides (Fisher Scientific, Pittsburgh, PA). Frozen slides were directly exposed to film in an imaging cassette for 4 hours at −80 °C in the dark. The distribution of radioactivity was visualized by a Fuji Bio-Imaging Analyzer FLA-7000 (Fuji Photo Film Co., Tokyo, Japan). Photo-stimulated luminescence (PSL) from the cerebellum was quantified using Multi Gauge v3.0 software (Fuji Photo Film Co., Tokyo, Japan). Data were background-corrected and expressed as photo-stimulated luminescence signals per square millimeter (PSL/mm2).
Brain microPET imaging was performed using two Siemens microPET scanners (Siemens Preclincial Solutions, Knoxville, TN, USA) - microPET-Focus-F220 and a microPET-Inveon MultiModality scanners. Imaging studies were done using 19 – 25 g SJL mice pretreated with Cyclosporin A. Animals were anesthetized using 2% isoflurane/oxygen and a tail vein catheter placed in the lateral tail vein. Gas anesthesia was maintained at <1.5% isoflurane during the imaging session; each mice was positioned on the scanner bed at least 30 min prior to the tracer injection. Body temperature was maintained using a warming lamp. A dose of 25 mg/kg CsA in saline administered i.v. 30 min prior to radiotracer injection was used for modulation of the BBB efflux transporters. Both CsA and the radiotracer (400-900 μCi of [11C]5a) were administered using an i.v. catheter placed in the lateral tail vein. The imaging sessions were carried out as 1 h dynamic scan using the MicroPET Focus 220 and Inveon scanners (Siemens Medical Solutions USA). Acquired list mode data were histogrammed into a 3D set of sinograms and binned to the following time frames: 1 × 3 s, 6 × 2 s, 9 × 5 s, 6 × 10 s, 4 × 30 s, 2 × 60 s, 2 × 2 min, 10 × 5 min. Sinogram data was then processed using filter back projection algorithm with attenuation and scatter corrections. Regions of interest (the cerebellum) were manually drawn with the software ASIPro (Acquisition Sinogram Image Processing, Siemens Medical Solutions, Malvern, PA, USA) using IDL's Virtual Machine (ITT Visual Information Solutions, Boulder, CO, USA) to calculate the average cerebellar uptake.
Supplementary Material
Acknowledgments
This work was financially supported by the USA DOE-Training Grant: #DESC0008432 and the Washington University, Mallinckrodt Institute of Radiology (MIR) Pilot Grant: #14-017, the USA National Institute of Mental Health (NIMH) of the National Institutes of Health (No. MH092797) and National Institute of Neurological Disorders and Stroke (NINDS, No. NS075527, NS061025, P01 NS059560), and National Multiple Sclerosis Society grants PP2043, RG 4632.
References
- 1.Mahad DH, Trapp BD, Lassmann H. The Lancet. Neurology. 2015;14:183–193. doi: 10.1016/S1474-4422(14)70256-X. [DOI] [PubMed] [Google Scholar]
- 2.Ransohoff RM, Hafler DA, Lucchinetti CF. Nature reviews. Neurology. 2015 DOI: 10.1038/nrneurol.2015.49. [Google Scholar]
- 3.Compston A. Elsevier/Churchill Livingstone; Philadelphia: 2006. [Google Scholar]
- 4.Compston A, Coles A. Lancet. 2008;372:1502–1517. doi: 10.1016/S0140-6736(08)61620-7. [DOI] [PubMed] [Google Scholar]
- 5.Handel AE, Jarvis L, McLaughlin R, Fries A, Ebers GC, Ramagopalan SV. PLoS ONE. 2011;6:e14606. doi: 10.1371/journal.pone.0014606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Orton SM, Herrera BM, Yee IM, Valdar W, Ramagopalan SV, Sadovnick AD, Ebers GC. The Lancet. Neurology. 2006;5:932–936. doi: 10.1016/S1474-4422(06)70581-6. [DOI] [PubMed] [Google Scholar]
- 7.Weinshenker BG, Bass B, Rice GP, Noseworthy J, Carriere W, Baskerville J, Ebers GC. Brain : a journal of neurology. 1989;112(Pt 6):1419–1428. doi: 10.1093/brain/112.6.1419. [DOI] [PubMed] [Google Scholar]
- 8.Tremlett H, Yinshan Z, Devonshire V. Multiple Sclerosis. 2008;14:314–324. doi: 10.1177/1352458507084264. [DOI] [PubMed] [Google Scholar]
- 9.Marsolais D, Rosen H. Nature reviews. Drug discovery. 2009;8:297–307. doi: 10.1038/nrd2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kappos L, Radue EW, O'Connor P, Polman C, Hohlfeld R, Calabresi P, Selmaj K, Agoropoulou C, Leyk M, Zhang-Auberson L, Burtin P. The New England journal of medicine. 2010;362:387–401. doi: 10.1056/NEJMoa0909494. [DOI] [PubMed] [Google Scholar]
- 11.Pelletier D, Hafler DA. The New England journal of medicine. 2012;366:339–347. doi: 10.1056/NEJMct1101691. [DOI] [PubMed] [Google Scholar]
- 12.Roberts E, Guerrero M, Urbano M, Rosen H. Expert opinion on therapeutic patents. 2013;23:817–841. doi: 10.1517/13543776.2013.783022. [DOI] [PubMed] [Google Scholar]
- 13.Cruz-Orengo L, Daniels BP, Dorsey D, Basak SA, Grajales-Reyes JG, McCandless EE, Piccio L, Schmidt RE, Cross AH, Crosby SD, Klein RS. The Journal of clinical investigation. 2014;124:2571–2584. doi: 10.1172/JCI73408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kunkel GT, Maceyka M, Milstien S, Spiegel S. Nature reviews. Drug discovery. 2013;12:688–702. doi: 10.1038/nrd4099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Inglese M, Petracca M. Prion. 2013;7:47–54. doi: 10.4161/pri.22650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Papenfuss TL, Rogers CJ, Gienapp I, Yurrita M, McClain M, Damico N, Valo J, Song F, Whitacre CC. J Neuroimmunol. 2004;150:59–69. doi: 10.1016/j.jneuroim.2004.01.018. [DOI] [PubMed] [Google Scholar]
- 17.Rivers TM, Sprunt DH, Berry GP. The Journal of experimental medicine. 1933;58:39–53. doi: 10.1084/jem.58.1.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li C, Chi XX, Xie W, Strong JA, Zhang JM, Nicol GD. Journal of neurophysiology. 2012;108:1473–1483. doi: 10.1152/jn.00825.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nam NL, Grandberg II, Sorokin VI. Chem. Heterocycl. Compd. 2003:937–942. [Google Scholar]
- 20.Fier PS, Hartwig JF. Science. 2013;342:956–960. doi: 10.1126/science.1243759. [DOI] [PubMed] [Google Scholar]
- 21.Rosenberg AJ, Liu H, Tu Z. Appl. Radiat. Isotopes. 2015;102:5–9. doi: 10.1016/j.apradiso.2015.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Adada M, Canals D, Hannun YA, Obeid LM. Febs J. 2013;280:6354–6366. doi: 10.1111/febs.12446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tu Z, Li S, Xu J, Chu W, Jones LA, Luedtke RR, Mach RH. Nuclear medicine and biology. 2011;38:725–739. doi: 10.1016/j.nucmedbio.2011.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Loscher W, Potschka H. NeuroRx : the journal of the American Society for Experimental NeuroTherapeutics. 2005;2:86–98. doi: 10.1602/neurorx.2.1.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Moran MD, Wilson AA, Elmore CS, Parkes J, Ng A, Sadovski O, Graff A, Daskalakis ZJ, Houle S, Chapdelaine MJ, Vasdev N. Bioorgan Med Chem. 2012;20:4482–4488. doi: 10.1016/j.bmc.2012.05.046. [DOI] [PubMed] [Google Scholar]
- 26.Wang CN, Moseley CK, Carlin SM, Wilson CM, Neelamegam R, Hooker JM. Bioorg Med Chem Lett. 2013;23:3389–3392. doi: 10.1016/j.bmcl.2013.03.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ishiwata K, Kawamura K, Yanai K, Hendrikse NH. J Nucl Med. 2007;48:81–87. [PubMed] [Google Scholar]
- 28.De Vries EFJ, Doorduin J, Vellinga NAR, Van Waarde A, Dierckx RA, Klein HC. Nuclear medicine and biology. 2008;35:459–466. doi: 10.1016/j.nucmedbio.2008.01.005. [DOI] [PubMed] [Google Scholar]
- 29.Kurdziel KA, Kiesewetter DO, Carson RE, Eckelman WC, Herscovitch P. J Nucl Med. 2003;44:1330–1339. [PubMed] [Google Scholar]
- 30.Syvanen S, Lindhe O, Palner M, Kornum BR, Rahman O, Langstrom B, Knudsen GM, Hammarlund-Udenaes M. Drug Metab Dispos. 2009;37:635–643. doi: 10.1124/dmd.108.024745. [DOI] [PubMed] [Google Scholar]
- 31.Gourraud PA, Harbo HF, Hauser SL, Baranzini SE. Immunological reviews. 2012;248:87–103. doi: 10.1111/j.1600-065X.2012.01134.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ascherio A, Munger KL. Annals of neurology. 2007;61:288–299. doi: 10.1002/ana.21117. [DOI] [PubMed] [Google Scholar]
- 33.Swenson RE. PCT Int. Appl. 2013 WO 2013148460 A1 20131003.
- 34.Tu Z, Dence CS, Ponde DE, Jones L, Wheeler KT, Welch MJ, Mach RH. Nuclear medicine and biology. 2005;32:423–430. doi: 10.1016/j.nucmedbio.2005.03.008. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.