

Abstract
The gastrointestinal (GI) transit behavior of and absorption from an amphotericin B (AmB) solid lipid nanoformulation (SLN) in rats was investigated. We aimed to estimate the gastric emptying time (GET) and cecal arrival time (CAT) of AmB SLN in rats as animal models. From these two parameters, an insight on the absorption window of AmB was ascertained. Three types of SLNs, AmB, paracetamol (PAR), and sulfasalazine (SSZ), were similarly formulated using beeswax/theobroma oil composite as the lipid matrix and characterized with regard to size, viscosity, density, migration propensity within agarose gel, in vitro drug release, morphology, gastrointestinal transit, and in vivo absorption. The GET and CAT were estimated indirectly using marker drugs: PAR and sulfapyridine (SP). All three types of SLNs exhibited identical properties with regard to z-average, viscosity, relative density, and propensity to migrate. PAR was absorbed rapidly from the small intestine following emptying of the SLNs giving the T50E (time for 50% absorption of PAR) to be 1.6 h. SP was absorbed after release and microbial degradation of SSZ from SLN in the colon with a lag time of 2 h post-administration, serving as the estimated cecal arrival time of the SLNs. AmB within SLN was favorably absorbed from the small intestine, albeit slowly.
KEY WORDS: absorption, gastric emptying, marker drugs, paracetamol, sulfasalazine
INTRODUCTION
Designing and developing pharmaceuticals aimed at achieving maximum bioavailability from the GI tract could best be ascertained through monitoring the GI transit properties of the carrier dosage form and absorption profiles of the drug payload from the GI tract. This is particularly crucial for drugs that have poor aqueous solubility.
AmB is a polyene antifungal (fungicidal) agent which was initially isolated from Streptomyces nodosus. It has a broad spectrum of activity and is efficacious in treating candidiasis, cryptococcosis, aspergillosis, histoplasmosis, blastomycosis, coccidioidomycosis, zygomycosis, sporotrichosis, fusariosis, and phaeohyphomycosis (1,2). Paradoxically, the clinical use of AmB is somewhat limited due to its systemic toxicity, which manifests in two main forms: acute or infusion-related toxicity and chronic (end organ) toxicity, which essentially affects the kidneys (nephrotoxicity) and could lead to permanent renal impairment, especially when co-administered with other nephrotoxic drugs. Strategies used to reduce the nephrotoxicity induced by AmB include saline loading, alternate day dosing, and dose reduction (3); however, there is evidence that when administered orally, the aforementioned side effects are potentially minimized to allow the correct therapeutic dose to be given (4–6).
AmB has poor aqueous solubility and, therefore, poorly absorbed from the GI tract when administered orally. In view of this, there is growing interest in developing alternative delivery systems that address the above constraints of absorption of AmB from the GI tract whilst minimizing the aforementioned side effects. One of such a delivery system that has received much attention lately is nanoparticle formulation, particularly solid lipid nanoparticles (SLN) (7,8). SLNs can be taken up by the Peyer’s patches along the GI tract when the former is administered orally, which drains into the lymph and then eventually empties into the systemic circulation (9). In the quest for a successful development of a delivery system with such potential, it is imperative to challenge the prototype to exhaustive physical characterization prior to studying GI transit properties of the dosage form. We have successfully developed an AmB-containing SLN and characterized the same with regard to surface composition, chemistry, charge, and stability (10,11).
In this paper, we describe further characterization and a pilot GI transit study tailored towards a subsequent full GI transit study. In the pilot GI transit study using Sprague-Dawley rats, paracetamol (PAR) was used as a marker drug for estimating the gastric emptying rate of the SLNs. This estimation was based on the rate of appearance of PAR in the blood following oral administration (12). On the other hand, sulfasalazine (SSZ) after arrival at the large bowel becomes reduced at the azo linkage by colonic bacteria to produce sulfapyridine (SP) and 5-aminosalicylic acid. SP is very rapidly and almost completely absorbed from the colon into the blood and, therefore, may serve as an estimate of the arrival time of an SSZ-containing SLN at the cecum (13,14).
These two compounds have been used as marker drugs to estimate the gastric emptying and orocecal transit times of several orally administered dosage forms (15–17) and provide a cheaper alternative to the use of gamma scintigraphy. Our review of the literature has not revealed any GI transit work carried out on AmB-containing SLNs. Prior to administration, AmB, PAR, and SSZ were formulated into SLNs using identical methods. These were then characterized to ensure similarity in their physical properties with the assumption that when administered simultaneously, the three types of SLNs would respond similarly to the hydrodynamics within the GI tract. Once this assumption was verified in vitro, a preliminary study was conducted on rats to ascertain the GI transit properties of the AmB SLN.
MATERIALS AND METHODS
Beeswax (Acros Organics, New Jersey, USA), theobroma oil (JB Cocoa Sdn Bhd, Johor, Malaysia), amphotericin B (Nacalai Tesque, Inc., Kyoto, Japan), sulfasalazine (Tokyo Chemical Industry Co. Ltd, Japan), agarose (1st Base Laboratories Sdn Bhd, Malaysia). Paracetamol, phosphate buffered saline (pH 7.4), and sulfapyridine were purchased from Sigma-Aldrich (Sigma-Aldrich Co. LLC., Missouri, USA), and lecithin soy and sodium cholate were obtained from MP Biomedicals (Illkirch, France). Chloroform, ethyl acetate, methanol, and hydrochloric acid were purchased from Fisher Scientific (Loughborough, UK). SYBR green and loading buffer were purchased from Thermo Scientific (Waltham, USA). All reagents used were of analytical grade.
Formulation of AmB, PAR, and SSZ SLNs
The three different types of SLNs were prepared in our lab (10). Briefly, the drug (AmB, PAR, or SSZ) and lecithin were first dissolved in a mixture of chloroform-methanol at a 1:1 ratio, along with theobroma oil (TO) and beeswax (BW) as indicated in Table I. The solvent was evaporated off using Rotavapor® (Büchi R-200/205) at 50°C, leaving the drug embedded within the lipid matrix. The lipid matrix was melted in 20 ml of ethyl acetate (previously equilibrated with water for 10 min) at 70°C. At the same time, 40 ml of 2.5% sodium cholate (SC) solution was heated at the same temperature. Both phases were homogenized using a high-speed (10,000 rpm) homogenizer (Ika-Turrax, T25) for 6 min. Sixty milliliter of water at 70°C was then added slowly into the mixture with continuous stirring for a further 20 min. The organic solvent was then evaporated off using the rotary evaporator at 70°C.
Table I.
Composition of the SLNs
| Drug | Amount (mg) |
TO (mg) |
BW (mg) |
Lecithin (mg) |
SC (mg) |
Water (mL) |
|---|---|---|---|---|---|---|
| AmB, PAR, or SSZ | 50 | 200 | 200 | 120 | 1000 | 60 |
TO theobroma oil, BW beeswax, SC sodium cholate, PAR paracetamol
Relative Density and Viscosity Determinations
The densities and viscosities of freshly prepared SLN dispersions were investigated using a density bottle and a Brookfield viscometer (DV-I prime), respectively. For the relative density determination, the mass of each formulation was divided by the mass of an equal volume of deionized water. The viscosity values, on the other hand, were obtained directly from the viscometer readings. All determinations were made in triplicate and results expressed as mean ± standard deviation (SD).
Gel Electrophoresis
In order to have an insight on the relative movement propensities of the three SLNs within a milieu as may be encountered in the GI tract, a gel electrophoresis was set up where the distances traveled by the three SLNs constrained within agarose gel were measured. Agarose was dissolved completely in 1× TBE (89 mM Tris base, 89 mM boric acid, and 2 mM EDTA) buffer solution at a concentration of 1% w/v using microwave heating to dissolve the agarose. The solution was allowed to cool slightly (before gelation), followed by the addition of 30 μL of SYBR® green dye, and the mixture was poured into a casting plate and a gel comb was inserted. The gel was left to set at room temperature for 30 min. The running buffer 1× TBE was then poured into the gel tank to submerge the gel. A 10-μL aliquot of SLN sample was mixed with 2 μL of the loading buffer (containing glycerol, bromophenol blue as dye, and water) and then loaded into the wells within the gel in triplicate. The gel electrophoresis unit was run at +45 V for 45 min and then +39 V for 15 min. The migration of the SLNs within the gel was viewed under UV light (Bio-Rad®) and distances measured using a calibrated ruler.
PCS
Photon correlation spectroscopy studies (PCS) on the SLNs were carried out using a Zetasizer Nano ZS® (Malvern, UK). The parameters measured were z-average diameter, zeta potential, and the polydispersity index (PDI). All the samples were diluted with Milli-Q water (18.2 MΩ·cm at 25°C) before analyses. Each analysis was carried out at 25°C and performed in triplicate, and the data obtained were expressed as mean ± standard deviation.
SEM
The topography and morphology of the SLNs were determined using a Quanta 400 FE-Scanning Electron Microscopy (SEM) (FEI). Prior to analysis, the samples were diluted with Milli-Q water and a drop placed on a carbon tape and air-dried at room temperature. Samples were then mounted on the stage and viewed under low vacuum mode at 20 kV.
DSC
Differential scanning calorimetry (DSC) thermograms on the SLNs were obtained using a DSC Q2000 (TA instruments, New Castle, USA) equipped with a TA Universal Analysis 2000 software. Approximately 5 mg samples were placed in standard aluminum pans, hermetically sealed, and subjected to the analyses using a scan rate of 5°C/min from −20 to 250°C. Analyses were run under a gentle stream of nitrogen (50 mL/ min) using an empty pan as the reference.
Encapsulation Efficiency (%EE)
The %EE for the three SLNs (AmB, PAR, and SSZ) was determined as follows using a slightly modified method (9). A few drops of a 0.1-M HCl were added to 1 ml of SLN dispersion to precipitate the nanoparticles, followed by centrifugation at 14,000 rpm for 45 min. The supernatant was decanted, and the precipitate washed with PBS to remove any free drug. The precipitate was then dissolved in 1000 μL of a mixture of methanol and DMSO at ratios of 1:3, 3:1, and 1:1 for AmB, PAR, and SSZ SLNs, respectively. The above mixtures were heated at 70°C and then cooled to room temperature, followed by centrifugation at 14,000 rpm for 10 min to separate the lipid. The amount of drug in the aqueous phase was determined using HPLC.
The %EE was calculated using the following:
| 1 |
Drug Release Study
In vitro drug release from the SLNs was conducted using 25 mL PBS (pH 7.4) as the release medium. SLNs containing 0.5 mg of drug were dispersed in the dissolution medium and incubated at 37°C in an orbital water bath operated at 100 rpm. At predetermined time intervals (0, 1, 2, 4, and 8 h), samples were withdrawn and the nanoparticles precipitated with 0.1 M HCl, followed by centrifugation at 14,000 rpm for 10 min at 37°C and the supernatant decanted. A 20-μL aliquot of the supernatant, containing the released drug, was then injected onto the HPLC to determine amount of drug released.
Pilot GI Transit and Pharmacokinetic Study
Pharmacokinetic studies on the AmB, PAR, and SP were performed using three male Sprague-Dawley rats (300–350 g) fasted overnight. The protocol used for the study was approved by an Animal Ethics Committee of the University of Science, Malaysia. The animals were obtained from the Animal Holding Unit of University of Science, Malaysia.
The three SLNs (AmB, PAR, and SSZ) were dispersed in 1 mL of distilled water to produce an equivalent dose of 10 mg/kg and administered by oral gavage to the rats.
Blood samples (300 μL) were collected from the tail of the rats before and at 1, 2, 4, 8, 12, and 24 h post-administration into heparinized microcentrifuge tubes. Plasma was immediately harvested from the blood samples by centrifugation and analyzed for drug content using a validated HPLC method (18).
RESULTS AND DISCUSSION
Relative Density and Viscosity
The mean values of relative densities ranged from 1.004 to 1.008 and were not significantly different with each other and with water. Furthermore, the mean viscosity values ranged from 1.04 ± 0.12 to 1.21 ± 0.06 cPs, being not statistically different from each other (p > 0.05). This indicates that the nanoformulations were just as viscous as purified water (used as the reference liquid). Since viscosity and density play a significant role in the GI transit of dosage forms (19–21), it is probable that the three SLNs would respond similarly to the hydrodynamics imposed by the GI tract.
Gel Electrophoresis
In order to further ascertain the relative propensities of the three SLNs to movement within a milieu, the SLNs were subjected to gel electrophoresis where the distances migrated by each type of SLN towards the positive electrode were measured. The agarose gel matrix contains pores which are formed from random packing of polymer (d-galactose and 3, 6-anhydro-l- galactopyranose). Although the pores in the gel are small and the gel is stationary in contrast to what would be expected in the GI tract, the analysis nonetheless gave an indication of the mobility propensities of the SLNs within a matrix such as chyme. The distances migrated by the three types of SLNs ranged from 22.2 to 22.4 mm, being statistically insignificant with each other (p > 0.05). This study along with the density and viscosity data suggests the possibility of similar dispositions amongst the three SLNs in response to GI hydrodynamics.
PCS
There was no statistically significant difference between the values of the z-average of the three SLNs (p > 0.05), albeit a slightly higher value of z-average diameter for SSZ (224.8 ± 3.31 nm) and PAR (210.1 ± 1.40 nm) compared to that for AmB (206.5 ± 1.71 nm). The PDI values obtained were well below 0.5, ranging from 0.161 ± 0.03 to 0.218 ± 0.008, indicating a mostly narrow particle size distribution. There were a negligible proportion of the SLNs with z-average diameters beyond 1000 nm.
The zeta potential values for all three SLNs showed that their surfaces were negatively charged, and this is attributable to the presence of sodium cholate employed in the formulation. The magnitude of zeta potential can be used to predict colloidal stability, with values greater than ±60 mV indicating very good stability of colloidal dispersions (22). The zeta potential for the three SLNs ranged from −61.90 ± 1.04 to −71.90 ± 0.93 mV indicating that particle aggregation during storage owing to van der Waal inter-particle attractions can be expected to be low.
SEM Analyses
The images obtained from the SEM are shown in Fig. 1, where the spherical morphologies of the three SLNs generally appear identical in outlook. The surfaces of the SLNs appear rough on closer observation. The SEM images corroborate the size data obtained in the PCS analysis. It appears that the drugs are dispersed within the matrix of the solid lipid comprised of the TO and BW.
Fig. 1.

SEM images for AmB, PAR, and SSZ SLNs
DSC
The melting peaks for pure AmB were 153.7, 170, and 204°C, with 170°C regarded as the melting temperature of AmB. The melting peaks for PAR were 169.6°C and 245.9°C for SSZ. These three melting peaks (170, 169.6, and 245.9°C) were absent in the thermograms of their respective SLNs (Fig. 2a). This shows that the drugs are molecularly dispersed in the SLNs as their amorphous forms, which are further suggested by the similarity of the thermograms for all the SLNs shown in Fig. 2b. This is likely to be initiated when the drugs are dissolved in solvents and, subsequently, dispersed and retained within the lipid melt during SLN formulation (23–25). There was a decrease in melting point and enthalpy in all the SLNs as compared with those for the bulk lipids (Table II), and this is attributable to the increase in surface area as a result of formation of the SLNs (26,27) and also the presence of the sodium cholate (28). Characteristic endotherms appear at about 33 and 61°C in all three SLNs which are related to the lipids (TO and BW) used. This strongly suggests that the presence of the drugs did not alter the chemical makeup of the lipids. The last three peaks in the thermograms for all the SLNs (at about 110, 115, and 195°C) are due to the SC.
Fig. 2.
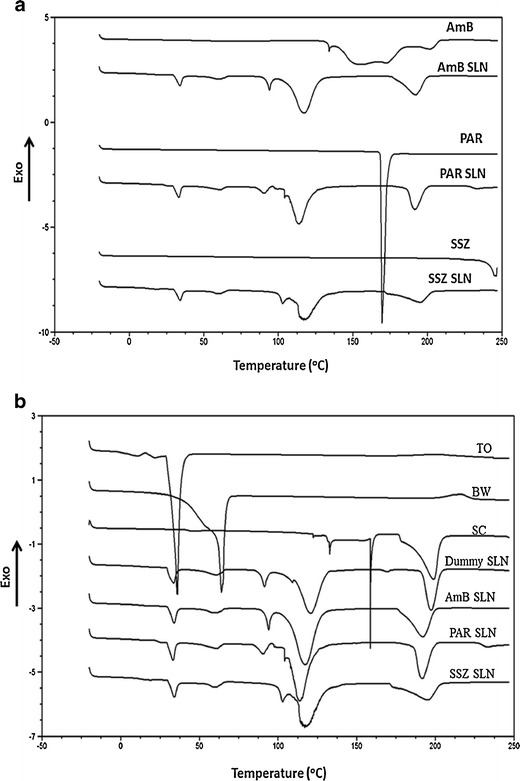
a DSC thermograms of pure drug (AmB, PAR, and SSZ) along with drug-loaded SLNs. b DSC thermograms of TO, BW, SC, dummy SLN, and drug-loaded SLNs
Table II.
Melting Peaks and Enthalpies of Bulk Lipids and SLNs
| Sample | TO | BW | ||
|---|---|---|---|---|
| Melting point (°C) |
Melting enthalpy (J/g) |
Melting point (°C) |
Melting enthalpy (J/g) |
|
| Bulk lipid | 35.1 | 156.1 | 63.3 | 176.7 |
| Drug-free SLN | 33.1 | 13.32 | 61.8 | 3.47 |
| AmB SLN | 33.1 | 5.95 | 61.5 | 3.76 |
| PAR SLN | 32.9 | 8.55 | 61.9 | 4.07 |
| SSZ SLN | 33.4 | 6.32 | 61.3 | 3.68 |
TO theobroma oil, BW beeswax, SLN solid lipid nanoformulation, SSZ salfasalazine
Encapsulation Efficiency (%EE)
The encapsulation efficiencies of the freshly prepared AmB, PAR, and SSZ SLNs were found to be 91.2 ± 3.04, 60.7 ± 0.26, and 78.4 ± 0.16%, respectively. The higher encapsulation efficiency of AmB is attributable to its lipophilicity, being compatible with the lipid matrices. On the other hand, the %EE for PAR was the lowest and this can be linked to its hydrophilic nature. Nonetheless, the %EE for all three drugs was appreciably high and suggests that the solvent diffusion method employed in preparing the SLNs is suitable for encapsulating AmB and the marker drugs.
Drug Release Study
Drug release study was conducted on the three SLNs, and Fig. 3 shows that only 11.8 ± 0.4% of AmB was released in 8 h whilst the maximum amount released from SSZ was 31.4 ± 1.95%. In contrast, 65.9 ± 0.04% of PAR was released within the study period. These are consistent with the fact that AmB and SSZ are biopharmaceutics classification system (BCS) class IV drugs while PAR belongs to class II. Furthermore, most of the PAR from the SLN are likely to be released prior to absorption of the SLNs because it partitions in favor of the aqueous environment (GI tract fluids) due to its lower log P value (0.46) as compared with that of AmB (0.8) and SSZ (2.5). This serves as a good predictor of gastric emptying of the three SLNs. Nonetheless, additional work is required to ascertain the consequence of a slow AmB release from the SLN when administered orally, but a slow release seems to be a favorable attribute in view of the possible uptake of the SLN by Peyer’s patches
Fig. 3.
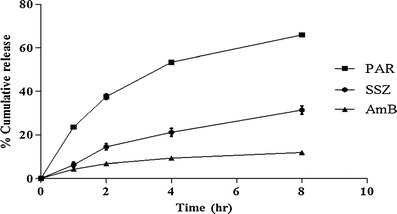
Cumulative drug release from SLNs as a function of time (mean ± SD; n = 3)
GI Transit and Pharmacokinetics Study
A pilot GI study was conducted on three male Sprague-Dawley rats to investigate the GI transit properties of the AmB-containing SLNs. Figure 4 and Table III show the plasma drug concentration-time profiles and pharmacokinetic data obtained respectively for AmB, PAR, and SP following the simultaneous oral administration of the three SLNs to the rats.
Fig. 4.
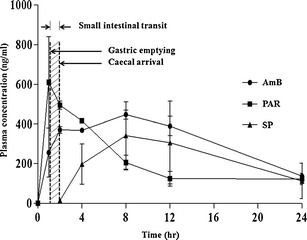
Plasma drug concentration-time profiles for AmB, PAR, and SP following simultaneous administration of AmB, PAR, and SSZ SLNs at a dose of 10 mg/kg each (mean ± SD; n = 3)
Table III.
Pharmacokinetic Parameters of AmB, PAR, and SP Following Simultaneous Oral Administration of AmB SLNs, PAR SLNs, and SSZ SLNs (mean ± SD; n = 3)
| Drug | Dose (mg/kg) |
Tmax
(h) |
Cmax
(ng/ml) |
AUC0–24
(ng h/ml) |
|---|---|---|---|---|
| AmB | 10 | 8 | 446.74 ± 23.6 | 7638 ± 440 |
| PAR | 10 | 1 | 609.85 ± 229.5 | 5146 ± 356 |
| SP | 10 | 8 | 341.78 ± 169.1 | 5095 ± 3186 |
SLN solid lipid nanoformulation, PAR paracetamol, SP sulfapyridine
PAR was rapidly absorbed and attained a Tmax of about 1 h. AmB was slowly absorbed reaching a Tmax in 8 h. This is suggestive of a slow SLN uptake process. The lipid moieties of SLNs have been reported to aid in prolonging the residence time of SLN when in contact with the GI epithelia, which possibly results in enhanced bioavailability (29).
There was a lag time of about 2 h prior to appearance of SP in blood, which then peaked at 8 h. The delay in the absorption of SP was mainly due to the transit time of the SLNs to the colon in addition to the delay resulting from degradation of SSZ to SP and 5-aminosalicylic acid by microbial flora in the colon prior to absorption of SP. The AUC0–24 was significantly high for AmB compared to PAR and SP, indicating a favorable, albeit slow uptake process. The high mean Cmax value for PAR is indicative of a rapid rate of absorption from the small intestine. The data from the released study above shows that PAR is rapidly released from the SLN, which, presumably, is the case in vivo. Therefore, the rapid rate of absorption of PAR may be attributed to rapid rate of release of PAR from the SLN. Figure 5 shows the mean PAR absorption-time profile from which the time taken for 50% of the PAR absorption to occur (T50E) was calculated (15). The value for T50E serves as the estimated time for 50% of the administered SLNs to be emptied from the stomach, which was found to be about 1.6 h. This relatively high gastric emptying time (1.6 h) can be attributed to the onset of the phase III motility pattern, where the “housekeeper” wave effectively empties the content of the stomach. The incidence of the housekeeper wave is variable and can be long, relative to the time of administration. On the other hand, the cecal arrival time (CAT) of the SLNs was found to be about 2 h, estimated from the initial detection of SP in the plasma. It follows that the estimated small intestinal transit time is 24 min, which is the difference between CAT and T50E. AmB absorption peaked at 8 h, and a significant concentration remained in circulation 12 h post-administration. This suggests that the absorption process is slow, and although more work may be required to confirm this, we believe that the SLNs are absorbed via the lymph after uptake by Peyer’s patches. Indeed, SLNs have been reported to increase the bioavailability of poorly soluble drugs after oral administration (9,30–32). This increase in bioavailability has been attributable to the uptake of the SLN from the GI tract followed by lymphatic transport into the systemic circulation. Therefore, this route of uptake is expected to be slow.
Fig. 5.
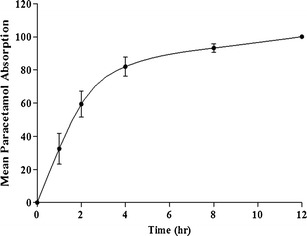
Mean PAR absorption-time curve (mean ± SD; n = 3)
CONCLUSION
AmB, PAR, and SSZ SLNs were successfully formulated with matched physical characteristics; thus, PAR and SSZ SLNs were deemed suitable for use as carriers for the marker drugs in the GI absorption study. The GI transit study indicates that AmB in SLN is absorbed slowly but significantly from the small intestines. Therefore, development of dosage forms for delivery of AmB via the oral route may exploit this attribute.
Acknowledgments
This work was supported by the Ministry of Science, Technology and Innovation (MOSTI) [02-02-12-SF0227].
References
- 1.Walsh TJ, Melcher GP, Rinaldi MG, Lecciones J, McGough DA, Kelly P, et al. Trichosporon beigelii, an emerging pathogen resistant to amphotericin B. J Clin Microbiol. 1990;28:1616–1622. doi: 10.1128/jcm.28.7.1616-1622.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Walsh M, White L, Atkinson K, Enno A. Fungal Pseudoallescheria boydii lung infiltrates unresponsive to amphotericin B in leukaemic patients. Aust NZ J Med. 1992;22:265–268. doi: 10.1111/j.1445-5994.1992.tb02123.x. [DOI] [PubMed] [Google Scholar]
- 3.Patel R. Amphotericin B, colloidal dispersion. Expert Opin Pharmacother. 2000;1:475–488. doi: 10.1517/14656566.1.3.475. [DOI] [PubMed] [Google Scholar]
- 4.Risovic V, Boyd M, Choo E, Wasan KM. Effects of lipid-based oral formulations on plasma and tissue amphotericin b concentrations and renal toxicity in male rats. Antimicrob Agents Chemother. 2003;47:3339–3342. doi: 10.1128/AAC.47.10.3339-3342.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Italia JL, Yahya MM, Singh D, Ravi Kumar MNV. Biodegradable nanoparticles improve oral bioavailability of amphotericin B and show reduced nephrotoxicity compared to intravenous Fungizone. Pharm Res. 2009;26:1324–1331. doi: 10.1007/s11095-009-9841-2. [DOI] [PubMed] [Google Scholar]
- 6.Jain S, Valvi PU, Swarnakar NK, Thanki K. Gelatin coated hybrid lipid nanoparticles for oral delivery of amphotericin B. Mol Pharm. 2012;9:2542–2553. doi: 10.1021/mp300320d. [DOI] [PubMed] [Google Scholar]
- 7.Fricker G, Kromp T, Wendel A, Blume A, Zirkel J, Rebmann H, et al. Phospholipids and lipid-based formulations in oral drug delivery. Pharm Res. 2010;27:1469–1486. doi: 10.1007/s11095-010-0130-x. [DOI] [PubMed] [Google Scholar]
- 8.Jawahar N, Meyyanathan SN, Reddy G, Sood S. Solid lipid nanoparticles for oral delivery of poorly soluble drugs. J Pharm Sci Res. 2012;4:1848–1855. [Google Scholar]
- 9.Li H, Zhao X, Ma Y, Zhai G, Li L, Lou H. Enhancement of gastrointestinal absorption of quercetin by solid lipid nanoparticles. J Control Release. 2009;133:238–244. doi: 10.1016/j.jconrel.2008.10.002. [DOI] [PubMed] [Google Scholar]
- 10.Tan SW, Billa N, Roberts CR, Burley JC. Surfactant effects on the physical characteristics of amphotericin B-containing nanostructured lipid carriers. Colloids Surf A. 2010;372:73–79. doi: 10.1016/j.colsurfa.2010.09.030. [DOI] [Google Scholar]
- 11.Tan SW, Billa N. Lipid effects on expulsion rate of amphotericin B from solid lipid nanoparticles. AAPS PharmSciTech. 2014;15:287–295. doi: 10.1208/s12249-013-0056-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Heading RC, Nimmo J, Prescott LF, Tothill P. The dependence of paracetamol absorption on the rate of gastric emptying. Br J Pharmacol. 1973;47:415–421. doi: 10.1111/j.1476-5381.1973.tb08339.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kellow JE, Borody TJ, Phillips SF, Haddad AC, Brown ML. Sulfapyridine appearance in plasma after salicylazosulfapyridine. Gastroenterology. 1986;91:396–400. doi: 10.1016/0016-5085(86)90574-3. [DOI] [PubMed] [Google Scholar]
- 14.Staniforth DH, Coates P, Clarke JGN. An HPLC assay for sulfapyridine in plasma and its use to assess small bowel transit time after the administration of sulfasalazine. Int J Clin Pharmacol Ther Toxicol. 1987;25:406–409. [PubMed] [Google Scholar]
- 15.Peh KK, Yuen KH. Indirect gastrointestinal transit monitoring and absorption of theophylline. Int J Pharm. 1996;139:95–103. doi: 10.1016/0378-5173(96)04602-9. [DOI] [Google Scholar]
- 16.Rahman NU, Yuen KH, Woei WJ. Gastrointestinal transit monitoring and absorption of controlled-release pellets of Diltiazem. Pharm Dev Technol. 2005;10:371–379. doi: 10.1081/PDT-65680. [DOI] [PubMed] [Google Scholar]
- 17.Sjödin L, Visser S, Al-Saffar A. Using pharmacokinetic modeling to determine the effect of drug and food on gastrointestinal transit in dogs. J Pharmacol Toxicol Methods. 2011;64:42–52. doi: 10.1016/j.vascn.2011.04.008. [DOI] [PubMed] [Google Scholar]
- 18.Amekyeh H. Development and validation of a simultaneous HPLC assay of paracetamol and sulfapyridine as markers for estimating GI transit of amphotericin B-containing nanoparticles in rat plasma [Dissertation]. Semenyih: University of Nottingham, Malaysia Campus; 2014.
- 19.Amidon GL, DeBrincat GA, Najib N. Effects of gravity on gastric emptying, intestinal transit, and drug absorption. J Clin Pharmacol. 1991;31:968–973. doi: 10.1002/j.1552-4604.1991.tb03658.x. [DOI] [PubMed] [Google Scholar]
- 20.Clarke GM, Newton JM, Short MD. Gastrointestinal transit of pellets of differing size and density. Int J Pharm. 1993;100:81–92. doi: 10.1016/0378-5173(93)90078-T. [DOI] [Google Scholar]
- 21.Tuleu C, Andrieux C, Boy P, Chaumeil JC. Gastrointestinal transit of pellets in rats: effect of size and density. Int J Pharm. 1999;180:123–131. doi: 10.1016/S0378-5173(98)00400-1. [DOI] [PubMed] [Google Scholar]
- 22.Riddick TM. Control of colloid stability through zeta potential. Pennsylvania: Livingston Publishing Company; 1968. [Google Scholar]
- 23.Cavalli R, Aquilano D, Carrlotti ME, Gasco MR. Study by X-ray powder diffraction and differential scanning calorimetry of two model drugs, phenothiazine and nifedipine, incorporated into lipid nanoparticles. Eur J Pharm Biopharm. 1995;41:329–333. [Google Scholar]
- 24.Cavalli R, Caputo O, Carlotti ME, Trotta M, Scarnecchia C, Gasco MR. Sterilization and freeze-drying of drug-free and drug-loaded solid lipid nanoparticles. Int J Pharm. 1997;148:47–54. doi: 10.1016/S0378-5173(96)04822-3. [DOI] [Google Scholar]
- 25.Venkateswarlu V, Manjunath K. Preparation, characterization and in vitro release kinetics of clozapine solid lipid nanoparticles. J Control Release. 2004;95:627–638. doi: 10.1016/j.jconrel.2004.01.005. [DOI] [PubMed] [Google Scholar]
- 26.Bunjes H, Koch MHJ, Westesen K. Effect of particle size on colloidal solid triglycerides. Langmuir. 2000;16:5234–5241. doi: 10.1021/la990856l. [DOI] [Google Scholar]
- 27.Unruh T, Bunjes H, Westesen K, Koch MHJ. Observation of size dependent melting in lipid nanoparticles. J Phys Chem B. 1999;103:10373–10377. doi: 10.1021/jp9912612. [DOI] [Google Scholar]
- 28.Westesen K, Bunjes H. Do nanoparticles prepared from lipids solid at room temperature always possess a solid lipid matrix? Int J Pharm. 1995;115:129–131. doi: 10.1016/0378-5173(94)00347-8. [DOI] [Google Scholar]
- 29.Zhuang CY, Li N, Wang M, Zhang XN, Pan WS, Peng JJ, et al. Preparation and characterization of vinpocetine loaded nanostructured lipid carriers (NLC) for improved oral bioavailability. Int J Pharm. 2010;394:179–185. doi: 10.1016/j.ijpharm.2010.05.005. [DOI] [PubMed] [Google Scholar]
- 30.Martins S, Silva AC, Ferreira DC, Souto EB. Improving oral absorption of Salmon calcitonin by trimyristin lipid nanoparticles. J Biomed Nanotechnol. 2009;5:76–83. doi: 10.1166/jbn.2009.443. [DOI] [PubMed] [Google Scholar]
- 31.Varshosaz J, Tabbakhian M, Mohammadi MY. Formulation and optimization of solid lipid nanoparticles of buspirone HCl for enhancement of its oral bioavailability. J Liposome Res. 2010;20:286–296. doi: 10.3109/08982100903443065. [DOI] [PubMed] [Google Scholar]
- 32.Cho HJ, Park JW, Yoon IS, Kim DD. Surface-modified solid lipid nanoparticles for oral delivery of docetaxel: enhanced intestinal absorption and lymphatic uptake. Int J Nanomedicine. 2014;9:495–504. doi: 10.2147/IJN.S56648. [DOI] [PMC free article] [PubMed] [Google Scholar]