

Abstract
This study aimed to develop films for potential delivery of omeprazole (OME) via the buccal mucosa of paediatric patients. Films were prepared using hydroxypropylmethylcellulose (HPMC), methylcellulose (MC), sodium alginate (SA), carrageenan (CA) and metolose (MET) with polyethylene glycol (PEG 400) as plasticiser, OME (model drug) and L-arg (stabiliser). Gels (1% w/w) were prepared at 40°C using water and ethanol with PEG 400 (0–1% w/w) and dried in an oven (40°C). Optimised formulations containing OME and L-arg (1:1, 1:2 and 1:3) were prepared to investigate the stabilisation of the drug. Tensile properties (Texture analysis, TA), physical form (differential scanning calorimetry, DSC; X-ray diffraction, XRD; thermogravimetric analysis, TGA) and surface topography (scanning electron microscopy, SEM) were investigated. Based on the TA results, SA and MET films were chosen for OME loading and stabilisation studies as they showed a good balance between flexibility and toughness. Plasticised MET films were uniform and smooth whilst unplasticised films demonstrated rough lumpy surfaces. SA films prepared from aqueous gels showed some lumps on the surface, whereas SA films prepared from ethanolic gels were smooth and uniform. Drug-loaded gels showed that OME was unstable and therefore required addition of L-arg. The DSC and XRD suggested molecular dispersion of drug within the polymeric matrix. Plasticised (0.5% w/w PEG 400) MET films prepared from ethanolic (20% v/v) gels and containing OME: L-arg 1:2 showed the most ideal characteristics (transparency, ease of peeling and flexibility) and was selected for further investigation.
KEY WORDS: buccal drug delivery, omeprazole, oral films, paediatric, plasticiser
INTRODUCTION
Amongst all the established routes of drug administration, the oral route is perhaps the most preferred for both patients and healthcare providers compared to other routes such as injections. However, this route of administration has disadvantages including enzyme degradation within the gastrointestinal tract which prohibits oral administration of certain classes of drugs such as peptides and proteins. Evidence has shown that the oral mucosa is relatively permeable with a rich supply of blood and shows a short recovery time after stress or damage. Further, it also lacks Langerhans cells which allow the oral cavity to be tolerant of any potential allergens [1]. Drug administration within the oral mucosa is generally classified into sublingual and buccal delivery. Amongst all the trans-mucosal routes, the buccal mucosa has excellent accessibility, an expanse of smooth muscle and relatively immobile mucosa, hence suitable for the administration of retentive dosage forms [2,3]. Direct access to the systemic circulation through the internal jugular vein bypasses hepatic first-pass metabolism leading to relatively high bioavailability compared to the GI tract. Additionally, the buccal mucosa has a high surface area (50.2 cm2) and a thin membrane (500–600 μm) which can contribute to rapid and extensive drug absorption [4].
Oral drug delivery systems have always been an important means of drug administration; however, many paediatric patients resist solid dosage forms such as tablets due to the bitter taste and fear of choking. Though sweetened liquid formulations are commonly used, they present many challenges including bitter after taste, unpleasant flavours, short half lives once opened and generally bulky to handle and store. Oral thin films offer easy administration and handling, rapid disintegration and dissolution, bypass first-pass metabolism, enhanced stability and taste masking for bitter drugs, local and systematic drug delivery, rapid onset of action and no trained or professional person is required for paediatric administration [5]. Due to the numerous advantages of buccal dosage forms, various technologies have been explored to manufacture oral films on a large scale as an alternative to traditional dosage forms such as tablets and capsules [6].
Numerous buccal delivery systems in the form of tablets, liquids and semi-solids have been reported in the past decades, yet only a limited number of these have reached the market [7]. The necessity of recurrent dosing might possibly arise due to the flushing activity of saliva, chewing and the ingestion of food materials which results in the rapid expulsion of drugs. Moreover, the drugs in the saliva may be unevenly distributed, which might consequently lead to lower amounts being absorbed by the mucosal tissues directly into the systemic circulation. Furthermore, the likely displacement of the formulation from the buccal area by tongue movements serves as an additional challenge [8]. The above notwithstanding, the buccal mucosal route is still considered a practical route to deliver a variety of active ingredients.
Hydrophilic polymers incorporating several hydrogen bonding groups make the formulation of bioadhesive buccal formulations feasible. Modified forms of such hydrogel polymers with better bioadhesivity create second-generation mucosal dosage forms [9]. In the present study, we report on the development of solvent cast films for buccal delivery in paediatric patients using various hydrogel polymers generally regarded as safe (GRAS) and used in mucosal formulations [10–13] including HPMC, MC, CA, MET, SA, plasticiser (PEG 400), OME (model drug) and L-arg (to stabilise OME). Various parameters such as drying times and temperatures, casting solvents as well as polymer and plasticiser concentrations were investigated and the films subsequently characterised as part of the development and optimisation.
METHODS
Materials
Carrageenan (CA) and sodium alginate (SA) were gifts from FMC BioPolymer and originally sourced from Cork (Republic of Ireland). Metolose (MET) was obtained from Shin Etsu (Stevenage, Hertfordshire). Hydroxypropylmethylcellulose (HPMC), methylcellulose (MC), polyethylene glycol (PEG 400), L-arginine (L-arg) were obtained from Sigma-Aldrich (Gillingham, UK). Ethanol was purchased from Fisher Scientific (Loughborough, UK) and Omeprazole (OME) obtained from TCI (Tokyo, Japan).
Formulation (Gel and Film) Development
Aqueous and ethanolic gels of the different polymers were prepared prior to film casting.
The aqueous gels were formulated by adding the required weight of polymers to the relevant solvent (deionised water) at laboratory temperature (22°C) to obtain 1% w/w gels. Following complete hydration (dissolution), the polymeric gels were heated to 40°C. Based on the total weight of polymers, various amounts of the plasticiser (PEG) were added to obtain different concentrations (0.00, 0.10, 0.25, 0.50, 0.75 and 1.00% w/w) in the final gels prepared. The resultant gels were left on a water bath with regulated temperature of 40°C (except for CAR which was prepared at 70°C) and stirring continued for 30 min to achieve a homogeneous dispersion. For ethanolic gels, the appropriate volume of ethanol (10 and 20% v/v) was added to yield the 1% w/w total concentration. The solution was left to cool to room temperature and stirred again for 30 min. The final solutions were left to stand overnight to remove entrapped air bubbles. After removal of the air bubbles, 20 g of each gel was poured into Petri dishes (86 mm diameter) and kept in a pre-heated oven at 60°C for 24 h. The dried films were then carefully peeled off from the Petri dish, images captured using a digital camera and transferred into poly bags and placed in a desiccator over silica gel at room temperature until required.
Formulation Development and Optimization of OME-Loaded Films
The main purpose for the development and optimization was to determine the optimised amount of the drug that could be incorporated into the solvent cast film whilst still maintaining the ideal physical characteristics in terms of flexibility, homogeneity and transparency [14]. The OME-loaded films were obtained by initially preparing MET gels as previously described above. However, the drug was added to the appropriate volume of water/ethanol to form an OME solution as shown in Table I. The polymer was then added slowly to the vigorously stirred drug solution at room temperature to obtain the drug-loaded gels. The resulting gels were covered with parafilm as above and left overnight to allow air bubbles to escape, and then 20 g was poured into Petri dishes and dried at 40°C [15].
Table I.
Drug Loaded MET Gels Formulated Using Different Solvent Systems and Containing Different Amounts of PEG 400 (0 and 0.5% w/w)
| Solvent systems | Water:ethanol (ml) | MET (g) | OME (g) | Plasticizers (g) | |
|---|---|---|---|---|---|
| 0% | 0.5% | ||||
| Water | 50:0 (1:0) | 0.50 | 0.10 | 0.00 | 0.25 |
| 10% v/v ethanol | 45:5 (9:1) | 0.50 | 0.10 | 0.00 | 0.25 |
| 20% v/v ethanol | 40:10 (4:1) | 0.50 | 0.10 | 0.00 | 0.25 |
MET metolose, OME omeprazole
Stabilisation of OME in Drug-Loaded MET Films Using L-Arg
Due to the breakdown of OME following gel formation, L-arg was used as a stabilising agent to prevent drug degradation. Table II shows the details for the different ratios of OME and L-arg in the gel formulations which were investigated. This step was performed by using different amounts of L-arg within the gel whilst keeping the original OME concentration (0.10% w/w) constant. The gels were prepared as above with L-arg and OME dissolved in the solvent before addition of MET and PEG 400.
Table II.
Different OME:L-arg Ratios in the MET Gel Formulations for Preparing Both Unplasticised and Plasticised Films (0 and 0.5% w/w (PEG 400), Respectively)
| Solvent systems | Water:ethanol (ml) | MET (g) | Drug (g) OME | OME:L-arg (g) | Plasticizers (g) | |||
|---|---|---|---|---|---|---|---|---|
| 1:1 | 1:2 | 1:3 | 0% | 50% | ||||
| Water | 50:0 (1:0) | 0.50 | 0.10 | 0.10 | 0.20 | 0.30 | 0.00 | 0.25 |
| 10% v/v ethanol | 45:5 (9:1) | 0.50 | 0.10 | 0.10 | 0.20 | 0.30 | 0.00 | 0.25 |
| 20% v/v ethanol | 40:10 (4:1) | 0.50 | 0.10 | 0.10 | 0.20 | 0.30 | 0.00 | 0.25 |
MET metolose, OME omeprazole
Characterisation of the Films
Tensile Characterisation by Texture Analysis
Texture analysis (TA) was used to measure tensile properties. A texture analyser (HD plus, Stable Micro System, Surrey, UK) equipped with 5-kg load cell was used. Thickness and width of the films were measured and stress and strain values were calculated based on these values. Data evaluation was performed by texture exponent-32 software program. The films free from any physical defects, with the average thickness of (0.07 ± 0.01 mm) were selected for testing. The films were cut into dumbbell-shaped strips and fixed between two tensile grips positioned 30 mm apart and stretched at a test speed of 1.0 mm/s to break point. The tensile strength (brittleness of the film), elastic modulus (rigidity) and percentage elongation (flexibility and elasticity) were determined using Eqs. 1, 2 and 3. Each testing was carried out in triplicate (n = 3).
| 1 |
| 2 |
| 3 |
Scanning Electron Microscopy (SEM)
SEM was used to investigate the surface morphology of the films and to cheque for film uniformity and the presence of any cracks. The films were analysed using a Hitachi Triple detector CFE-SEM SU8030 (Roland Schmidt, Hitachi High-Technologies Europe GmbH) scanning electron microscope. Films were mounted onto 12 mm aluminium pin stubs (G301, Agar Scientific) with double-sided adhesive carbon tapes (G3347N, Agar scientific) and chrome coated (Sputter Coater S150B, 15 nm thickness). The coated films were analysed at 2 kV accelerating voltage.
Differential Scanning Calorimetry (DSC)
DSC was used to characterise the thermal behaviour of selected optimised MET and SA films and pure materials to investigate changes in their properties with introduction of PEG and drug within the films. Analysis of the films and starting materials were carried out on a Q2000 (TA Instruments) calorimeter. About 2.5 mg of each sample was placed into hermetically sealed Tzero aluminium pans with a pin hole in the lid and heated from −40 to 180°C at a heating rate of 10°C/min under constant purge of nitrogen (100 ml/min).
Thermogravimetric Analysis (TGA)
TGA studies were performed using a Q5000 (TA instrument) thermogravimetric analyser. About 2.5 mg of sample (films and starting materials, MET and SA) was placed into hermetically sealed Tzero aluminium pans with a pin hole in the lid. Samples were heated under nitrogen atmosphere at a flow rate of 25 ml/min from ambient temperature to 600°C at a heating rate of 2°C/min.
X-Ray Diffraction (XRD)
XRD was used to investigate the physical form (crystalline or amorphous) of the selected optimised films and starting materials (MET, SA and PEG). XRD patterns of films and starting materials were obtained with a DIFFRAC plus instrument (Bruker Coventry, UK) equipped with an XRD commander programme. A Goebel mirror was used as monochromator which produced a focused monochromatic CuKα1&2 primary beam (λ = 1.54184 Å) with exit slits of 0.6 mm and a Lynx eye detector for performing the experiment. The operating conditions during the experiment were 40 kV and 40 mA. Film samples were prepared by cutting into 2 cm2 square strips, mounted on the sample cell and scanned between 2 theta of 0° to 70° and counting time of 0.1 s step size.
RESULTS AND DISCUSSION
Formulation Development and Optimisation
Omeprazole is an ideal candidate for buccal drug delivery using polymeric films as the delivery system, as it degrades readily in acidic medium and undergoes first-pass metabolism [16]. The polymers used in this study were chosen because of their hydrophilic nature. Stirring was applied during gel formulation to prevent formation of lumps which could occur through incomplete hydration especially for polymers with high viscosity. The heat (40 or 70°C) reduced the viscosity of the final gels and helped to facilitate the escape of entrapped air bubbles caused by stirring and also allowed ease of pouring into the casting Petri dishes. Ethanol was used as solvent in addition to deionised water because some polymers/drugs are more soluble in ethanol than water and the former also helped to increase the rate of drying. The removal of the air bubbles entrapped inside the gel was essential to avoid any empty gaps, which could lead to non-uniform distribution of various film components. The drying process for unplasticised gels was shorter (12 h) compared to plasticised gels (18–24 h) due to the known water affinity of most plasticisers [17].
Visual Evaluation of Films
The MET and SA films were transparent, uniform and easy to peel from the Petri dishes. However, though HPMC, MC and CA films were transparent, they were not uniform due to the presence of air bubbles during drying, and were difficult to peel off without damaging the films (Fig. 1). Further, HPMC and MC films showed excessive elasticity at high concentrations of PEG which made them sticky. As a result, films prepared using CAR, HPMC and MC were discontinued from further investigations and only MET and SA films were taken forward for further development and drug loading.
Fig. 1.
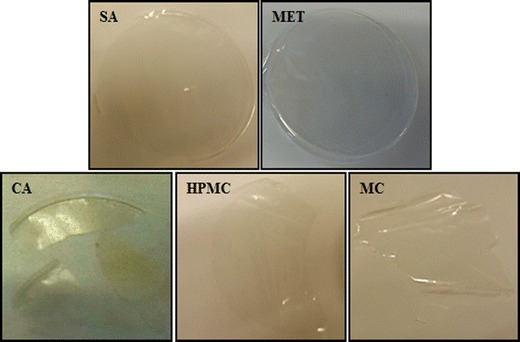
Physical appearance (digital photographs) of films prepared using different polymers, i.e. sodium alginate (SA), metolose (MET), carrageenan (CA), hydroxypropyl methylcellulose (HPMC) and methylcellulose (MC)
Further development of MET and SA films, during the preliminary experiments, involved preparing films with and without plasticiser. The main purpose of using plasticiser is to provide flexibility and to overcome the brittleness in films. Unplasticised MET and SA films were brittle whilst films plasticised with PEG showed reduced brittleness and desirable flexibility [18]. Optimum plasticiser concentration(s) for further formulation development was however, investigated by using texture analysis to determine film tensile properties which provided more reliable data for accurate evaluation.
Tensile Properties of Films
Generally, soft and weak polymers have low tensile strength, low elastic (Young’s) modulus and low percent elongation at break. On the other hand soft and strong polymers display acceptable strength, low elastic modulus and high percent elongation at break [17]. The films showed significant differences in the tensile strength (brittleness) based on the PEG concentration. The initial linear portion of the stress-strain curve was used to estimate the elastic modulus and tensile strength [19]. The effects of PEG concentration on the tensile strength values of the MET and SA films are shown in Fig. 2a, b, respectively. The percent elongation at break point of MET gradually increased with increased concentration of PEG. It has been suggested that the average percent elongation at break point should ideally be between 30 and 50% [17] which indicates a good balance between flexibility and elasticity. MET films prepared from gels containing 0.5 and 0.75% w/w of PEG satisfied this criteria. MET films prepared from aqueous and ethanolic (water, 10 and 20% v/v of EtOH) gels containing 0.50% w/w PEG, showed percent elongation of break values between 27 and 57%. Unplasticised films prepared using water as the casting solvent showed a very low percent elongation at break (Fig. 2a), whilst films obtained from EtOH (10 and 20% v/v) gels showed a significantly higher values of percent elongation. There was also a general increase in percent elongation with increasing concentration of PEG for all films. At the concentration of 0.75% w/w of PEG, all the films showed elongation at break point of 55–58% which was deemed high. Compared to MET films, SA films demonstrated low values in the overall percent in elongation at break.
Fig. 2.
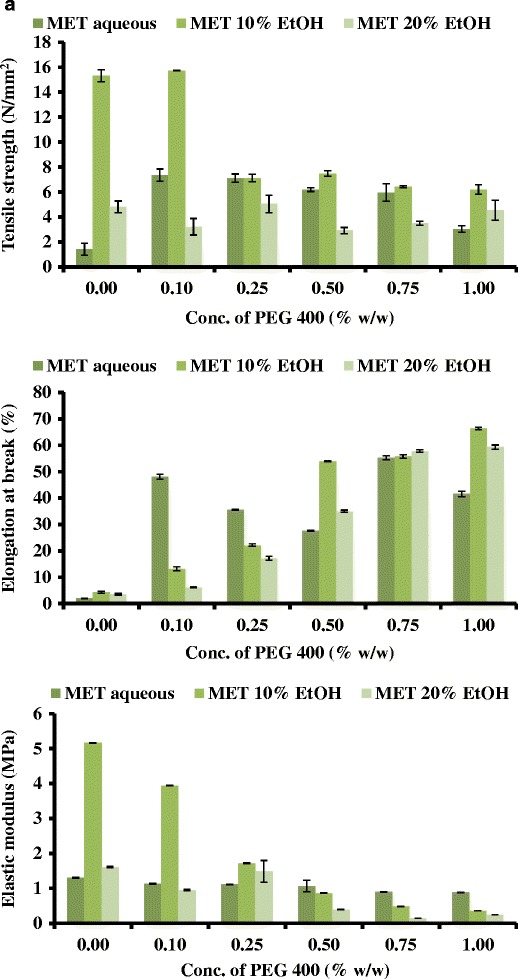
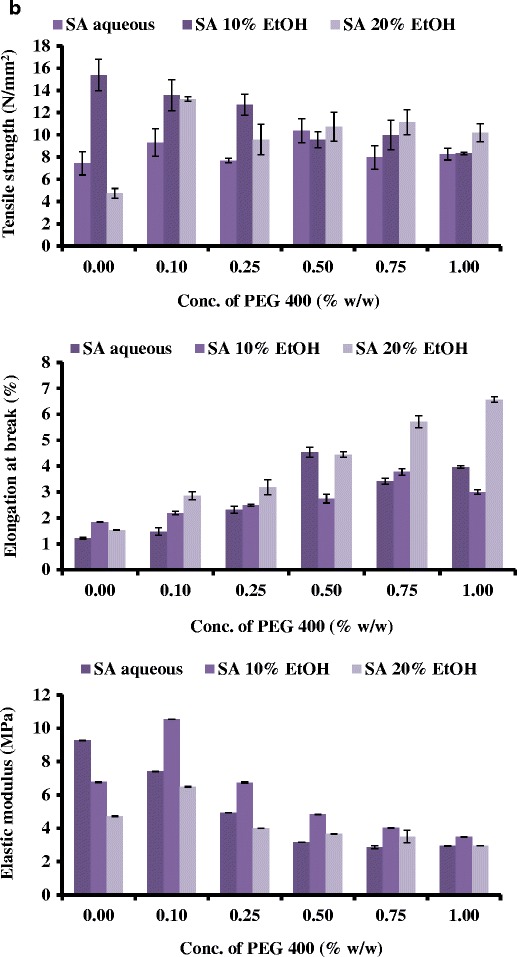
Tensile (tensile strength, percent elongation at break and elastic modulus) profiles of a MET films and b SA films containing different concentrations of PEG and cast from different solvent systems
Generally, plasticisers such as PEG in the system increase the free volume between the polymeric chains and allow them to slide past each other and subsequently produced appropriate flexibility and consequent decrease in tensile strength and elastic modulus [20]. Based on these observations all subsequent gels for drug loading were prepared using only MET at two concentrations (0.00 and 0.50% w/w (original gel) of PEG, with the unplasticised films being used as a control.
Physical Evaluation of Drug-Loaded Films
When OME is added to water, it dissolves quickly to produce a clear solution. After adding polymer and desired amount of plasticiser in solution for gel formation, the stability of OME plays a vital role in the overall stability of the gel [21]. However, it was observed that OME degraded within 20 min and changed the colour of the gel to red as can be seen in Fig. 3a. This resulted in a completely opaque and brown coloured film as shown in Fig. 3b. OME can only be stable in alkaline solution with pH of eight and stability can be achieved in two main ways: (i) introducing cyclo-dextrin or (ii) L-arg to the drug-loaded gel. However, because of the toxicity of cyclo-dextrin for paediatric patients, use of L-arg was the preferred option [22]. To determine the optimum concentration of L-arg required to stabilise the drug and determine its effect on MET film properties, different amounts relative to the drug were added to the original gels before drying as shown in Table II above. Blank MET films showed complete transparency similar to that shown in Fig. 1; whereas drug-loaded films containing L-arg were slightly cream in colour as shown in Fig. 3c.
Fig. 3.

a Degradation of OME in aqueous gel as evidenced by change in colour to red within 20 min of preparation, b films prepared from gels containing OME without L-arg showing OME degradation and c films prepared from gels containing OME stabilised with L-arg
Generally, plasticised drug-loaded films containing OME and L-arg (1:1; 1:2 and 1:3) showed a significant difference in their visual appearance compared to unplasticised films with the former showing better transparency and uniformity. Another difference observed between the different formulations was that the films prepared from aqueous only gels were difficult to peel off from the Petri dish due to their thin nature. Further, the distribution of OME and L-arg was more uniform in the films prepared from the ethanolic gels (10 and 20% v/v EtOH). It was therefore concluded that films prepared from ethanolic gels (EtOH 20%) were the most transparent and uniform which could be due to complete molecular dispersion of drug (OME) and L-arg within the polymeric matrix.
Based on the visual observation and the expected characteristics for an ideal film in terms of flexibility, uniformity and transparency, films prepared from ethanolic gels (20% v/v EtOH) containing 1:2 ratio of OME: L-arg and 0.5% w/w PEG400 was the most appropriate for further investigations. It was also obvious that the addition of L-arg helped to stabilise the drug within the films as can be seen by comparing Fig. 3b and c, with the latter showing desired homogeneity, transparency and uniform drug distribution. Figueiras et al. [23] suggested that when combined together, the H atom of the L-arg was observed to be in closer proximity to the nitrogen atom of OME. They also observed that the distance between the H (L-arg) and the N (OME) is relatively small which increases the chances of formation of hydrogen bonds between the two compounds.
Scanning Electron Microscopy (SEM)
SEM images of the MET films cast from gels prepared with different solvents (water, 10% EtOH and 20% EtOH) with or without PEG (0.50% w/w) are shown in Fig. 4a. The microscopic appearance of all MET films, showed continuous sheets with relatively smooth and homogeneous surfaces and suggest that all the components were uniformly mixed during gel formation. The plasticised films showed smooth and homogeneous surfaces whilst unplasticised films showed rougher surfaces with some lumps. The surface topography of the SA films was dependent on the solvent used during gel preparation. Films prepared from aqueous gels showed considerably rougher surfaces than films prepared using 10% v/v EtOH, which in turn showed uneven surfaces than films prepared using 20% v/v EtOH as shown in Fig. 4b. This could be related to the more rapid drying of ethanolic gels during film formation. Such differences in surface topography could influence the uniformity of the films, because any pores or lumps in the film could affect the subsequent functional performance of different formulations with respect to hydration capacity/swelling studies, mucoadhesion and drug release characteristics.
Fig. 4.

Scanning electron microscope images of a MET films cast from aqueous and ethanolic (10 and 20% v/v) gels containing different concentrations of PEG 400 (0 and 0.50% w/w) and b SA films cast from aqueous and ethanolic (10 and 20% v/v) gels containing no PEG 400
Differential Scanning Calorimetry (DSC)
The thermogram for pure MET and SA can be seen in Fig. 5a, showing a broad endothermic peak at between 80 and 95°C, caused by evaporation of water and no definite melt or glass transition peaks. In general, the thermograms of the films shown in Fig. 5b were similar to the pure MET powder. Figure 5b further shows the different MET films [aqueous and ethanolic (10 and 20% EtOH)] which were prepared using different percentages of PEG 400. All the films can be characterised as amorphous, as only the broad endothermic peak can be observed between 40 and 100°C which is attributed to water loss.
Fig. 5.
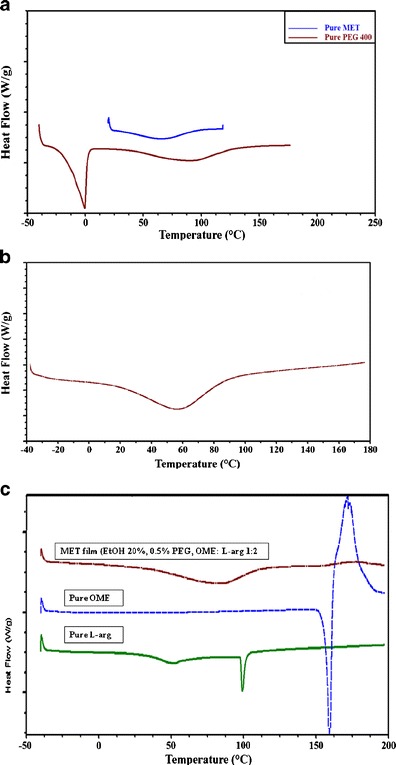
DSC thermograms of a pure PEG and pure MET, b representative optimum blank, plasticized (0.50% w/w PEG 400) MET films cast from ethanolic (20% v/v) gels and c pure L-arg, pure OME and drug loaded MET film prepared from ethanolic (20% v/v) gels containing OME: L-arg (1:2) and PEG 400 (0.50% w/w)
The DSC thermograms for pure OME, L-arg and drug-loaded MET (OME: L-arg 1:2, 0.50% PEG, EtOH 20% v/v) films are shown in Fig. 5c. It can be observed that OME has a melting point at 158°C and L-arg at 100°C and broad endothermic peak which can be seen at 80°C for the drug (L-arg)-loaded film representing water loss and a complete absence of the melt peaks for both OME and L-arg. This suggests amorphous drug formation or molecular dispersion of both OME and L-arg within the MET film matrix.
Thermogravimetric Analysis (TGA)
The TGA results of blank films (aqueous and ethanolic) are shown in Table III indicating the percentage loss with heating, attributed to residual water present within the film matrix. Due to PEG having hydrophilic characteristics, it was expected that the residual moisture content will increase for all films with increasing PEG 400 concentration. However, this was not the case except at higher concentrations (0.50 and 0.75% w/w of PEG) where the percent of water content increased. It also appears that the residual water was generally lower for films prepared using ethanolic gels than those from aqueous gels which is to be expected as there was less water in the original gel and ethanol generally allows faster drying than pure water on its own. In addition, the moisture content of less than 3% in all films was considered low enough to sustain drug stability during storage though this will need to be investigated with an accelerated stability study.
Table III.
Weight Loss Observed for MET Films Cast from Water, Ethanol (10% v/v) and Ethanol (20% v/v) Gels Containing Different Concentrations of PEG 400 (0, 0.25, 0.50 and 0.75% w/w)
| MET Blank Films | |
|---|---|
| Films | Weight loss (%) |
| MET, 0.00% PEG, aqueous | 2.77 |
| MET, 0.25% PEG, aqueous | 1.74 |
| MET, 0.50% PEG, aqueous | 2.03 |
| MET, 0.75% PEG, aqueous | 2.75 |
| MET, 0.00% PEG, 10% EtOH | 2.26 |
| MET, 0.25% PEG, 10% EtOH | 1.60 |
| MET, 0.50% PEG, 10% EtOH | 2.12 |
| MET, 0.75% PEG, 10% EtOH | 2.47 |
| MET, 0.00% PEG, 20% EtOH | 2.64 |
| MET, 0.25% PEG, 20% EtOH | 1.80 |
| MET, 0.50% PEG, 20% EtOH | 1.99 |
| MET, 0.75% PEG, 20% EtOH | 2.17 |
MET metolose, PEG polyethylene glycol, EtOH ethanol
X-Ray Diffraction (XRD)
To investigate the crystalline-amorphous characteristics of all initial compounds and of the films, XRD was used. Amorphous compounds generally show very broad peaks, in comparison to the sharp peaks belonging to the crystalline form. XRD can also give information about the crystalline-amorphous ratios for the various starting materials and the formulated films [24]. Figure 6 shows XRD diffractograms of pure MET and PEG 400, generally indicating the amorphous nature of MET and plasticiser. Figure 6 also shows the diffractogram of blank plasticised MET films with broad peaks indicating amorphous characteristics as was observed in the pure polymers as well as the diffractograms of pure OME, L-arg and drug-loaded film (20% v/v EtOH, 0.5% w/w PEG, OME: L-arg 1:2). As can be seen, the results demonstrate that the drug-loaded film was also amorphous suggesting possible molecular dispersion of the drug. This is interesting as it confirms the DSC results previously discussed and also the fact the MET together with L-arg were able to successfully maintain the stability of OME in amorphous form within the film matrix during formulation and storage prior to analysis. These results are interesting, however, it is well known that the amorphous forms are generally unstable and have the tendency to convert back to the amorphous forms. Therefore, further physical and chemical stability studies under controlled conditions of temperature and humidity (both normal and accelerated) are required over a longer period of time (over 1 month) for firm confirmation of its long term stability in the current physical state.
Fig. 6.
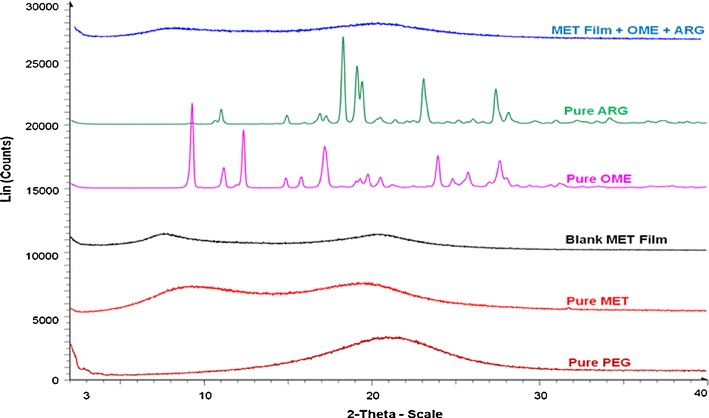
XRD diffractograms for pure MET, pure PEG, pure OME, L-arg, blank MET films, and drug loaded MET films, showing amorphous drug distribution in the drug loaded films
CONCLUSIONS
Due to the poor stability of OME in aqueous environment, L-arg was required in drug-loaded films as a stabilising agent. The most promising characteristics were observed in plasticised MET films (0.50% PEG 400) prepared from ethanolic (20% v/v) gels and containing OME: L-arg ratio of 1:2. These characteristics include transparency, ease of peeling and flexibility of the films for further investigation. It was also confirmed that OME originally loaded in crystalline form was molecularly dispersed (amorphous) within the MET film matrix. The MET films have potential for paediatric buccal administration and will be further functionally characterised to determine its in vitro cell culture, ex vivo and in vivo performance.
Footnotes
Sajjad Khan and Joshua S. Boateng are joint first authors.
References
- 1.Garg SK, Danodia A, Dangi V, Dhakar RC. Buccal adhesive drug delivery system: safer delivery of biotherapeutics. Drug Del Ther. 2011;1(2):35–45. [Google Scholar]
- 2.Sudhakar Y, Kuotsu K, Bandyopadhyay A. Buccal bioadhesive drug delivery—A promising option for orally less efficient drugs. J Contr Rel. 2006;114:15–40. doi: 10.1016/j.jconrel.2006.04.012. [DOI] [PubMed] [Google Scholar]
- 3.Boateng JS, Okeke O. Chitosan-based films for sustained release of peptides: a new era in buccal delivery? Ther Del. 2014;5(5):497–500. doi: 10.4155/tde.14.21. [DOI] [PubMed] [Google Scholar]
- 4.Sohi H, Ahuja A, Ahmad FJ, Khar RK. Critical evaluation of permeation enhancers for oral mucosal drug delivery. Drug Dev Ind Pharm. 2010;36(3):254–82. doi: 10.3109/03639040903117348. [DOI] [PubMed] [Google Scholar]
- 5.Dixit RP, Puthli SP. Oral strip technology: overview and future potential. J Contr Rel. 2009;139(2):94–107. doi: 10.1016/j.jconrel.2009.06.014. [DOI] [PubMed] [Google Scholar]
- 6.Siddhiqui N, Garg G, Sharma P. A short review on “A Novel Approach in Oral Fast-Dissolving Drug Delivery System and Their Patents”. Adv Biol Res. 2011;5(6):291–303. [Google Scholar]
- 7.Yehia SA, El-Gazayerly ON, Basalious EB. Design and in vitro/in vivo evaluation of novel mucoadhesive buccal discs of an antifungal drug: relationship between swelling, erosion, and drug release. AAPS PharmSciTech. 2008;9(4):1207–17. doi: 10.1208/s12249-008-9166-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Birudaraj R, Mahalingam R, Li X, Jasti B. Advances in buccal drug delivery. Crit Rev Ther Drug Carrier Syst. 2005;22(3):295–330. doi: 10.1615/CritRevTherDrugCarrierSyst.v22.i3.20. [DOI] [PubMed] [Google Scholar]
- 9.Salamat-Miller N, Chittchang M, Johnston TP. The use of mucoadhesive polymers in buccal drug delivery. Adv Drug Deliv Rev. 2005;57(11):1666–91. doi: 10.1016/j.addr.2005.07.003. [DOI] [PubMed] [Google Scholar]
- 10.Kianfar F, Chowdhry B, Antonijevic M, Boateng J. Formulation development of a carrageenan based delivery system for buccal drug delivery using ibuprofen as a model drug. J Biomater Nano Biotech. 2011;2(5A):582–95. [Google Scholar]
- 11.Pawar H, Tetteh J, Boateng J. Preparation and characterization of novel wound healing film dressings loaded with streptomycin and diclofenac. Coll Surf B: Biointerf. 2013;102:102–10. doi: 10.1016/j.colsurfb.2012.08.014. [DOI] [PubMed] [Google Scholar]
- 12.Rai D, Maniruzzaman M, Boateng J. Development and characterisation of sodium alginate and HPMC films for mucosal drug delivery. Int J Biotech. 2010;11(3–4):169–81. doi: 10.1504/IJBT.2010.036594. [DOI] [Google Scholar]
- 13.Boateng J, Mani J, Kianfar F. Improving drug loading of mucosal solvent cast films using combination of hydrophilic polymers with amoxicillin and paracetamol as model drugs. BioMed Res Int. 2013;2013:8. doi: 10.1155/2013/198137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kianfar F, Antonijevic M, Chowdhry B, Boateng J. Lyophilized wafers comprising carrageenan and pluronic acid for buccal drug delivery using model soluble and insoluble drugs. Coll Surf B: Biointerf. 2013;103:99–106. doi: 10.1016/j.colsurfb.2012.10.006. [DOI] [PubMed] [Google Scholar]
- 15.Morales J, McConville J. Manufacture and characterization of mucoadhesive buccal films. Eur J Pharm Biopharm. 2011;77(2):187–99. doi: 10.1016/j.ejpb.2010.11.023. [DOI] [PubMed] [Google Scholar]
- 16.Choi H-G, Jung J-H, Yong CS, Rhee C-D, Lee M-K, Han J-H, et al. Formulation and in vivo evaluation of omeprazole buccal adhesive tablet. J Contr Rel. 2000;68(3):405–12. doi: 10.1016/S0168-3659(00)00275-3. [DOI] [PubMed] [Google Scholar]
- 17.Boateng J, Stevens H, Eccleston G, Auffret A, Humphrey J, Matthews K. Development and mechanical characterization of solvent-cast polymeric films as potential drug delivery systems to mucosal surfaces. Drug Dev Ind Pharm. 2009;35(8):986–96. doi: 10.1080/03639040902744704. [DOI] [PubMed] [Google Scholar]
- 18.Lim K, Kim D, Paik U, Kim S. Effect of the molecular weight of poly(ethylene glycol) on the plasticization of green sheets composed of ultrafine BaTiO3 particles and poly(vinyl butyral) Mater Res Bull. 2003;38(1):1021–32. doi: 10.1016/S0025-5408(03)00071-0. [DOI] [Google Scholar]
- 19.Lehrsch G, Sojka R, Koehn A. Surfactant effects on soil aggregate tensile strength. Geoderma. 2012;189–190:199–206. doi: 10.1016/j.geoderma.2012.06.015. [DOI] [Google Scholar]
- 20.Alexander A, Ajazuddin M, Swarna M, Sharma M, Tripathi D. Polymers and permeation enhancers: specialized components of mucoadhesives. Stamford J Pharm Sci. 2011;4(1):91–5. [Google Scholar]
- 21.Wang Y, Dave R, Pfeffer R. Polymer coating/encapsulation of nanoparticles using a supercritical anti-solvent process. J Supercrit Fluids. 2004;28(1):85–99. doi: 10.1016/S0896-8446(03)00011-1. [DOI] [Google Scholar]
- 22.Abruzzo A, Bigucci F, Cerchiara T, Cruciani F, Vitali B, Luppi B. Mucoadhesive chitosan/gelatin films for buccal delivery of propranolol hydrochloride. Carbo Polym. 2012;87(1):581–8. doi: 10.1016/j.carbpol.2011.08.024. [DOI] [PubMed] [Google Scholar]
- 23.Figueiras A, Sarraguça J, Pais A, Carvalho R, Veiga F. The role of L-arginine in inclusion complexes of omeprazole with cyclodextrins. AAPS PharmSciTech. 2010;11(1):233–40. doi: 10.1208/s12249-009-9375-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kumar A, Negi Y, Bhardwaj N, Choudhary V. Synthesis and characterization of methylcellulose/PVA based porous composite. Carbo Polym. 2012;88(4):1364–72. doi: 10.1016/j.carbpol.2012.02.019. [DOI] [Google Scholar]