

Abstract
The present work was undertaken with the objectives of improving the dissolution velocity, related oral bioavailability, and minimizing the fasted/fed state variability of repaglinide, a poorly water-soluble anti-diabetic active by exploring the principles of nanotechnology. Nanocrystal formulations were prepared by both top-down and bottom-up approaches. These approaches were compared in light of their ability to provide the formulation stability in terms of particle size. Soluplus® was used as a stabilizer and Kolliphor™ E-TPGS was used as an oral absorption enhancer. In vitro dissolution profiles were investigated in distilled water, fasted and fed state simulated gastric fluid, and compared with the pure repaglinide. In vivo pharmacokinetics was performed in both the fasted and fed state using Wistar rats. Oral hypoglycemic activity was also assessed in streptozotocin-induced diabetic rats. Nanocrystals TD-A and TD-B showed 19.86 and 25.67-fold increase in saturation solubility, respectively, when compared with pure repaglinide. Almost 10 (TD-A) and 15 (TD-B)-fold enhancement in the oral bioavailability of nanocrystals was observed regardless of the fasted/fed state compared to pure repaglinide. Nanocrystal formulations also demonstrated significant (p < 0.001) hypoglycemic activity with faster onset (less than 30 min) and prolonged duration (up to 8 h) compared to pure repaglinide (after 60 min; up to 4 h, respectively).
KEY WORDS: diabetes mellitus, fasted and fed state variability, nanocrystal, oral hypoglycemic activity, repaglinide
INTRODUCTION
Diabetes mellitus (DM) is among the most common diseases of the world. It is stated that more than 366 million people are suffering from DM and this statistics is expected to reach over 552 million by 2030 (1). Hence, development of a cost-effective anti-diabetic therapy becomes a thrust research area in the field of academia as well as industry.
Repaglinide (RPG), S (+) 2-ethoxy-4(2((3-methyl-1-(2-(1-piperidinyl) phenyl) butyl) amino)-2-oxoethyl) benzoic acid is chemically unrelated to the sulfonylurea, oral insulin secretagogues. It is indicated in adults with type 2 DM whose hyperglycemia is unsatisfactorily controlled by diet, exercise, and weight reduction alone. It exerts insulinotropic action by inhibiting adenosine tri-phosphate dependent potassium ion channels in the pancreatic β-cell membrane (2). RPG was the first commercially available meglitinide analogue for use in patients with type 2 DM. It acts by lowering post-prandial glucose by targeting early-phase insulin release. The advantage of this effect is thought to be important in reducing long-term cardiovascular complications of diabetes. RPG is devoid of adverse effects such as hypoglycemia, secondary treatment failure, and cardiovascular complications associated with other anti-diabetics (3). Despite of these advantages, the poor solubility (34.6 μg/mL at 37°C) and high lipophilicity (log p = 3.97) (4) of RPG are the main concerns for the administration through oral route as the mean absolute bioavailability is only 45–65% (5). It undergoes significant first-pass metabolism by the cytochrome P450 system (6) and possess a significant affinity towards permeability glycoprotein (P-gp) which is located in the apical membranes of intestinal absorptive cells (7). Reports also suggest that co-administration of 2 mg RPG with a high-fat meal resulted in a decrease in peak plasma concentration (Cmax) by 19.92% and the area under the plasma drug concentration (AUC) by 12.36% (8). Hence, in order to provide an efficient delivery system of RPG, design of an intelligent dosage form is necessary which will help to address all these problems associated with oral administration.
Previously, few formulation strategies had been explored to enhance the oral bioavailability of RPG primarily by improving the aqueous solubility such as co-amorphisation with saccharin (9), solid dispersion (10), HP-β-CD inclusion complex (11), or ultra rapid freezing (12). Although these approaches were found to be providing improved in vitro release profile, the in vivo performance has not been established. Hence, it was decided to engineer an improved dosage form that will overcome the problem of poor aqueous solubility, low oral bioavailability, and variability in fed-fasted state bioavailability. Engineering of nanocrystals formulation is thus hypothesized to be a promising approach.
Over the past two decades, nanosizing becomes a scientifically proven platform to address the issues of drug molecules with poor aqueous solubility. Since the beginning of the 1990s, Elan Pharma International Ltd. (San Francisco, CA, USA) has proven the significance of nanocrystals over the microcrystals to improve the oral absorption of poorly water-soluble drug. The drug nanocrystals are the crystals with a size in the nanometer range, typically below 500 nm (13,14). According to Noyes-Whitney equation, the dissolution is a function of surface area, so formulating nanocrystals will benefit to enhance the oral bioavailability, where absorption is dissolution rate limited. Nanocrystals attracted the attention of many formulation scientists owing to their superior attributes such as 100% drug loading, carrier free, stable, reduced fasted/fed state variability, and applicability of administration by various means of routes etc. over existing approaches used to enhance aqueous solubility (15).
In the present work, bottom-up and top-down approaches were employed to prepare a stable nanocrystal formulation using Soluplus® (SLPS) as a stabilizer. The effect of addition of oral absorption enhancer such as Kolliphor™ E-TPGS (TPGS) along with SLPS on the oral bioavailability of repaglinide was also evaluated. SLPS is a relatively novel graft copolymer that has been introduced in the pharmaceutical industry as a solubilizer for poorly soluble drug molecules (16). Unlike existing hydrophilic polymers, it has amphiphilic nature owing to the presence of hydrophobic polyvinyl caprolactam moiety linked via polyvinyl acetate to the long hydrophilic polyethylene glycol chain. In this study, we have evaluated its role as a stabilizer to prevent nanocrystal aggregation while processing or storage.
TPGS (D-alpha-tocopheryl polyethylene glycol 1000 succinate) is a water-soluble D-alpha vitamin-E ester derived from natural vitamin-E. It improves oral absorption of poorly soluble drugs by increasing solubility as well as by modulating P-gp dependent drug efflux mechanism (17–20). TPGS also exhibits inhibitory effects on cytochrome P450 3A (CYP3A) (21,22).
The key objective of the present research work was, therefore, to investigate the feasibility of bottom-up and top-down approaches to prepare stable RPG nanocrystals, in order to improve the solubility and related bioavailability. The second objective was to investigate the influence of food on in vivo pharmacokinetic profile of pure RPG and formulated RPG nanocrystals and to compare the in vivo pharmacodynamics of pure RPG with its nanocrystals in experimental animals. To the best of our knowledge, preparation of RPG nanocrystals has not yet been reported in the literature.
MATERIALS AND METHODS
Materials
Repaglinide was obtained as a generous gift from USV Limited (Mumbai, India). Indomethacin was kindly gifted by Emcure Pharmaceuticals Ltd. (Pune, India). Soluplus® (SLPS) and Kolliphor™ E-TPGS (TPGS) were kindly donated by BASF Corporation (Minden, Germany). Streptozotocin and glucose estimation kit (GOD/POD) were purchased from Sigma Chemical Co. USA and Accurex Biomedical Pvt. Ltd. (Mumbai, India), respectively. Pluronic F68, Pluronic F127, sodium lauryl sulfate (SLS), Tween 80, sodium taurocholate, egg lecithin, and pepsin were purchased from Sigma-Aldrich (Mumbai, India). Acetonitrile (HPLC grade), sodium chloride, and sodium acetate were purchased from Merck (Mumbai, India). Milli Q water was used in all formulations. All other chemicals and reagents used were of analytical grade.
Methods
Preparation of Simulated Gastric Fluids
Bio-relevant media simulating the gastric conditions in pre-prandial and post-prandial states (23,24) were utilized to predict equilibrium solubility, in vivo dissolution, and effect of food on RPG absorption. Fasted state simulated gastric fluid pH 1.6 (FaSSGF) contained sodium taurocholate (80 μM), lecithin (20 μM), pepsin (0.1 mg/mL), and sodium chloride (34.2 mM). Fed state simulated gastric fluid (FeSSGF) pH 5.0 composed of glacial acetic acid, sodium chloride in concentrations of 17.12, 29.75, and 237.02 mM, respectively, as a blank medium which was then mixed with ultra heat treated (UHT)-milk in the ratio of 1:1.
Selection of Stabilizer
Solubility of the drug in aqueous stabilizer solution was used as criteria to select an appropriate stabilizer (25,26). RPG solubility was determined in SLPS, Pluronic F68, Pluronic F127, Tween 80, sodium lauryl sulfate (SLS), and TPGS at 1% w/v aqueous solutions and combination of SLPS (0.5 and 1% w/v) with TPGS in the concentration range of 0.1–1.0% w/v. An excess amount of drug was added in vials containing aqueous stabilizer solution followed by continuous shaking using a mechanical shaker at room temperature (RT) for 24 h. Resulting suspensions were centrifuged at 25,000 rpm for 30 min; the supernatants were separated and filtered using 0.1 μm PTFE syringe filter (Whatman Inc., Clifton, NJ, USA). Filtrates were analyzed for RPG content after recording their absorbance at 241 nm using UV-Visible spectrophotometer (V-630, JASCO, Japan). All solubility experiments were performed in triplicate, and the data obtained was expressed as mean ± standard deviation (SD) of three measurements.
Preparation of RPG Nanocrystal Dispersions
Bottom-Up Approach
Nanocrystal dispersions were prepared by anti-solvent precipitation–ultrasonication method, a bottom-up approach. Briefly, solvent phase composed of 50 mg drug dissolved in 2 mL of isopropyl alcohol (IPA). Solvent phase was added to anti-solvent phase composed of 1.0% w/v SLPS (BU-A nanocrystals) and 0.5% w/v SLPS with 0.5% w/v TPGS (BU-B nanocrystals) solutions prepared in 50 mL of Milli Q water, respectively. Continuous sonication was applied during the addition of solvent phase using high intensity ultrasonicator (Vibra-Cell, Sonics and Material, Inc. Newtown, CT; at amplitude 75%, for 4 min, with 30:05 on: off cycle). Resulting dispersions were kept under vacuum at RT to remove IPA for 2 h. Plain precipitated drug was obtained by adding the drug solution to plain water without stabilizer.
Top-Down Approach
Wet media milling, a well proved top-down approach was employed to prepare RPG nanocrystals. Prior to beginning of the milling process, 50-mg drug powder was dispersed in 50-mL aqueous stabilizer solution containing 1.0% w/v SLPS (TD-A nanocrystals) and SLPS with TPGS 0.5% w/v (each) (TD-B nanocrystals) using a mechanical stirrer to form microsuspensions, followed by homogenization using ultra-turrax (ULTRA-TURRAX, T25 basic, IKA® India Private Limited, Bangalore, India) at 11000 RPM for 10 min to break lumps of drug present if any. Further, these microsuspensions were subjected to wet milling with grinding media (2.0 mm diameter balls) using a planetary ball mill model PM 100 (Retsch Inc., Newtown, PA, USA), equipped with a single zirconia milling chamber of 250-mL volume capacity. Milling process was performed with the rotational frequency of 100 rpm, for 14 h, at RT to obtain unimodal size distribution. Plain wet milled RPG crystals were obtained by adding the pure RPG to plain water without stabilizer.
Downstream Processing of Nanocrystal Dispersions
In order to retrieve the prepared nanosuspension in powder form, prepared nanocrystal dispersions were freeze-dried. Nanocrystal dispersions snap-frozen previously in a deep freezer at −80°C were subjected to freeze drying (FD) process using FreeZone® 2.5 L Freeze Dry System (Labconco Corporation Model 7670560, Kansas city, USA). The FD was carried out for 12 h, at −40°C condenser temperature with 0.45 mbar vacuum. Mannitol was used as cryoprotectant in the 2% w/v concentration. The resulting lyophilised powder was used for solid state characterization of nanocrystals.
Characterization of Nanocrystals
Equilibrium Solubility Measurement
The equilibrium/saturation solubility of pure/unprocessed RPG and lyophilized nanocrystals were determined in various media, including distilled water (DW), FaSSGF pH 1.6, FeSSGF pH 3.0, and pH 5.0, phosphate buffer solution (PBS) pH 6.8 and pH 7.4. The solubility measurement was carried out as per the method described above. All solubility experiments were performed in triplicate and the data obtained was expressed as mean ± SD of three measurements.
Drug Content
Lyophilised nanocrystals equivalent to 10 mg of RPG were weighed accurately and dissolved in 10 mL of methanol. The resulting solution was vortexed for 5 min and filtered through 0.1 μm PTFE syringe filter (Whatman Inc., Clifton, NJ, USA). The filtrate was then diluted suitably with methanol, and RPG content was determined by measuring the absorbance at 241 nm using UV-Visible spectrophotometer. All the samples were analyzed in triplicate, and the data obtained was expressed as mean values ± SD of three measurements.
Crystal Size, Size Distribution, and Zeta Potential
The crystal size and size distribution of nanocrystal dispersions were measured by laser diffractometer by Mastersizer 2000 SM version 2.0 (Malvern Instruments Ltd., Malvern, UK.) equipped with small volume dispersion unit (Hydro 2000 μ Precision). The principle of Mie theory (dispersant refractive index (RI) = 1. 33, the real particle RI = 1. 57 and imaginary part of the particle RI = 0. 001) was used for particle size calculation. Particle size values were recorded as volume-based 90% (d 0.9) diameter percentiles.
Zeta potential was measured using Zetasizer Nano ZS 90 (Malvern Instruments Ltd., Malvern, UK) upon dilution with Milli Q water.
All the measurements were carried out in triplicate, and the data was expressed as mean values ± SD of three measurements.
Surface Morphology using Scanning Electron Microscopy
The shape and surface morphology of the pure RPG and prepared nanocrystals were examined using scanning electron microscope (SEM) (JEOL JSM-6360-A). Samples were placed on a carbon specimen holder and then coated with platinum in an auto fine coater (JEOL JFC 1600).
Fourier Transform Infrared Spectroscopy
Jasco FTIR-4100 Emission spectrophotometer was used to record the Fourier transform infrared spectroscopy (FTIR) spectrum of pure RPG, SLPS, TPGS, and nanocrystals. The sample (2–3 mg) was grounded with dry potassium bromide, and the prepared sample was scanned from 4000 to 400 cm−1 wave number.
Differential Scanning Calorimetry
The thermal characteristics of pure RPG and prepared nanocrystals were analyzed using differential scanning calorimetry (DSC). DSC analysis was performed using a Mettler Toledo Star 821e thermal analyzer (Mettler Toledo, Switzerland) in a dry nitrogen atmosphere. Indium was used as a reference to calibrate the enthalpy scale and DSC temperature. Approximately 5–10 mg of each sample was sealed in pierced aluminum pan, and the heating curves were recorded at a scan rate of 10°C/min from 30 to 150°C, under a stream of nitrogen.
Powder X-ray Diffraction
Powder X-ray diffraction (PXRD) diffractograms of pure RPG and prepared nanocrystals were recorded using X-ray diffractometer PW 1729 (Philips, Eindhoven, Netherland) with a monochromatized CuKα line (1.5442Ǻ) as the source of radiation. Standard runs using a 40Kv voltage, a 40 mA current, and a scanning rate of 0.02° min−1 over a 2θ range of 5–50° were used.
In vitro Dissolution Study
In vitro dissolution studies of the nanocrystals equivalent to 2 mg of RPG was performed and compared with pure RPG using a USP Type II dissolution test apparatus (Lab India DS-8000, India). DW, FaSSGF pH 1.6, and FeSSGF pH 5.0 (400 mL each) maintained at 37 ± 0.5°C were used as three different dissolution media and stirred at 75 rpm. The sampling port was fitted with a 0.1 μm filter, which was retained in the dissolution medium throughout the dissolution studies. Samples (3 mL) were collected periodically (5, 10, 15, 30, 45, 60, 90, and 120 min) and replaced with a fresh dissolution medium. The dissolved RPG content was determined using UV-Visible spectrophotometer by measuring absorbance at 241 nm vs. respective blanks. All dissolution experiments and sample analysis were carried out in triplicate.
Physical Stability and Crystal Growth during Storage
The physical stability and the ability of nanocrystal dispersions to retain the crystal size (i.e., absence of crystal growth) were assessed for 3 months at 25°C/60% RH, 40°C/75% RH as mentioned in ICH guidelines Q1A (R2) (27). During this study, nanocrystal dispersions were stored in glass bottles (type I glass) with polypropylene caps. Samples were withdrawn periodically (0, 1, and 3 months) and analyzed for the crystal size and zeta potential (28).
Fasted and Fed State Bioavailability Study in Male Wistar Rats
Experimental Animals and Research Protocol Approval
A research proposal was prepared according to the guidelines of the Committee for the Purpose of Control and Supervision of Experiments of Animals (CPCSEA). The experimental protocol was approved by the Institutional Animal Ethics Committee (IAEC) of Poona College of Pharmacy, Pune (CPCSEA/15/2014). Male Wistar rats (200–250 g) were purchased from the National Toxicology Centre (Pune, India). Animals were housed together under standard conditions of temperature (24 ± 1°C), relative humidity (55 ± 10%), and 12-h light/dark cycles throughout the experiment. Animals had free access to commercially available standard pellet diet (4% carbohydrates, 22.15% protein, 4.15% fat, 2.46% glucose, 1.8% vitamins, 3600 Kcal) (Pranav Agro Industries, Sangli, Maharashtra, India) and filtered water ad libitum unless otherwise stated. Animals were acclimatized for 1 week prior to the initiation of treatment. During this acclimatization period, the health status of the animals was monitored daily.
Single-Dose Oral Administration
Single-dose oral pharmacokinetic studies were carried out for two different physiological conditions viz. fasted and fed state. Twenty-four male Wistar rats were used in the study. Animals were divided into four groups (n = 6). For the fasted state study, animals were fasted for 23 h before dosing and for 10 h post-dosing as a fasted condition (29). The dosing was done as follows
-
Group I
Vehicle control group
-
Group II
Pure RPG (2 mg/Kg body weight, RPG dispersed in DW)
-
Group III
TD-A (2 mg/Kg body weight) wet milled RPG nanocrystal formulation containing SLPS as a stabilizer
-
Group IV
TD-B (2 mg/Kg body weight) wet milled RPG nanocrystal formulation containing SLPS along with TPGS
Fed states pharmacokinetic studies were carried out on the same group of animals after 2 week wash out period. For the fed condition, the animals were fasted for 22 h until 30 min prior to the dosing and were then provided with 10–15 g of manipulated normal pelleted diet to high-fat diet (composed of ghee, groundnut oil, water, tween 80, and propylene glycol with 40, 20, 25, 10, and 5% of calories in mL, respectively) (30). Prior to dosing the complete consumption of food was ensured. The dosing schedule was same as described above. Blood samples were collected from the retro-orbital plexus at pre-determined time intervals (0-h pre-dose and 0.25, 0.5, 1, 1.5, 2, 4, 8, 12 and 24-h post-dose) in EDTA tubes to prevent clotting. Samples were centrifuged at 7000 rpm for 20 min at 4°C. Separated plasma was collected and stored in refrigerator at −20°C until analysis.
Bioanalytical Method
RPG in plasma was determined by RP-HPLC method reported previously (31), with slight modification. Chromatographic separation was carried out with a Thermo Scientific ODS Hypersil C-18 column (4.8 mm × 150 mm; 5 μm) attached with guard column. The mobile phase used during HPLC analysis consisted of acetonitrile and 0.01 M ammonium formate pH 2.7 adjusted using formic acid 90% in the ratio of 75:25 (v/v). The flow rate was kept at 0.7 mL/min. Indomethacin (IND) was used as an internal standard. The detector consisted of UV/VIS (Jasco UV 2075) model operated at a 241-nm wavelength.
Preparation of Standard Solutions and Plasma Samples
Standard solutions of RPG and IND were prepared by dissolving 10 mg of standard drug in 10 mL of methanol (1000 μg/mL). Further dilutions were prepared from stock solutions with methanol.
Liquid-liquid extraction process was used for further processing of plasma samples. A 50 μL of internal standard solution (10 μg/mL in mobile phase) was added to the 200 μL plasma sample, mixed well with vortex mixer, and kept at RT for 20 min. To the above mixture, 200 μL extraction solvent (methanol) was added and vortexed for 10 min. The mixture is then centrifuged (AllegraTM 64 R Centrifuge, Beckman-Coulter India Pvt. LTD, Mumbai, India) at 7000 rpm for 20 min at 4°C. The supernatant was collected and 20 μL volume was injected into the HPLC system.
Data Analysis
The pharmacokinetics parameters such as Cmax, Tmax, AUC0-24, AUC0-∞, AUMC0-∞, T1/2, and MRT were calculated for each individual set of data by non-compartmental analysis using WinNonLin version 4.0 (Pharsight, USA).
Hypoglycemic Activity in Streptozotocin-Induced Diabetic Rats
Induction of Experimental Diabetes and Determination of the Serum Glucose Level
Diabetes was induced by a single intra-peritoneal injection of a freshly prepared streptozotocin (STZ) solution (50 mg/Kg in cold sodium citrate buffer 0.1 M, pH 4.5) to overnight-fasted rats (32). Diabetes was identified by polydipsia and polyuria along with measuring the non-fasting plasma glucose levels after 48 h of injection of STZ (33). Animals, which did not develop more than 300 mg/dL glucose levels, were rejected. The rats were classified into five different groups (n = 6). The first and second group served as control and consisted of non-diabetic (Normal) and diabetic rats (vehicle control), respectively. Group III was administered with pure RPG (2 mg/Kg, RPG dispersed in DW), while Group IV and Group V received nanocrystals TD-A and TD-B, respectively (dose 2 mg/Kg body weight).
Oral samples were administered intra-gastrically using a feeding tube. Blood samples were collected pre-dose (0 min) and at 5, 15, 30, 60, 120, 240, 480, 720, and 1440-min post-dose. Blood samples were allowed to clot at RT, centrifuged at 7000 rpm for 20 min at 4°C, and the serum was separated. The resultant serum was diluted with a working solution (GOD-POD method), and the absorbance was measured at 505 nm. The percent glucose in milligrams per decilitre (mg/dL) was determined using Eq. (1).
| 1 |
Statistical Analysis
All the data reported are expressed as mean ± standard error of the mean (S.E.M), and statistical analysis was performed by two-way analysis of variance (ANOVA) followed by Bonferroni post hoc test, performed using Graph Pad Prism 5.0 software. The values were considered to be significantly different when the p < 0.05 compared to the vehicle control and pure RPG group.
RESULTS AND DISCUSSION
Preparation of Nanocrystals
Commercial pure RPG used in this study was characterized by relatively larger crystal size; 206 ± 4.12 μm (Table I). Size reduction using plain DW as anti-solvent by anti-solvent precipitation–ultrasonication and wet media milling of pure RPG lead to significant (p < 0.05) reduction in crystal size (14.51 and 7.61-fold, respectively) (Table I). The resulted crystal size was undesirable (>500 nm) and had a wide range of size distribution. Furthermore, visual observation of formulated dispersions showed signs of instability (aggregation) after 2 h storage at RT from the time of preparation. This might be due to the dramatically increased surface energy attributed to the smaller crystal size. Therefore, the key of formulating stable nanocrystal dispersions by either of approach was compensating the extra free energy associated with newly exposed crystal surfaces.
Table I.
Crystal Size, Size Distribution, and Zeta Potential of Pure Drug Repaglinide and Nanocrystals Prepared Without and With the Stabilizer by Bottom-Up (BU) and Top-Down (TD) Approach
| Dispersions | Crystal size (μm ± SD) | Uniformity | Zeta potential (mV ± SD) |
|---|---|---|---|
| Pure RPG | 206.55 ± 4.12 | 0.91 ± 0.05 | – |
| Plain precipitated RPG | 14. 23 ± 1.26 | 0.81 ± 0.02 | – |
| Plain wet milled RPG | 27.12 ± 2.45 | 0.98 ± 0.05 | – |
| BU-A | 0.141 ± 0.86 | 0.24 ± 0.01 | −9.06 ± 7.29 |
| BU-B | 0.192 ± 0.22 | 0.28 ± 0.06 | −5.64 ± 4.34 |
| TD-A | 0. 304 ± 0.06 | 0.65 ± 0.00 | −4.41 ± 3.36 |
| TD-B | 0.331 ± 0.34 | 0.57 ± 0.04 | −1.54 ± 1.88 |
The use of an appropriate ionic or steric stabilizer at optimized concentration is vital to restrain aggregation of thermodynamically unstable dispersions. Unfortunately, there was no systematic empirical way or theoretical guideline for appropriate stabilizer selection and optimization. However, few studies developed an empirical relationship between stabilizer efficacy and the drug properties to assist stabilizer selection. We have screened various stabilizers based on their influence on the intrinsic solubility of the drug, and concentration was optimized on trial and error basis.
Selection of Stabilizer
Stable nanocrystal formulation results with the use of stabilizers having a minimal/negligible effect on drug solubility (25). The solubility of the formulated drug in the stabilizer solution plays a significant role in increasing the particle size on storage owing to Ostwald ripening. As a stabilizer of the formulated nanocolloidal system, it should possess little effects on the drug solubility. Otherwise, it may induce the agglomeration and/or Ostwald ripening of the nanocolloidal system.
The suitable stabilizer was screened based on saturation solubility studies in aqueous stabilizer solution. Table II depicts that SLPS (1% w/v) minimally affects the intrinsic aqueous solubility of the RPG (0.022 mg/mL) whereas other stabilizers (SLS, Tween 80, Pluronic F127 & Pluronic F68) affect it significantly (p < 0.05). The results demonstrated that the combination of SLPS and TPGS (except 0.5% w/v of each) significantly (p < 0.05) increased RPG solubility. The minimum effect of 1% w/v SLPS and its combination with TPGS at 0.5% w/v each on the intrinsic solubility of RPG might be attributed to a pH of the solution. The pH of an aqueous 1% w/v SLPS and SLPS in combination with TPGS (0.5% w/v each) solution was found in the range of 4.2–5.4 and 4.6–5.8, respectively. Whereas pH values for 1% aqueous solution of Pluronic F127, Pluronic F68, SLS, and Tween 80 were in the range of 6.0–8.0, 6.50–7.10, 7.5–9.5, and 6.0–7.0, respectively (determined using laboratory digital pH meter). Various studies have reported that compound/molecules had lower solubility at pH values corresponding to its isoelectric point (pHI); however, solubility is higher case by case at pH values above or lower than the pHI. Since the pH values for aqueous SLPS (1% w/v) and SLPS in combination with TPGS (0.5% w/v each) solutions are closer to or nearly equivalent to that of RPG (5.0) (4), this lead to least influence on RPG intrinsic solubility. Hence, SLPS was selected and investigated for its role as a stabilizer for nanocrystals and TPGS was used as an oral absorption enhancer.
Table II.
Solubility of Pure Repaglinide in Various Stabilizer Solutions
| Surfactant/polymeric stabilizer (w/v) | Drug concentration (mg/L ± SD)* |
|---|---|
| 1% SLPS | 140.22 ± 0.66 |
| 1% Pluronic F 68 | 836.15 ± 00.21 |
| 1% Pluronic F127 | 768.36 ± 00.84 |
| 1% Tween 80 | 516.48 ± 00.15 |
| 1% SLS | 982.54 ± 00.32 |
| 1% TPGS | 254.27 ± 00.11 |
| 1% SLPS + 0.1% TPGS | 369.64 ± 00.09 |
| 1% SLPS + 0.25% TPGS | 391.18 ± 00.56 |
| 1% SLPS + 0.5% TPGS | 453.09 ± 00.00 |
| 1% SLPS + 0.75% TPGS | 516.74 ± 00.43 |
| 1% SLPS + 1.0% TPGS | 561.94 ± 00.97 |
| 0.5% SLPS + 0.5% TPGS | 312.86 ± 00.50 |
*n = 3
Characterization of Nanocrystals
Crystal Size, Size Distribution, and Zeta Potential
The average crystal size, size distribution, and zeta potential of RPG nanocrystals prepared by bottom-up and top-down approach are shown in Table I. Both methods showed significant (p < 0.05) size reduction with the unimodal size distribution. It was observed that nanocrystals prepared by the bottom-up approach yielded smaller mean crystal size of 0.141 ± 0.86 μm and 0.192 ± 0.22 μm for BU-A and BU-B, respectively, than the top-down approach, where crystal size was found to be 0.304 ± 0.06 μm and 0.331 ± 0.34 μm, for TD-A and TD-B, respectively. The difference in crystal size obtained through both the techniques could be attributed to the principle involved in the actual crystal size reduction.
The mechanism of nanocrystal formation by ultrasonication involves rapid transformation of less ordered (amorphous form) to highly ordered lattice structure (crystal form). This usually takes place by disrupting the crystal agglomerates and by intensifying adsorption of polymer on the newly formed crystal surface, thereby reducing the surface energy (34) resulting in smaller crystal size with uniform size distribution (35,36). In case of wet media milling, size reduction (formation of new surface) is achieved by collision between adjacent crystal or crystal and wall of the milling chamber or compression of crystals between adjacent grinding balls. The process is comparatively slower with resultant larger crystal size than that achieved by ultrasonic waves.
Comparable zeta potential (ZP) values were observed for formulations prepared using either processing method (Table I). Prepared nanocrystals exhibited lower ZP values (within 0 to –10 mV), indicating that they are neutral in nature and had neither net positive charge (ZP > + 30 mV) nor negative charged (ZP > − 30 mV). As SLPS has both hydrophobic and hydrophilic parts in its structure, its hydrophobic part probably gets adsorbed over hydrophobic drug nanocrystals, whereas long loops or hydrophilic tails extends out into an aqueous phase. This long polymeric chains shift the plane of shear at which the ZP is measured at a larger distance from the particle surface. This leads to an increase in thickness of the diffusion double layer, and as a consequence, these measured ZP values get lowered. ZP values close to zero are indicative of complete coverage of the RPG nanocrystals by SLPS. The steric stabilization offered by SLPS will contribute to enhance stability during storage.
To investigate which approach yielded the best stable nanocrystals, the experimental storage stability study was performed. The study involved analyzing formulations stored at RT periodically (0, 15, and 30 days) for change in mean crystal size as a function of time. The increase in crystal size was observed with both nanocrystals BU-A and BU-B prepared by the bottom-up approach; in contrast, insignificant (p > 0.05) crystal growth was observed in top-down approach at the end of 30 days (Fig. 1a). Agglomeration, crystal growth due to Ostwald ripening, or both may be the contributing factors for the observed crystal size growth. The higher increase in crystal size observed in formulations made by bottom-up approach suggests that Ostwald ripening might be the key driving force. Ultrasonic waves increase the RPG solubility, and according to the Lifshitz-Slyozov-Wagner (LSW) theory (37), the rate of Ostwald ripening is directly proportional to the concentration of the dispersed phase in the system. Another possible reason for crystal size growth in formulation prepared by the bottom-up approach may be the incomplete transformation of amorphous (thermodynamically unstable) particles to the crystals (thermodynamically stable) of nanometer size range, and consequently solubilization of fractional amorphous particles in the medium, owing to higher saturation solubility. Under such super saturation conditions, some of the dissolved drug gets re-precipitated/re-deposited on to the larger crystals with a lower surface energy leading to crystal size growth.
Fig. 1.
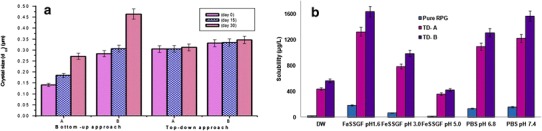
a Experimental storage stability of preliminary batches. b Saturation/equilibrium solubility of RPG in different dissolution media
The results concluded that the nanocrystal dispersions prepared by wet media milling were more stable than that prepared using the anti-solvent precipitation ultrasonication method at the same stabilizer concentration. Hence, further studies were carried out using nanocrystals prepared via top-down approach.
Downstream Processing of Nanocrystal Dispersion
RPG nanocrystals being formulated as aqueous dispersions, water needs to be removed to obtain powder state nanocrystals for solid state characterization. Owing to better stability of nanocrystals at a low temperature, FD is a popular method to convert them into a solid form (38). FD involves removal of water from frozen samples through sublimation. Three different concentrations of mannitol viz. 2, 4, and 6% were explored and compared with the quality of the product obtained by freeze drying the nanocrystal dispersion as such. The yield for both the processed nanocrystal dispersions was nearly 100% for all the three mannitol concentrations. Freeze-dried cakes were extremely fluffy and presented very high volumes when mannitol was used at a high concentration (4 and 6%) whereas FD without mannitol produced sticky cakes. The fluffy and sticky nature of these freeze-dried products could make difficulty in further utilization. FD with mannitol at a concentration of 2% produced free flowing powder and also exhibited excellent re-dispersibility in DW.
The drug content analysis of FD product showed that formulated nanocrystal dispersions had 98.18 ± 1.10 and 99.92 ± 3.02 (% ± SD) of RPG content in TD-A and TD-B, respectively. As this process gives higher yield than other downstream processes such as spray drying, it is also important for the solidification of expensive drugs.
Saturation/Equilibrium Solubility
Determination of solubility in a medium of wide pH range and DW gives the complete idea with regards to drug behavior in various pH conditions. Data obtained from the solubility study also helps to predict the drug absorption and to select the appropriate dissolution medium. The saturation/thermodynamic solubility of pure RPG in DW (12.02 μg/mL) was enhanced nearly by 19.86 and 25.67 times as compared with nanocrystal TD-A and TD-B, respectively (Fig. 1b). The results obtained were in good agreement with Ostwald-Freundlich equation and signifies the importance of particle size reduction to improve the aqueous solubility of poorly soluble drug candidates. This significantly improved (p < 0.05) aqueous solubility may benefit the in vivo dissolution and absorption of the drug.
Saturation solubility profile in media with different pH values showed that RPG nanocrystals exhibit significantly higher solubility in all the media as compared to pure RPG. The solubility of pure RPG was 2.70 and 12.24 times lower in FeSSGF pH 3.0 and pH 5.0, respectively, as compared to FaSSGF pH 1.6 (Fig. 1b). Higher solubility in FaSSGF to that in water or FeSSGF could be attributed to the pH of the medium as well as the micellar solubilization due to presence of Na taurocholate and lecithin (29). However, nanocrystals exhibit superior solubility than pure RPG in all the media, but the highest solubility was observed in FaSSGF pH 1.6 as well as FeSSGF pH 5.0.
The solubility data suggest that RPG had characteristic pH dependent solubility with minimum solubility in FeSSGF pH 5.0. Since chemically, RPG is a benzoic acid derivative and it possesses one weakly basic and one weakly acidic group in aqueous solutions; RPG exists as an ampholytic neutral molecule. Two neutral forms of RPG (zwitterionic and uncharged form) exist at pHI (5.0) (4), resulting in low solubility at pH 5.0.
Surface Morphology of Nanocrystals
The shape and surface morphology of the pure RPG and lyophilised RPG nanocrystals were observed by SEM (Fig. 2). The pure RPG powder exhibited as long, sharp, irregular, needle shaped crystals (Fig. 2a). The lyophilised nanocrystals (TD-A and TD-B) had smoother edges with slightly spherical structure (Fig. 2b and c). The spherical shape in case of nanocrystals can be attributed to the mechanism involved in size reduction by wet media milling. The process typically involves the friction between the crystal surface and with the wall of grinding chamber or between two crystals rather than crystal breaking. Another contributing factor could be the adsorption of SLPS on the hydrophobic drug particles which further help to control the crystal growth.
Fig. 2.

Surface morphology of pure RPG (a), nanocrystals TD-A (b), and TD-B (c)
Drug-Stabilizer Interaction Study
Typical FTIR spectrum of pure RPG was recorded and compared with SLPS, TPGS, and freeze-dried nanocrystals (Fig. 3a) to interpret the drug-stabilizer interaction at the molecular level. Pure RPG showed prominent, sharp peaks (Fig. 3a (a)) at 3306 cm−1 (strong) (-N-H stretching in secondary amine group), 1690 cm−1 (strong) (>C = O stretching in amide functional group), 1710 cm−1 (strong) (>C = O stretching in carboxylic acid group), 2500–3000 cm−1 (weak) (characteristic peaks due to -O-H stretching), 2943 cm−1 (medium) (-C-H stretching) ,and 1450–1650 cm−1 (weak) (>C = C < stretching in aromatic ring). Spectra corresponding to SLPS (Fig. 3a (b) and TPGS (Fig. 3a (c)) showed characteristic peaks confirming the pure form.
Fig. 3.
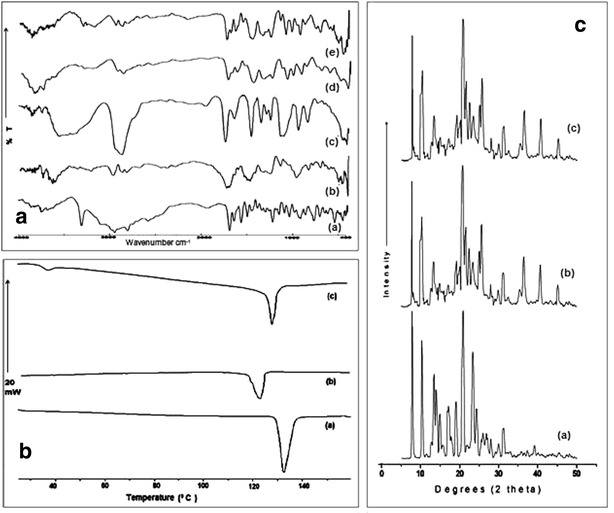
a FTIR spectra of pure RPG a, SLPS b, TPGS c, freeze-dried nanocrystals TD-A d, TD-B e. b DSC thermograms of pure RPG a, nanocrystals TD-A b, TD-B c. c PXRD diffractograms of pure RPG a, freeze-dried nanocrystals TD-A b, TD-B c
In case of nanocrystals, all characteristic peaks of RPG were observed, indicating no chemical instability between drug and stabilizer. However, few peaks appeared with reduced intensity or found to be disappeared in the case of nanocrystals. This might be due to the presence of higher drug: polymer ratio (1:10) in prepared nanocrystals. The characteristic broad peaks observed in the region from 3300 cm−1 to 3500 cm−1 may be due to the formation of H-bonding between RPG secondary amine group and >C = O group in stabilizer. In addition to this, peak due to > C = O stretch of acid functional moiety (at 1710 cm−1) shifted towards lower frequency at 1695 and 1680 cm−1 with TD-A (Fig. 3a (d)) and TD-B (Fig. 3a(e)), respectively. This hypsochromic shift further confirms the H-bonding of RPG carboxyl groups with numerous -OH groups in stabilizer. Thus, the appearance of broad peaks and hypsochromic shift in drug peaks signifies molecular level interaction between RPG and stabilizer used, more specifically the H-bonding. This confirms the adsorption of stabilizer over the drug crystal surface through the H-bonding.
Solid State Characterization
Thermal Behavior of Nanocrystals
The DSC thermogram of pure RPG exhibited a sharp endothermic peak at 132.64°C (Tm) with fusion enthalpy (∆H) of −123.16 J/g indicating the melting of RPG crystals (Fig. 3b (a)). The appearance of endothermic peaks with both nanocrystals TD-A and TD-B indicated that the crystalline form of RPG was retained (Fig. 3b (b) and 3b (c)). In the thermogram of the freeze-dried RPG nanocrystals TD-A, (Fig. 3b (b)), the melting temperature (Tm) and fusion enthalpy (∆H) were reduced to 123.13°C and −85.68 J/g, respectively, as compared to pure RPG. Similarly, in case of TD-B (Fig. 3b (c)) also, the Tm and ∆H were reduced to 129.84°C and −93.66 J/g, respectively, than that of the pure RPG. One additional characteristic endothermic peak was observed in TD-B thermogram at 38°C owing to the presence of TPGS (Fig. 3b (c)). However, the endothermic peak of RPG nanocrystals drifted 9 and 3°C to the left as compared to pure RPG along with lowered fusion enthalpy, suggesting reduced crystallinity due to size reduction of the crystals.
Powder X-ray Diffraction Study
PXRD diffractograms confirm that the wet media milling operation does not interfere with the RPG crystalline state as the diffraction pattern for the RPG is conserved for nanocrystals (Fig. 3c). The PXRD pattern of the pure RPG (Fig. 3c (a)) showed characteristic high energy diffraction peaks at 2 theta values between 8° and 32° indicating the crystalline structure of RPG. The only difference observed between unmilled/pure RPG and RPG nanocrystals TD-A and TD-B ((Fig. 3c (b) and 3c (c)) lie in peak intensities. The differences in the relative intensities of their peaks might be attributed to reduced crystallinity with nanocrystals. This seems to be in agreement with the DSC data. Another interesting feature represented by the PXRD diffractograms is that mannitol used during freeze drying operation showed high energy diffraction peaks at 2 theta values between 9° and 45°, which masked the characteristic diffraction peaks of RPG in both freeze-dried formulation. Such characteristic diffraction pattern indicates mannitol exists in crystalline state rather than amorphous that shall be of relevant importance when considering long-term stability of nanocrystals.
In vitro Dissolution Profile
Dissolution in Distilled Water
The in vitro dissolution studies of pure RPG was performed in DW and compared with formulated nanocrystals. The pure microcrystals did not achieve complete dissolution during the 120-min test period and only 32.88 ± 4.36% of the drug was dissolved over 120 min; in contrast, nanocrystals TD-A and TD-B showed 80.38 ± 6.21 and 90.80 ± 4.53 (% ± SD) of cumulative drug dissolution at 15 min, respectively (Fig. 4a).
Fig. 4.
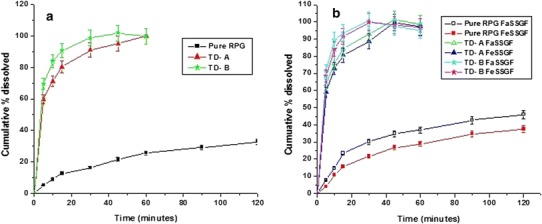
a In vitro dissolution profiles of pure drug, nanocrystals TD-A, and TD-B in distilled water. b In vitro dissolution profiles of pure drug, nanocrystals TD-A and TD-B in the fasted and fed state bio-relevant dissolution media
Since the nanocrystal has smaller crystal size, this leads to an increased saturation concentration around the nanocrystals (Freundlich–Ostwald equation), the decreased diffusion layer thickness (Prandtl equation), and an increase in surface area exposed to dissolution medium (39). All these parameters contribute to an increased dissolution rate (Noyes and Whitney equation). Although TD-B nanocrystals had slightly larger crystal size (Table I), it showed faster dissolution rate (90.80 ± 4.53%) than TD-A (80.38 ± 6.21%) at 15 min. This might be attributed to additional wetting and interfacial activity of TPGS adsorbed over the surface of nanocrystals in case of TD-B. Since the saturation solubility is increased, the concentration gradient across gut lumen and blood would also increase, resulting in increased absorption by passive diffusion (40).
Dissolution in Bio-relevant Media
The in vitro dissolution study of pure RPG and nanocrystals was also carried out in FaSSGF and FeSSGF, in order to mimic in vivo dissolution and to investigate the influence of food on in vivo absorption (Fig. 4b). Data obtained from this study showed that the dissolution rates of pure RPG in FeSSGF was similar to and those in FaSSGF was faster than those obtained in DW. Dissolution rates of both the nanocrystal formulations in FaSSGF and FeSSGF were higher as compared to pure RPG. The amount of pure RPG dissolved was not more than 50% over a period of 120 min in both FaSSGF (45.85%) and FeSSGF (39.48%). In contrast, the nanocrystals TD-A and TD-B showed more than 60% dissolution at the end of 5 min. Cumulative percent dissolution of pure RPG was 13.05% lower in FeSSGF compared to FaSSGF. However, such variation in dissolution rate was negligible for the RPG nanocrystals. This could be due to the faster dissolution rate of nanocrystals owing to their smaller particle size. These promising results encouraged us to perform further in vivo studies in order to investigate the food effect on in vivo absorption.
Oral Bioavailability Study
The in vivo bioavailability studies of pure RPG and RPG nanocrystals were carried out in male Wistar rats. In fasted as well as fed state, the nanocrystal formulations showed significant (p < 0.05) improvement in the pharmacokinetics profiling compared to the pure drug (Fig. 5). In the fasted state, the pharmacokinetic parameters mainly, Cmax and AUC0-24 were found to be increased by 2.77 and 10.00 times for TD-A and 4.30 and 14.90 times for TD-B, respectively, as compared to pure RPG (Table III). Both the nanocrystal formulations showed reduction in time required to reach the peak plasma concentration by 30 min and significant (p < 0.05) increase in mean residence time attributed to their faster dissolution rate and higher plasma concentration achieved. Significant difference between Cmax and AUC0-24 values was observed between TD-A and TD-B. The Cmax and AUC0-24 values of TD-B were higher by 1.55 and 1.46 times, respectively, as compared to TD-A. These results were in good accordance with the results obtained from the in vitro dissolution study.
Fig. 5.
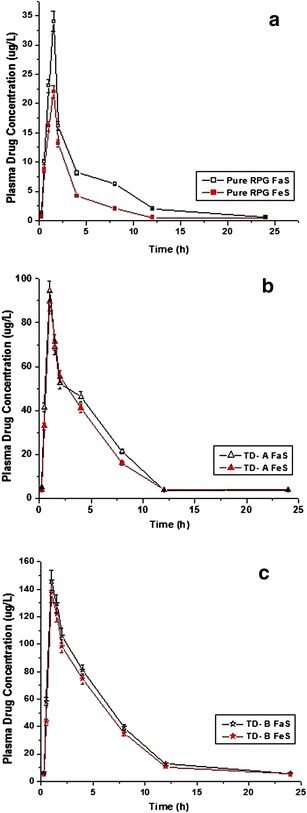
Plasma concentration-time profiles resulting from single oral administration at a dose of 2 mg/kg body weight in male Wistar rats; a pure RPG, b nanocrystals TD-A, and c nanocrystals TD-B
Table III.
Pharmacokinetic Parameters Resulting from Single Oral Administration of Pure Repaglinide and Nanocrystals TD-A and TD-B with Fasted and Fed Food Conditions in Male Wistar Rats
| Food state | Parameters | Group I Pure RPG | Group II nanocrystals TD-A | Group III nanocrystals TD-B |
|---|---|---|---|---|
| Fasted | Cmax (μg/L) | 34.01 ± 02.96 | 94.41 ± 04.54* | 146.41 ± 14.42*** |
| Tmax (h) | 01.5 ± 00.50 | 01.00 ± 00.50 | 01.00 ± 00.00 | |
| AUC0-24 (μg.h/L) | 3.08 ± 0.98 | 24.11 ± 3.27 | 33.81 ± 4.78 | |
| AUC0-∞ (μg.h/L) | 25.76 ± 04.17 | 257.75 ± 07.32*** | 384.07 ± 09.54*** | |
| AUMC0-∞ | 70.49 ± 11.08 | 779.16 ± 40.93 | 1192.58 ± 57.92 | |
| MRT (h) | 02.74 ± 01.11 | 03.02 ± 01.25 | 03.11 ± 01.35 | |
| Vd (L) | 236.78 ± 17.15 | 27.43 ± 05.71 | 19.05 ± 06.24 | |
| Kel (L/h) | 00.14 ± 00.02 | 00.11 ± 00.06 | 00.10 ± 00.02 | |
| Cl (L/h) | 00.013 ± 00.00 | 00.13 ± 00.02 | 00.19 ± 00.03 | |
| T 1/2 (h) | 04.96 ± 00.89 | 06.35 ± 01.39 | 06.75 ± 02.73 | |
| Fed | Cmax (μg/L) | 22.01 ± 03.65 | 89.72 ± 07.32* | 137.36 ± 09.32*** |
| Tmax (h) | 01.5 ± 00.25 | 01.00 ± 00.00 | 01.00 ± 00.02 | |
| AUC0-24 (μg.h/L) | 2.54 ± 0.78 | 22.22 ± 3.12 | 30.84 ± 6.12 | |
| AUC0-∞ (μg.h/L) | 18.57 ± 06.13 | 241.63 ± 13.51*** | 355.88 ± 10.69*** | |
| AUMC0-∞ | 48.48 ± 16.01 | 735.86 ± 35.51 | 1112.95 ± 80.22 | |
| MRT (h) | 02.61 ± 01.50 | 03.05 ± 01.33 | 03.13 ± 01.02 | |
| Vd (L) | 301.64 ± 41.58 | 29.55 ± 04.24 | 20.74 ± 06.43 | |
| Kel (L/h) | 00.16 ± 00.050 | 00.11 ± 00.05 | 00.10 ± 00.02 | |
| Cl (L/h) | 00.01 ± 00.006 | 00.12 ± 00.00 | 00.18 ± 00.01 | |
| T1/2 (h) | 04.34 ± 01.437 | 06.46 ± 02.66 | 06.85 ± 01.24 |
Data were analyzed by one-way analysis of variance followed by Bonferroni post hoc test, with p < 0.05 considered to indicate significance
*p < 0.05 vs. pure drug repaglinide
**p < 0.01 vs. pure drug repaglinide
***p < 0.001 vs. pure drug repaglinide
The highest bioavailability with nanocrystals TD-B, compared to the pure RPG and nanocrystals TD-A can be attributed to the inhibitory effect of adsorbed TPGS on the P-gp efflux transporter (41) as well as on CYP3A4 the main enzyme (21,22) involved in the extensive first-pass metabolism of RPG. TPGS has a low critical micelle concentration (0.02%). The concentration of TPGS present in TD-B nanocrystals is 5 mg/mL. Hence, it can be concluded that the concentration of TPGS that can be achieved after oral administration of TD-B nanocrystals to rats is sufficient to exert these inhibitory effects. Earlier Dintaman et al. and Parsa et al. have concluded that the enhanced oral bioavailability of various drugs co-administered with TPGS may be due to inhibition of P-gp in the intestine (17,42). TPGS acts by competing for drug binding sites on P-gp, followed by rigidization of lipid bilayer leading to an indirect destabilization of efflux transporter P-gp (43).
In the fed state, pure RPG showed decrease in Cmax by 35.27% and AUC0-24 by 27.90% as compared to the fasted state (Table III) indicating that the rate and extent of RPG gets affected negatively in the presence of food. In case of nanocrystal formulations such fasted/fed state, variation was not statistically significant (p > 0.05). These results suggest that such negative food effect on the oral bioavailability of the RPG can be minimized or eliminated by developed nanocrystal formulation. Nanocrystals decrease the food effect owing to their faster dissolution rate even in the presence of food (15,29). In such cases, the absorption might be a permeability rate limited rather than dissolution and the variation in absorption/bioavailability resulting from difference in dissolution rate between fasted/fed conditions can thus be eliminated (41).
Therefore, it can be concluded that the extensive particle size reduction leads to improvement in the oral bioavailability of poorly soluble drugs. Improved absorption and associated bioavailability of nanocrystals is thus attributed to their higher saturation solubility and dissolution velocity in gastrointestinal fluid. It results in higher drug concentration gradient across the biological membrane.
Hypoglycemic Activity
The STZ-induced experimental animals were used as a type 2 DM model. STZ acts by selective destruction of pancreatic β-cells that are responsible for insulin secretion. Irregular functioning of pancreatic β-cells results in a DM (44,45). RPG exerts its acute hypoglycemic action by promoting insulin release from pancreatic β-cells. RPG pharmacodynamics was characterized by determining serum glucose levels in diabetes induced experimental animals. In pure RPG treated rats, a slight decrease in serum glucose level was observed after 1 h of dosing (Fig. 6) and the significant (p < 0.001) hypoglycemic effect was observed from the second to the fourth hour after administration compared to the vehicle control group. However, after 4 h of administration, the decrease of serum glucose levels was insignificant (p > 0.05). In case of experimental animals treated with nanocrystals TD-A, a significant (p < 0.05) decrease in serum glucose level was observed at 30 min. However, it dramatically attenuated the serum glucose level from the first hour to the eighth hour after administration (p < 0.001). In case of animal group treated with nanocrystals TD-B, the onset of action was much faster (15 min) than that of the pure drug treated group (p < 0.001) and the significant hypoglycemic effect was observed up to 8 h. Nanocrystals TD-A and TD-B showed significant (p < 0.05) decline in serum glucose level compared to pure RPG from 1st h to the 8th h and from 30 min to the 8th h after administration, respectively. Pure RPG showed slower onset of action (1 h), for a short duration of time (4 h). In contrast, TD-A and TD-B showed faster onset of action (30 min and 15 min, respectively) which was retained for a prolonged period of time (8 h) as compared to pure RPG. This faster onset and longer lasting hypoglycemic effect of RPG nanocrystal formulations can be attributed to their higher rate of absorption and plasma drug concentration achieved as compared to pure RPG. Thus, the superiority of both the nanocrystal formulations than that of the pure RPG was confirmed in terms of significant (p < 0.001) hypoglycemic activity that was observed up to 8 h.
Fig. 6.
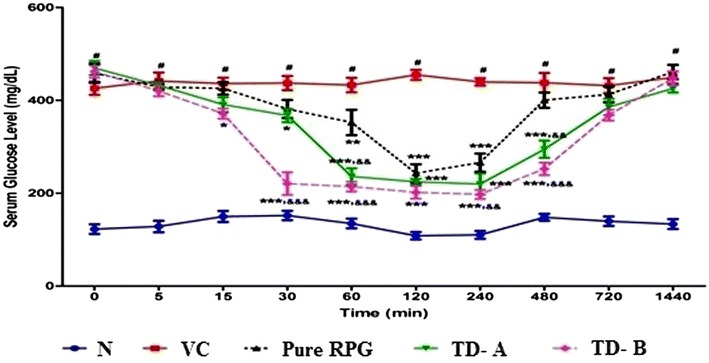
Mean serum glucose-time profiles in streptozotocin induced diabetic male Wistar rats after single oral administration of pure RPG and nanocrystals at dose of 2 mg/kg body weight
Long-Term Physical Stability on Storage
The stability study of both the nanocrystal dispersions was carried out over a period of 90 days. During the study, these samples were analyzed for change in crystal size and ZP as a function of time and temperature (Fig. 7). Samples were also examined by visual observation, for signs of flocculation if any. Both the nanocrystal formulations showed insignificant (p > 0.05) growth of crystal size and ZP throughout the stability period at 25°C/60% RH and 40°C/75% RH.
Fig. 7.
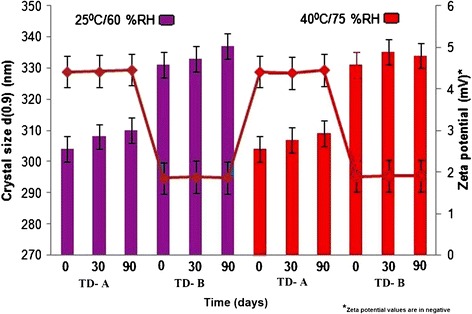
Long-term physical stability during storage
CONCLUSION
Repaglinide is the commercially available most promising anti-diabetic agent. The current study involved the formulation of pure repaglinide in the form of nanocrystals to improve its saturation solubility and dissolution rate and to study the effect of nanoformulation on the fasted/fed state bio variability. Among the two different approaches that were explored for nanocrystals formation, top-down approach was found to be a promising approach. Developed nanocrystals helped to reduce fasted and fed state variability in oral absorption associated with pure repaglinide. Repaglinide nanocrystals also showed prominent hypoglycemic activity compared to pure repaglinide. Steric stabilization with SLPS was sufficient to stabilize RPG nanocrystal dispersions for a period of 90 days. In conclusion, engineering of repaglinide nanocrystals was an effective approach in improving its oral bioavailability and in reducing the influence of food on the pharmacokinetics profile which may prove beneficial in the treatment of diabetes mellitus. However, it could be ascertained only after clinical investigation of developed nanocrystal formulations. The future work shall consist of incorporation of prepared nanocrystals in solid oral dosage form such as fast dissolving tablets.
Acknowledgments
The authors thank USV Limited (Mumbai, India) for providing a gift sample of repaglinide. The authors also thank BASF Corporation (Germany) for providing gift samples of the excipients. Rahul Gadadare would also like to thank AICTE (New Delhi, India) for providing financial support in the form of Junior Research Fellowship.
Declaration of Interest
The authors report no conflict of interest.
References
- 1.International Diabetes Federation. 2011: 5th ed.; www.idf.org/diabetesatlas/papers. Accessed 28 April 2014
- 2.Hatorp V. Clinical pharmacokinetics and pharmacodynamics of repaglinide. Clin Pharmacokinet. 2002;41:471–83. doi: 10.2165/00003088-200241070-00002. [DOI] [PubMed] [Google Scholar]
- 3.Nattrass M, Lauritzen T. Review of prandial glucose regulation with repaglinide: a solution to the problem of hypoglycaemia in the treatment of Type 2 diabetes. Int J Obes Relat Metab Disord. 2000;24(Suppl 3):S21–31. doi: 10.1038/sj.ijo.0801422. [DOI] [PubMed] [Google Scholar]
- 4.Mandic Z, Gabelica V. Ionization, lipophilicity and solubility properties of repaglinide. J Pharm Biomed Anal. 2006;41:866–71. doi: 10.1016/j.jpba.2006.01.056. [DOI] [PubMed] [Google Scholar]
- 5.Hatorp V, Oliver S, Su CA. Bioavailability of repaglinide, a novel anti-diabetic agent, administered orally in tablet or solution form or intravenously in healthy male volunteers. Int J Clin Pharmacol Ther. 1998;36:636–41. [PubMed] [Google Scholar]
- 6.Bidstrup TB, Bjornsdottir I, Sidelmann UG, Thomsen MS, Hansen KT. CYP2C8 and CYP3A4 are the principal enzymes involved in the human in vitro biotransformation of the insulin secretagogue repaglinide. Br J Clin Pharmacol. 2008;56:305–14. doi: 10.1046/j.0306-5251.2003.01862.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chang C, Bahadduri PM, Polli JE, Swaan PW, Ekins S. Rapid identification of P-glycoprotein substrates and inhibitors. Drug Metab Dispos. 2006;34:1976–84. doi: 10.1124/dmd.106.012351. [DOI] [PubMed] [Google Scholar]
- 8.Hatorp V, Bayer T. Repaglinide bioavailability in the fed or fasting state [abstract] J Clin Pharmacol. 1997;37:875. [Google Scholar]
- 9.Yuan G, Jiao L, Xuan Q, Jianjun Z. Co-amorphous repaglinide—saccharin with enhanced dissolution. Int J Pharm. 2013;450:290–5. doi: 10.1016/j.ijpharm.2013.04.032. [DOI] [PubMed] [Google Scholar]
- 10.Kavitha R, Sathali AAH. Enhancement of solubility of repaglinide by solid dispersion technique. Int J Chem Sci. 2012;10:377–90. [Google Scholar]
- 11.Nicolescu C, Arama C, Nedelcu A, Monciu CM. Phase solubility studies of the inclusion complexes of repaglinide with β-cyclodextrin and β-cyclodextrin derivatives. Farmacia. 2010;58:620–8. [Google Scholar]
- 12.12)Troy P, Michal E, Mattucci M, Todd C, Keith P, Williams RO. Rapidly dissolving repaglinide powders produced by the ultra-rapid freezing process. AAPS PharmSciTech 2007; 8: Article 58. doi: 10.1208/pt0803058 [DOI] [PubMed]
- 13.Junghanns AH, Muller RH. Nanocrystal technology, drug delivery and clinical applications. Int J Nanomedicine. 2008;3:295–309. doi: 10.2147/ijn.s595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Peltonen L, Hirvonen J. Pharmaceutical nanocrystals by nanomilling: critical process parameters, particle fracturing and stabilization methods. J Pharm Pharmacol. 2010;62:1569–79. doi: 10.1111/j.2042-7158.2010.01022.x. [DOI] [PubMed] [Google Scholar]
- 15.Liversidge GG, Cundy KC. Particle size reduction for improvement of oral bioavailability of hydrophobic drugs 1: absolute oral bioavailability of nanocrystalline danazol in beagle dogs. Int J Pharm. 1995;125:91–7. doi: 10.1016/0378-5173(95)00122-Y. [DOI] [Google Scholar]
- 16.Hardung H, Djuric D, Ali S. Combining HME and solubilisation-soluplus—the solid solution. Drug Deliv Technol. 2010;10:20–7. [Google Scholar]
- 17.Dintaman JM, Silverman JA. Inhibition of P-glycoprotein by of d-α-tocopheryl PEG 1000 succinate (TPGS) Pharm Res. 1999;16:1550–6. doi: 10.1023/A:1015000503629. [DOI] [PubMed] [Google Scholar]
- 18.Yu L, Bridgers A, Polli J, Vickers A, Long S, Roy A. Vitamin E TPGS increases absorption flux of an HIV protease inhibitor by enhancing its solubility and permeability. Pharm Res. 1999;16:1812–7. doi: 10.1023/A:1018939006780. [DOI] [PubMed] [Google Scholar]
- 19.Wu SHW, Hopkins WK. Characteristics of d-α-tocopheryl PEG 1000 succinate for applications as an absorption enhancer in drug delivery systems. Pharm Technol. 1999;23:52–68. [Google Scholar]
- 20.Verma VSM, Panchagnula R. Enhanced paclitaxel oral absorption with Vitamin E-TPGS: effect on solubility and permeability in vitro, in situ, in vivo. Eur J Pharm Sci. 2005;25:445–53. doi: 10.1016/j.ejps.2005.04.003. [DOI] [PubMed] [Google Scholar]
- 21.Christiansen A, Backensfeld T, Denner K, Weitschies W. Effects of non-ionic surfactants on cytochrome P450-mediated metabolism in vitro. Eur J Pharm Biopharm. 2011;78:166–72. doi: 10.1016/j.ejpb.2010.12.033. [DOI] [PubMed] [Google Scholar]
- 22.Johnson BM, Charman WN, Porter CJH. An in vitro examination of the impact of polyethylene glycol 400, pluronic P85 and vitamin E D-α-tocopheryl polyethylene glycol 1000 succinate on P-glycoprotein efflux and enterocyte-based metabolism in excised rat intestine. AAPS PharmSciTech. 2002;4:E40. doi: 10.1208/ps040440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dressman JB, Amidon GL, Reppas C, Shah VP. Dissolution testing as a prognostic tool for oral drug absorption: immediate release dosage. Pharm. Res. 1998;15:11–22. doi: 10.1023/A:1011984216775. [DOI] [PubMed] [Google Scholar]
- 24.Jantratid E, Janssen N, Chokshi H, Tang K, Dressman J. Designing bio-relevant dissolution tests for lipid formulations: case example—lipid suspension of RZ-50. Eur J Pharm Biopharm. 2008;69:776–85. doi: 10.1016/j.ejpb.2007.12.010. [DOI] [PubMed] [Google Scholar]
- 25.Verma S, Gokhale R, Burgess DJ. A comparative study of top-down and bottom-up approaches for the preparation of micro/nanosuspensions. Int J Pharm. 2009;380:216–22. doi: 10.1016/j.ijpharm.2009.07.005. [DOI] [PubMed] [Google Scholar]
- 26.He W, Lu Y, Qi JP, Chen LY, Hu FQ, Wu W. Food proteins as novel nanosuspension stabilizers for poorly water-soluble drugs. Int J Pharm. 2013;441:269–78. doi: 10.1016/j.ijpharm.2012.11.033. [DOI] [PubMed] [Google Scholar]
- 27.ICH Harmonised Tripartite Guideline. Stability testing of new drug substances and products Q1A (R2), version 4 dated 6 February, 2003
- 28.Cerdeira AM, Mazzotti M, Bruno G. Miconazole nanosuspensions: influence of formulation variables on particle size reduction and physical stability. Int J Pharm. 2010;396:210–8. doi: 10.1016/j.ijpharm.2010.06.020. [DOI] [PubMed] [Google Scholar]
- 29.Jinno J, Kamada N, Miyake M, Yamada K, Mukai T, Odomi M. Effect of particle size reduction on dissolution and oral absorption of a poorly water-soluble drug, cilostazol, in beagle dogs. J Control Release. 2006;111:56–64. doi: 10.1016/j.jconrel.2005.11.013. [DOI] [PubMed] [Google Scholar]
- 30.Rahigude A, Bhutada P, Kaulaskar S, Aswar M, Otari K. Participation of antioxidant and cholinergic system in protective effect of naringenin against type-2 diabetes-induced memory dysfunction in rats. Neuroscience. 2012;226:62–72. doi: 10.1016/j.neuroscience.2012.09.026. [DOI] [PubMed] [Google Scholar]
- 31.Ruzilawati AB, Wahab MSA, Imran A, Ismail Z, Gana SH. Method development and validation of repaglinide in human plasma by HPLC and its application in pharmacokinetic studies. J Pharm Biomed Anal. 2007;43:1831–5. doi: 10.1016/j.jpba.2006.12.010. [DOI] [PubMed] [Google Scholar]
- 32.Amrani FE, Rhallab A, Alaoui T, Badaoui KE, Chakir S. Hypoglycaemic effect of Thymelaea hirsuta in normal and streptozotocin-induced diabetic rat. J Med Plants Res. 2009;3:625–9. [Google Scholar]
- 33.Andrade-Cetto A, Wiedenfeld H, Revilla MC, Islas S. Hypoglycaemic effect of Equisetum myriochaetum aerial parts on streptozotocin diabetic rats. J Ethnopharmacol. 2000;72:129–33. doi: 10.1016/S0378-8741(00)00218-X. [DOI] [PubMed] [Google Scholar]
- 34.Kipp JE, Wong T, Chung J, Doty MJ, Werling J, Rebbeck CL. Method for preparing submicron particle suspension. U S Patent. 2005;6:884,436. [Google Scholar]
- 35.Guo Z, Zhang M, Li H, Wang J, Kougoulos E. Effect of ultrasound on antisolvent crystallisation process. J Cryst Growth. 2005;273:555–63. doi: 10.1016/j.jcrysgro.2004.09.049. [DOI] [Google Scholar]
- 36.Dhumal RS, Biradar SV, Paradkar AR, York P. Particle engineering using sonocrystallization: salbutamol sulphate for pulmonary delivery. Int J Pharm. 2009;368:129–37. doi: 10.1016/j.ijpharm.2008.10.006. [DOI] [PubMed] [Google Scholar]
- 37.Yao JH, Elder KR, Guo H, Grant M. Theory and simulation of Ostwald ripening. Physical review B. 1993;47(21):110–25. doi: 10.1103/PhysRevB.47.14110. [DOI] [PubMed] [Google Scholar]
- 38.Teeranachaideekul V, Junyaprasert V, Souto E, Muller R. Development of ascorbyl palmitate nanocrystals applying the nanosuspension technology. Int J Pharm. 2008;354:227–34. doi: 10.1016/j.ijpharm.2007.11.062. [DOI] [PubMed] [Google Scholar]
- 39.Kesisoglou F, Panmai S, Wu Y. Nanosizing—oral formulation development and biopharmaceutical evaluation. Adv Drug Del Rev. 2007;59:631–44. doi: 10.1016/j.addr.2007.05.003. [DOI] [PubMed] [Google Scholar]
- 40.Gao L, Liu G, Ma J, Wang X, Zhou L, Li X. Drug nanocrystals: in vivo performances. J Control Release. 2012;160:418–30. doi: 10.1016/j.jconrel.2012.03.013. [DOI] [PubMed] [Google Scholar]
- 41.Li Xi G, Xu Y, Wang Y. Preparation of fenofibrate nanosuspension and study of its pharmacokinetic behaviour in rats. Drug Dev Ind Pharm. 2009;35:827–33. doi: 10.1080/03639040802623941. [DOI] [PubMed] [Google Scholar]
- 42.Parsa A, Saadati R, Abbasian Z, Aramaki SA, Dadashzadeh S. Enhanced permeability of etoposide across everted sacs of rat small intestine by vitamin E-TPGS. Iranian J Pharm Res. 2013;12:37–46. [PMC free article] [PubMed] [Google Scholar]
- 43.Rege BD, Kao JPY, Polli JE. Effects of nonionic surfactants on membrane transporters in Caco-2 cell monolayers. Eur J Pharm Sci. 2002;16:237–46. doi: 10.1016/S0928-0987(02)00055-6. [DOI] [PubMed] [Google Scholar]
- 44.Ivora MD, Paya M, Villar A. A review of natural products and plants as potential anti-diabetic drugs. J Ethnopharmacol. 1989;27:243–75. doi: 10.1016/0378-8741(89)90001-9. [DOI] [PubMed] [Google Scholar]
- 45.Sharma SR, Dwivedi SK, Swarup D. Hypoglycaemic, anti hyperglycaemic and hypolipidemic activities of Cesalpinia bounducella seeds in rats. J Ethnopharmacol. 1997;58:39–44. doi: 10.1016/S0378-8741(97)00079-2. [DOI] [PubMed] [Google Scholar]