

Abstract
This study investigated the potential use of mesoporous silica nanoparticles (MSNs) as a carrier for duloxetine hydrochloride (DX), which is prone to acid degradation. Sol–gel and solvothermal methods were used to synthesize the MSNs, which, after calcination and drug loading, were then characterized using X-ray diffraction (XRD), Brunauer-Emmett-Teller (BET) technique, thermogravimetric analysis (TGA), Fourier transform infrared (FT-IR) spectroscopy, scanning electron microscopy (SEM), differential scanning calorimetry (DSC), and diffuse reflectance ultraviolet-visible (DRS-UV-Vis) spectroscopy. Releases of DX from the MSNs were good in pH 7.4 (90%) phosphate buffer but poor in acidic pH (40%). In a comparative release study between the MSNs in phosphate buffer, TW60-3DX showed sustained release for 140 h, which was higher than the other nanoparticles. The mechanism of DX release from the MSNs was studied using Peppas kinetics model. The “n” value of all three MSNs ranged from 0.45 to 1 with a correlation coefficient (r2) >0.9, which indicated that the release of the drug from the system follows the anomalous transport or non-Fickian diffusion. The results supported the efficacy of mesoporous silica nanoparticles synthesized here as a promising carrier for duloxetine hydrochloride with higher drug loading and greater pH-sensitive release.
Electronic supplementary material
The online version of this article (doi:10.1208/s12249-014-0273-x) contains supplementary material, which is available to authorized users.
KEY WORDS: controlled release, duloxetine hydrochloride, meso silica nanoparticles, sol–gel synthesis
INTRODUCTION
Modern medications research over the past two decades involved in the development of new drug delivery tools for the effective targeting of disease and to reduce the toxicity. Mesoporous silica nanoparticles (MSNs) have attracted increasing attention for their potential biomedical applications because of their tailored mesoporous structure and high surface area. MSNs as drug delivery systems (DDSs) show significant advantages over traditional drug nanocarriers (1–3). These mesoporous systems are designed to carry and release their loaded drugs to a specific location in the body at a controllable release rate, without compromising the patient’s health. According to the literature, these materials are biodegradable and biocompatible (4,5). Many authors have reported that release of the drug can be easily controlled by manipulating pore size and surface chemistry of the MSN (6,7). Porosity can be induced in the inorganic silica part by removal of the surfactant through thermal or chemical means (8,9).
Recent advances in the morphology control and surface modification of MSNs, as well as an increased knowledge of the physiological factors affecting a favorable DDS, have opened new possibilities for more efficient treatment via this promising area of research (10,11). The mesoporous materials offer high loading ability for therapeutic molecules, proteins, and enzymes while protecting these molecules from premature release and degradation (12). In recent studies, many authors have reported the use of MSNs for enhancing dissolution and oral bioavailability of poorly soluble drugs (13,14). The improvement in the dissolution rate of poorly water-soluble drugs was due to the high specific area of the carrier material, the lack of crystalline phase, and the weak interactions between the silica surface and the drug (15). The density of surface silanol groups in the pore walls affects the drug loading. The pharmaceuticals interact via hydrogen bonds with surface silanol groups located inside the pores making the MSNs promising candidates for use as carriers in drug delivery systems (16,17).
Oral sustained/controlled and pH-sensitive release of loaded drug from MSNs is well documented (18–21). Sustained release of therapeutic agents such as ibuprofen, duloxetine, antipyrine, griseofulvin, ranitidine, furosemide, and captopril was enhanced by MSNs (22–25). Thermal carbonization and thermal oxidation of silica resulted in improved particle stability and modified surface properties which significantly affected the compound affinity toward the particles and subsequent drug release (24). Electrostatic interactions, i.e., attractive, repulsive, and hydrophobic forces between the drug and the silica surface, play an important role in controlling drug release. Ng et al reported that cationic molecules have a different release profile than anionic drug molecules from mesoporous silica materials due to the high affinity between the negatively charged silica material and the positively charged drug molecules (25).
Duloxetine hydrochloride (DX) is an antidepressant drug. According to BCS classification, DX is a class II drug; it has limited aqueous solubility (26). The degradation of this antidepressant drug in the acidic environment of the stomach leads to subtherapeutic levels; hence, it has low and variable bioavailability (ranging from 30 to 80%). Currently, the typical carriers used for DX consist of organic materials that are highly water-soluble and/or polymeric in nature (27). The method like probe sonication was not suitable commercial scale. Further, the main drawback of polymeric carriers is that the polymer should not interact with the drug molecules and the completeness of the device degradation must coincide with the completion of the drug release (28). Recently, Patel et al reported the usefulness of the solid lipid nanodelivery system to protect and control the delivery of DX. In their conclusion, they reported that formulations prepared by probe sonication produced better nanoparticles than other (29). Mesoporous silica materials can function as a carrier system for pharmaceuticals with effective loading and dissolution to the surrounding media.
In our previous study, we synthesized mesoporous silica material using a combination of dual templates, such as Triton X-100 together with Tween 20 and Tween 40, and found success in forming mesoporous silica (30). Potential biomedical applications of the synthesized MSNs were evaluated by loading with the poorly water-soluble drug DX HCl. The loading capacity and sustained release of the newly developed MSNs were found improved, and the mechanism of the drug release from MSNs could follow Higuchi diffusion kinetics (20).
In the present study, duloxetine hydrochloride entrapped mesoporous silica nanoparticles were prepared using Triton X-100 and Tween 60 as combined templating agent. The MSNs were characterized by X-ray diffraction (XRD), Brunauer-Emmett-Teller (BET) technique, thermogravimetric analysis (TGA), Fourier transform infrared (FT-IR) spectroscopy, differential scanning calorimetry (DSC), and diffuse reflectance ultraviolet-visible (DRS-UV-Vis) spectroscopy. The morphology of the MSNs was studied by scanning electron microscopy (SEM). Finally, we examined the in vitro release of the loaded drug from MSNs at different pHs. Here, we report our initial studies of this system knowing that further work has to be carried out to better understand how the silica surface and pH affect the release of the drug from MSNs.
MATERIALS AND METHODS
Materials
The materials included tetraethyl orthosilicate (TEOS), as a silica source (Sigma-Aldrich, GmbH, Germany), template surfactants polyethylene glycol p-(1,1,3,3-tetramethylbutyl)-phenyl ether (Triton X-100), and polyethylene glycol sorbitan monostearate (Tween 60) ( Sigma-Aldrich, GmbH, Germany), acetonitrile (ACN), ammonium fluoride, DX (reference standard from Sigma-Aldrich, GmbH, Germany), methylene dichloride (MDC) (Dae Jung Chemicals, Korea), and buffering agents such as disodium hydrogen phosphate, potassium dihydrogen ortho phosphate, and HCl (Acros Chemicals, Korea). All the chemicals were used as received, unless otherwise noted.
Methods
Synthesis of MSNs
The MSNs were synthesized according to a procedure adopted from our earlier report using Triton X-100 and Tween 20, 40, and 60 (20,30). In a typical procedure, 6.4 g of Triton X-100 and various amounts of Tween 60 were added to 300 g of DI water kept under constant stirring between 35 and 40°C to obtain a clear solution. While stirring, 16.6 g of TEOS was added dropwise to this solution. The solution was stirred until a gel was obtained by the addition of 0.083 g of ammonium fluoride. The gel composite was aged at room temperature for 24 h. Likewise, another set of gels were prepared and aged at 100°C for 24 h in a static hot air oven to obtain the solvothermaly derived product. The MSNs obtained by both methods were recovered by filtration, washed repeatedly with water, and dried overnight at 70°C. The obtained materials were designated as TW60-1-AS, TW60-2-AS, and TW60-3-AS. The templates occluded in the materials were removed by calcination at 450°C (heating rate 1°C min−1) for 5 h in air. The calcined materials were designated as TW60-1, TW60-2, TW60-3, TW60-1ST, TW60-2ST, and TW60-3ST where T represents Triton X-100; TW60 represents Tween 60; -1, -2, and -3 represent the content of Tween 60 used (0.0009, 0.0018, and 0.0027 mol, respectively), and ST represents the silicas synthesized under solvothermal condition (100°C).
Characterization of MSNs
The pore size and surface properties of the materials were characterized by physicochemical methods. A Rigaku Miniflex diffractometer was used to study the powder XRD patterns using Cu Kα radiation (λ = 1.54 Å at the 2θ range, 0.9–10° with a 1° step size and a 1-s step time). The pore structure and specific surface area of the materials were evaluated by a BELSORP mini II volumetric adsorption analyzer using nitrogen adsorption–desorption isotherm measurements at −196°C. Prior to the measurements, the samples were evacuated at 150°C. The specific surface area was determined from the linear part of the BET. A SCINCO N-1000 thermogravimetric (TG) analyzer was used to measure the weight loss of the as-synthesized, calcined MSNs, and the drug-loaded MSNs. A sample weighing ca. 10 mg was loaded onto an alumina sample pan of the TG unit, and the temperature was programmed to attain 800°C at a heating rate of 10°C min−1 in nitrogen atmosphere. Nicolet IR 200 FT-IR was used to record the FT-IR spectra of the samples. KBr pelleting technique was used for the FT-IR recording. Each sample was set for 20 scans with 4 cm−1 resolution over the range of 4000–400 cm−1. A JEOL-JSM 5600 SEM was used to study the morphology of MSNs after they were sputter-coated with gold plasma via a sputter coater (Cressington, Sputter Coater-108 auto). Solid-state NMR experiments were recorded using Bruker Avance II 500 spectrometers at 75.47 MHz for 13C using 7 mm ZrO2 rotors. For the 13C magic angle spinning (MAS; spinning frequency = 5–6 kHz), 90° pulses (5–6.5 μs) were used. The recycle delays were adjusted depending on the sample from 10 up to 500 s. The 13C CP MAS NMR spectra were recorded with an MAS frequency of 5 kHz, a contact time of 1 ms, and a recycle delay of 5 s. All the 13C NMR experiments were conducted with proton decoupling. A SCINCO DSC N 650 differential scanning calorimeter was used to record the DSC traces of the sample with a heating rate of 10°C min−1 under a helium purge of 40 mL min−1 to determine whether the DX was dispersed over the MSNs in crystalline or amorphous form. Further, to confirm inclusion of the drug into the pores of the silica matrices, the DRS-UV spectra of the samples were recorded using a SCINCO Neosys-2000 DRS-UV by comparing the maximum absorbance of the free drug and the drug-loaded MSNs.
Drug Loading
Drug loading was carried out by an adsorption procedure similar to that reported in our earlier study (20,21). In a typical procedure, about 50 mL of MDC solution of DX (10 mg mL−1) was sealed in an air-tight screw-capped container, after which a quantity of MSN, equivalent to that of the total quantity of DX, was slowly added at ice-cold temperature. The temperature was kept constant, and the solution was stirred continuously for another 3 h. The mixture was kept at room temperature for 3 days to ensure equilibrium, then filtered through a membrane filter (0.05 μm) and washed gently three times with a little additional MDC to remove the surface-adhered DX. The obtained filtrate was tested for DX by UV-visible spectrophotometer (Shimadzu, Japan) at 288 nm (31). The entrapment efficiency (EE) of MSNs was determined by the following (Eq. 1):
| 1 |
The drug-loaded MSNs were designated as TW60-1DX, TW60-2DX, and TW60-3DX, where DX represents the drug. The samples were dried overnight at room temperature and then dried more at 100°C in vacuum to remove MDC adsorbed with drug. The dried DX-loaded MSN were pressed into 0.3 g disks (13 × 3 mm in size) using uniaxial pressure (5.0 MPa) before the drug release studies (6). To evaluate the drug entrapment efficiency by the MSNs, MSNs containing an equivalent of 10 mg of DX were weighed accurately and dispersed in 25 mL acetonitrile, sonicated for 30 min, and stirred for 24 h. They were then centrifuged by HANIL, Fletas centrifuge at 5000 rpm for 30 min. The supernatant liquid was taken and diluted suitably, and the drug content was estimated by a UV spectrophotometer at 288 nm. This was repeated three times for statistical evaluation.
In Vitro Drug Release Studies
A phosphate buffer of pH 7.4 and 0.1 N HCl (pH 2) was chosen as the media for drug release. The tablet disks prepared using different MSNs (TW60-1DX, TW60-2DX, and TW60-3DX) were dropped into about 100 mL of the medium in a beaker that was maintained at 37 ± 2°C while being stirred at 100 rpm (1). A predetermined amount of solution was withdrawn at different times and was replaced with the same amount of fresh medium immediately after each sampling. The withdrawn samples were then filtered through a 0.1-μm filter, and the amount of the drug was estimated spectrophotometrically at 288 nm. Three samples were measured for each time point, and the results were reported as average values in terms of standard deviation (SD). The in vitro drug release data in pH 7.4 obtained by the above procedure were subjected to the analysis of Ritger and Peppas to elucidate the release mechanism (32,33). The initial 60% cumulative release data were used to estimate the diffusion exponent, n, by Eq. 2:
| 2 |
where Mt is the amount of drug released at time t, M∞ is the nominal total amount of drug released, k is the kinetic constant, and n is the diffusion exponent that is used to characterize the release mechanism.
RESULTS AND DISCUSSION
Nitrogen Adsorption–Desorption Isotherms of MSNs Before and After Drug Loading
Figure 1a, b depicts the results of nitrogen adsorption–desorption isotherms of MSNs before and after drug loading, respectively. All the synthesized MSNs exhibited a type IV N2 adsorption isotherm for mesoporous materials. In all cases, the capillary condensation of nitrogen started at p/p0 = 0.2 and extended up to 0.8, proving the wide pore distribution in the synthesized MSNs. The nitrogen desorption curve showed hysteresis, which confirmed the mesoporous nature. In the case of sol–gel synthesized silica, smooth polymerization of silica matrices formed over the surfactant micelles; at room temperature, the micelles structure was not disturbed. The possibility of smooth polymerization of SiO2 was higher in sol–gel procedure. Whereas, at high temperature the concentration of surfactant micelle, its structure was lost due to decreasing in viscosity of surfactant solution. This leads to destruction of the micelle assembly into smaller micelles. As the surface areas of the solvothermaly synthesized MSNs were much lower than those of the sol–gel-synthesized MSNs, the sol–gel-derived silica was selected for drug loading and release experiments. The nitrogen adsorption isotherm after DX loading showed similar behavior to that of free mesoporous silica. The results showed that the mesoporous characteristics of MSNs were not affected by the DX loading. The results (Table I) show that the surface area and pore volume of the sol–gel-synthesized MSNs were higher than that of the solvothermaly synthesized MSNs. Further, after DX loading, the surface area and pore volume decreased, which confirmed the ability of the synthesized silica to be a reservoir for drug storage and release.
Fig. 1.
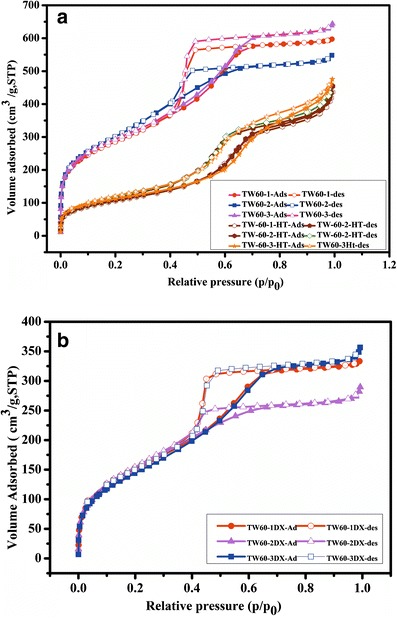
N2 adsorption–desorption isotherms of a calcined MSNs and b DX-loaded MSNs
Table I.
BET Data of Pure and Drug-Loaded Particles
| MSNs | BET surface area (m2 g−1) | Pore volume (cm3 g−1) | Average pore diameter (nm) |
|---|---|---|---|
| TW60-1 | 1024.3 | 0.9229 | 3.6038 |
| TW60-2 | 1056.2 | 0.9904 | 3.7509 |
| TW60-3 | 1083.6 | 0.8468 | 3.1257 |
| TW60-1ST | 383.69 | 0.6603 | 6.8839 |
| TW60-2ST | 395.97 | 0.6842 | 6.9122 |
| TW60-3ST | 402.58 | 0.7274 | 7.2272 |
| TW60-1DX | 600.14 | 0.4950 | 3.6895 |
| TW60-2DX | 554.24 | 0.4396 | 3.1037 |
| TW60-3DX | 512.06 | 0.5085 | 4.3032 |
X-Ray Diffraction Analysis
The XRD patterns of the calcined MSNs synthesized by sol–gel (TW60-1, TW60-2, and TW60-3) are shown in Fig. S1(supplementary file). The spectra of the sol–gel-synthesized MSNs show a second-order peak (110) at 1.75 (2θ), which is characteristic of mesopores. Similar to SBA-15 (Santa Barbara Amorphous) silica, the first-order peak (100) at 0.6 (2θ) was also seen in all the MSNs synthesized here. The patterns of the solvothermaly synthesized MSNs (TW60-1ST, TW60-2ST, and TW60-3ST) did not show any characteristic reflections at low angle (figure not shown). Therefore, the sol–gel method was considered a better route for producing ordered MSNs with the choice of templates used herein. The patterns of the corresponding drug-loaded samples (TW60-1DX, TW60-2DX, and TW60-3DX) are shown in Fig. S1(supplementary file) with their respective labels. Even after the drug loading, the second-order peaks appeared in all drug-loaded silica but with lower intensity than that of the parent MSNs. This confirms the loading of the drug inside the silica pores. Further, the absence of higher order peaks above 10 (2θ) proves that DX was dispersed in the matrices in an amorphous form which remains even after storage at 20% RH for 6 months.
Thermogravimetric Analysis
The TGA of the as-synthesized silica is given in Fig. S2a (supplementary file), which shows an initial 5–6% weight loss due to loss of physically adsorbed water occluded in the pores. In addition, a major weight loss due to loss of template surfactants was noted at around 400–450°C in all cases of silica. Hence, 450°C was used as calcination temperature. Further, the TGA of the calcined silicas is given in Fig.S2b, which shows only a small weight loss at around 100°C, which may be due to the loss of moisture captured during the storage of silica. Hence, to remove the moisture, the MSNs were completely vacuum-dried for 12 h at 150°C before the drug was loaded. Figure 2 presents the thermograms of drug-loaded MSNs. The DX-loaded MSNs showed a similar two-step weight loss pattern as that of pure DX around 100–200 and 350–400°C (20,27). These results exhibited a DX loading of 20, 26, and 31% into TW60-1DX, TW60-2DX, and TW60-3DX, respectively. However, the results were lower when compared to the UV-estimated drug loading of 24, 30, and 39%. This difference between the two estimation methods was due to DX degradation in the N2 environment of the TGA experiments, which may have induced the formation of carbonized drug particles inside the silica pores; the drug inside the pores was not completely oxidized in N2 atmosphere. This loading was higher than that observed in our earlier studies where the MSN was synthesized using Tween 20 and 40 as cotemplate. In addition, the amount of loaded DX was higher than that of ibuprofen loading reported in our previous preliminary report with these MSNs. This may be due to the strong ionic interaction between the surface silanol group and the positively charged DX (compared to ibuprofen which was discussed in detail in the later section on drug release) (21). Further, the loading increased little with an increased surface area of the MSNs. Even though the difference in surface area and pore volume was little, the loading of DX on TW60-3DX was highest. The mechanism of increased loading is unknown but might be possible that the presence of large number of surface silanol group in TW60-3, due to use of increased quantities of surfactants during the synthesis, contributing increased numbers of surface –Si–OH groups. The negative surface –Si–OH may retain the positively charged drug in high quantity.
Fig. 2.
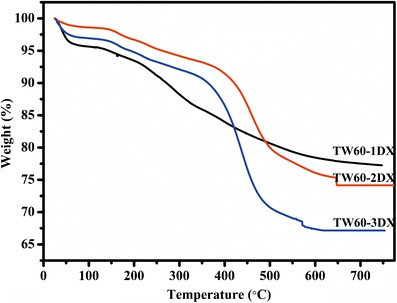
Thermograms of DX-loaded MSNs
From the results of UV estimation for the drug content in about 9.46 ± 0.095, 9.36 ± 0.097, and 9.51 ± 0.087 mg DX was present in TW60-1DX, TW60-2DX, and TW60-3DX MSNs, respectively. This proves that the entire drug contained in the matrices get released from the matrices by the extraction procedure followed here. The percent entrapment efficiency of all three MSNs were 24 ± 0.34, 30 ± 0.45, and 39 ± 0.56% for TW60-1DX, TW60-2DX, and TW60-3DX, respectively.
FT-IR Spectroscopy
The FT-IR spectra of the calcined MSNs, TW60-1, TW60-2, and TW60-3, are shown in Fig. S3a (supplementary file). All three spectra showed similar features to mesoporous silicas reported earlier. The intense broad O–H stretching vibration of water appearing 3500 cm−1 is due to water that is hydrogen-bonded to defective Si–OH groups of the silica matrix. The band at 1620 cm−1 is attributed to the bending vibration of water. The broad peak observed between 1000 and 1200 cm−1 is due to Si–O–Si asymmetric stretching. The peak observed at around 960 cm−1 indicates defective Si–O–H. The shoulder peak immediately following this is due to symmetric Si–O–Si stretching. Si–O–Si bending vibration is observed at the end of spectrum. The peaks for template –CH2– were absent in all calcined silica, which proves the expulsion of templates from the silica after the calcinations.
The FT-IR spectrum of the DX is shown in Fig. S3b (supplementary file). The N–H stretching vibration is weak (close to 3400 cm−1) due to hydrogen bonding. The peak above 3000 cm−1 is due to aromatic C–H stretching, and the band below 3000 cm−1 is attributed to alkyl CH2 stretching. The other bands for hydrogen-bonded N–H groups of the drug were observed between 2200 and 2800 cm−1. The CH2 bending vibrations are shown at 1450 and 1375 cm−1. The phenoxy C–O vibration appears at 1200 cm−1. The peaks attributable to thiophene and naphthalene rings as seen between 1600 and 1450 cm−1. The group of peaks around 700 cm−1 is due to aromatic C–H bending modes of the naphthalene and thiophene rings.
The FT-IR spectra of the drug-loaded MSNs, TW60-1DX, TW60-2DX, and TW60-3DX, are shown in Fig. 3. The peak for meso silica was completely resolved with good intensity, although compared to pure silica, the intensities are slightly reduced. This is due to the inclusion of the drugs in the pores of the silica. In addition, the CH2 vibrations of the drug are partly seen just below 3000 cm−1 and its bending modes are also seen between 1300 and 1500 cm−1. Further, the aromatic –CH– bending modes for the naphthalene and thiophenyl rings of DX are also observed at around 700 cm−1 as a small hump. All other remaining peaks for DX are not well resolved here, because of the fine dispersion of DX into the pores rather than on the surface.
Fig. 3.
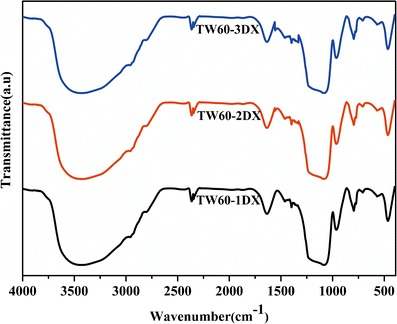
FT-IR spectra of DX-loaded MSNs
13C-NMR Spectroscopy
The 13C-NMR spectrum of the drug-loaded samples (Supplementary Fig. S4) was compared with that of pure DX given by Marjo et al (34). The peaks for Ar–H and thiophenyl CH appear at 129.65 and 127.53 ppm (the actual region for solid-state NMR is from 130 to 120 ppm), respectively. Due to the fine dispersion of DX inside the pores, the peaks were not split into separate peaks as like in the reference. Some of the shoulder peaks appear above major peaks are attributed to Ar–H splits for naphthalene ring. The split near 79 ppm was attributed to –O–CH– but was slightly shifted downfield compared to the reference, which might be because of the measurement in the reference was carried out at 6–15 MHz, compared to 5 MHz in our study. The resulting difference in frequencies may have caused shifts in peak position. The peaks at 45.31 ppm (46.5 ppm in the reference) were assigned to –CH2–N–, and the peak at 30.41 ppm was assigned to the N–CH3– group of the amine. In combination, the NMR data further verified the presence of the drug in the pores of the silica. The shifts in the positions of the peaks are due to interference from the silica matrix as well as the difference in frequency of radiation used (32).
Scanning Electron Microscopy
SEM images of the MSNs and DX-loaded MSNs are shown in Fig. 4. These images reveal the spherical morphology of the particles with smooth surface texture and little deposits; the formation of such deposits may due to incomplete silica polymerization during synthesis (Fig. 4a–c). The images of the DX-loaded MSNs (Fig. 4d–f) show morphology with slightly disturbed smoothness and little deposits on the surfaces possibly caused by some remaining surface-adhered drug particles. In the case of TW60-1DX, the particles were broken into smaller ones. This contrasted with the case of ibuprofen loading (21) on the same MSNs in our previous study, in which a large amount of drug was deposited over the surface of the particles, increasing the particles’ surface diameter. However, in the case of higher pore volume and BET pore size, TW60-2DX and 3DX were not greatly disturbed; this was also observed from their XRD reflections for the ordered peaks at 1.8–2.3 (2θ). Further, due to the high concentration of surfactants during synthesis, micelles thus formed may get stabilized, and this enhances the smooth polymerization of silica in the case of TW60-2 and 3. Hence, particles TW60-2 and 3 were observed to be good for loading the drug DX. This suggests that the smooth polymerization of silica and the formation of mesopores required a high concentration of Tween 60 surfactant. These results of XRD and SEM were good with respect to their XRD reflections and morphology when compared to our previous reports on silica synthesized using Triton X-100 together with Tween 20 and Tween 40 where the particles clumped together to form aggregates; Tween 60 formed smooth and individual particle.
Fig. 4.

SEM images of MSNs a TW60-1, b TW60-2, c TT60-3 (Calcined), and drug-loaded MSNs, d TW60-1DX, e TW60-2DX, and f TW60-3DX
Differential Scanning Calorimetry Analysis
DSC analysis was carried out to determine the dispersion of DX on the silica matrix, as crystalline or amorphous form Fig. S5 (supplementary file). Melting of the free drug is evidenced by the sharp peak of the free drug at around 160°C. The degradation temperature appears at 300°C, which is further supported by TGA results. Traces with similar features were also observed in the physically blended DX (TW60-1DP, TW60-2DP, and TW60-3DP) and silicas, due to poor drug dispersion on the silica surface because of physical mixing. During mixing, the trituration technique that was used reduced the crystalline size of DX, leading to an endotherm at low temperature with a less intense peak than that of the free drug. The endotherm responsible for the decomposition was shifted to a slightly lower temperature than that of the free DX but was slightly broadened due to the size reduction of DX crystals. This is due to the hydrogen bonding interaction of the organic that over the solid matrix (35). These results matched those of our previous report with Triton X-100 with Tween 20 and 40 (20,30). Such interaction was also seen in DSC traces of samples obtained by the usual solvent-aided loading which did not show melting peaks (TW60-1DX, T60-2DX, and T60-3DX); this illustrates the complete dispersion of the drug into the pores of matrix in amorphous form. The broad endotherm also matched that in the physically blended samples. Further, the fine dispersion and decreased concentration of drug in the matrix made it difficult to measure the melting temperature exactly.
DRS-UV-Vis Spectral Studies and Drug Content/Entrapment Efficiency
The solid-state DRS-UV-Vis spectra of the MSNs and drug-loaded MSNs are shown in Fig. S6 (supplementary file). This analysis was carried out to confirm the inclusion of the DX in the MSNs. The absorbance of the bare MSNs was zero from 400 to 200 nm. The pure DX and DX-loaded MSNs showed two broad absorptions near the UV region due to the transition of π-electron of the naphthalene ring (36). The λ-max of DX in MSNs was shifted toward a shorter wavelength when compared to that of the pure drug (hypsochromic shift) due to the suppression of the free resonance caused by the oxygen lone pair of the 1-naphthol moiety of the drug. The electrons’ dispersion prevents the electron pair of the oxygen of the 1-naphthol ring from being delocalized over the aromatic rings. Furthermore, restricted free rotation is caused by the formation of weak hydrogen bonds between both the lone pair electron of the positively charged nitrogen of DX and the loan pair electrons in the 1-napthol moiety with that of the defective/negatively Si–OH groups of MSNs. The absorbance due to thiophenyl moiety close to 210 nm was also not clearly resolved because of peak broadening (37).
In Vitro Drug Release
Three-hundred-milligram disks of each MSNs containing 120, 90, and 72 mg of DX for TW60-1DX, TW60-2DX, and TW60-3DX, respectively, were put into release media and studied under the conditions given in experimental sections using a UV-vis spectrophotometer at 288 nm. The release profiles of DX from the three different MSNs (TW60-1DX, TW60-2DX, and TW60-3DX) at different pH conditions are shown in Fig. 5. All the MSNs released the DX in pH 7.4 phosphate buffer, but the release was very poor in acidic conditions. First, it can reasonably be assumed that partial charge interactions between the drugs and the silica surface contributed to the differential release behavior. In acidic conditions (pH 1.2), the maximum release was only 40% due to affinity between the silica surface and drug, in general the silanol group of MSNs, which carries a net negative charge, bonded tightly to the positively charged DX (pKa 9.34). At pH 7.4, the drugs and the silica surface both carry a negative charge as the buffer causes ionic replacement of HCl by the Na+ ions. This causes the drug to be ionized into free base with loan pairs of electrons at the nitrogen center and gives partial negative charge to the DX molecule. This partial negative charge will cause charge repulsion between the silica surface and DX, which likely promotes desorption. These results demonstrated that the release of the drug from this carrier is strongly pH dependent. To summarize, MSNs may be suitable for selective drug release in higher pH conditions while protecting the drug from degradation in acidic pH conditions.
Fig. 5.
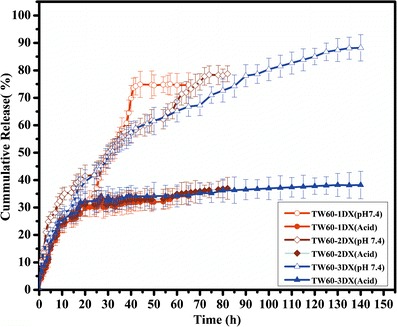
Cumulative drug release pattern of TW60-1DX, TW60-2DX, and TW40-3DX
The release data of the formulations were fitted to models representing Higuchi’s square-root of time model and Ritiger and Peppas’ model to predict the drug release kinetics and mechanism. The regression coefficient (r2) of the n value was determined, and the data are shown in Table II. Higuchi’s kinetics model was also applied to all the three MSNs (38). A straight line in the Higuchi plot corresponds to a pure Higuchi type of diffusion-driven release for a carrier. Further, the mechanism of DX release from the MSNs was studied using the n values of all three MSNs; these values ranged from 0.45 to 1 with a correlation coefficient of r2 > 0.9 at 95% confidence level using the initial 60% cumulative release data. The n value range indicated that the drug release from the system followed the anomalous transport or non-Fickian diffusion. Fickian diffusion release occurs by the usual molecular diffusion of the drug due to the chemical potential gradient (39–41). The results might be attributed to the influence of weak interactions between the drug and the silica surface and the eroding silica matrix in basic conditions (pH 7.4). While in comparison, the TT60-3DX showed prolonged sustained release in the phosphate buffer than other two nanoparticles. This may be due to higher loading, which will cause reduced pore size and may inhibit the faster penetration dissolution media. Further, the erosion silica should alter the release and change the kinetics to Peppa’s case II. The silica was preferentially eroded from the regions where DX had been released from, since the rate of MSN (TW60-3DX) dissolution was clearly slower for DX compared to the other MSNs. The in vitro study showed that the MSNs protected the release of DX in acidic conditions while releasing the DX in basic conditions. This is significant for oral sustained release of duloxetine hydrochloride.
Table II.
In Vitro Drug Release Kinetic Data
| Kinetic models | Correlation coefficient (r) and n values | TW60-1DX | TW60-2DX | TW60-3DX |
|---|---|---|---|---|
| Higuchi | r 2 | 0.897 | 0.968 | 0.987 |
| Ritger and Peppas | r 2 | 0.923 | 0.924 | 0.988 |
| n | 0.641 | 0.639 | 0.787 |
CONCLUSIONS
Mesoporous silica nanoparticles were synthesized successfully and evaluated for their efficacy as a carrier for DX HCl. In extensive characterization of the MSNs, SEM and XRD results revealed slight morphological changes in the silica after loading the drug but no destruction of the mesopore order. The drug-loading experimental results revealed that the MSNs were capable of engulfing DX, as verified by the TGA results. In vitro release data showed that the MSNs released the drug in basic conditions and that they protected the drug from degradation in acidic environment. The mechanism of drug release followed non-Fickian diffusion. The results highlighted porous silica’s potential as a carrier that allows higher drug loading, greater pH sensitivity, and prolonged sustained release of DX HCl.
Electronic supplementary material
Below is the link to the electronic supplementary material.
(DOC 7874 kb)
Acknowledgments
This work was financially supported by grants from the Korea CCS R&D Center, funded by the Ministry of Education, Science and Technology of the Korean Government. The authors extend their thanks to National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT and Future Planning (Grant No. 2014-004694) for their partial financial support.
References
- 1.Vallet-Regi M, Ra’mila A, del Real RP, Pe’rez-Pariente JA. New property of MCM-41: drug delivery system. Chem Mater. 2001;13:308–311. doi: 10.1021/cm0011559. [DOI] [Google Scholar]
- 2.Salonen J, Kaukonen AM, Hirvonen J, Lehto VP. Mesoporous silicon in drug delivery applications. J Pharm Sci. 2008;97:632–653. doi: 10.1002/jps.20999. [DOI] [PubMed] [Google Scholar]
- 3.Colilla M, González B, Vallet-Regí M. Mesoporous silica nanoparticles for the design of smart delivery nanodevices. Biomater Sci. 2013;1:114–134. doi: 10.1039/C2BM00085G. [DOI] [PubMed] [Google Scholar]
- 4.Tang F, Li L, Chen D. Mesoporous silica nanoparticles: synthesis, biocompatibility and drug delivery. Adv Mater. 2012;24:1504–1534. doi: 10.1002/adma.201104763. [DOI] [PubMed] [Google Scholar]
- 5.Hirvonen J, Laaksonen T, Peltonen L, Santos H, Lehto V-P, Heikkilä T, et al. Feasibility of silicon-based mesoporous materials for oral drug delivery applications. Dosis. 2008;24:129–149. [Google Scholar]
- 6.Horcajada P, Ramila A, Perez-Pariente J, Vallet-Regi M. Influence of poresize of MCM-41 matrices on drug delivery rate. Micro Meso Mater. 2004;68:105–109. doi: 10.1016/j.micromeso.2003.12.012. [DOI] [Google Scholar]
- 7.Friedrich H, Fussnegger B, Kolter K, Bodmeier R. Dissolution rate improvement of poorly water-soluble drugs obtained by adsorbing solutions of drugs inhydrophilic solvents onto high surface area carriers. Eur J Pharm Biopharm. 2006;62:171–177. doi: 10.1016/j.ejpb.2005.08.013. [DOI] [PubMed] [Google Scholar]
- 8.Chen C-Y, Li H-X, Davis ME. Studies on mesoporous materials. I. Synthesis and characterization of MCM-41. Micro Meso Mater. 1993;2:17–26. doi: 10.1016/0927-6513(93)80058-3. [DOI] [Google Scholar]
- 9.Tanev PT, Pinnavaia TJ. A neutral templating route to mesoporous molecular sieves. Science. 1995;267:865–867. doi: 10.1126/science.267.5199.865. [DOI] [PubMed] [Google Scholar]
- 10.Shahbazi M-A, Herranz B, Santos HA. Nanostructured porous Si-based nanoparticles for targeted drug delivery. Biomatter. 2012;2:296–312. doi: 10.4161/biom.22347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ukmar T, Planinek O. Ordered mesoporous silicates as matrices for controlled release of drugs. Acta Pharm. 2010;60:373–385. doi: 10.2478/v1007-010-0037-4. [DOI] [PubMed] [Google Scholar]
- 12.Mehrdad M. Synthesis and characterization of pH-sensitive silica nanoparticles for oral-insulin delivery. Current Drug Deliv. 2011;8:607–611. doi: 10.2174/156720111797635522. [DOI] [PubMed] [Google Scholar]
- 13.Kumar D, SailajaChirravuri SV, Shastri NR. Impact of surface area of silica particles on dissolution rate and oralbioavailability of poorly water soluble drugs: a case study with aceclofenac. Int J Pharm. 2014;461:459–468. doi: 10.1016/j.ijpharm.2013.12.017. [DOI] [PubMed] [Google Scholar]
- 14.Charnay C, Begu, Tourn-Pateilh C, Nicole L, Lerner DA, Devoisselle JM. Inclusion of ibuprofen in mesoporous templated silica: drug loading and release property. Eur J Pharm Biopharm. 2004;57:533–540. doi: 10.1016/j.ejpb.2003.12.007. [DOI] [PubMed] [Google Scholar]
- 15.Ambrogi V, Perioli L, Marmottini F, Accorsi O, Pagano C, Ricci M, et al. Role of mesoporous silicates on carbamazepine dissolution rate enhancement. Micro Meso Mater. 2008;113:445–452. doi: 10.1016/j.micromeso.2007.12.003. [DOI] [Google Scholar]
- 16.Zhu Y, Shi J, Shen W, Dong X, Feng J, Ruan M, et al. Uniform magnetic hollow spheres with a magnetic core/mesoporous silica shell structure. Angew Chem Int Ed Engl. 2005;4:5083–5087. doi: 10.1002/anie.200501500. [DOI] [PubMed] [Google Scholar]
- 17.Mal N, Fujiwara M, Tanaka Y. Photocontrolled reversible release of guest molecules from coumarin-modified mesoporous silica. Nature. 2003;421:350–353. doi: 10.1038/nature01362. [DOI] [PubMed] [Google Scholar]
- 18.He Q, Gao Y, Zhang L, Zhang Z, Gao F, Ji X, et al. A pH-responsive mesoporous silica nanoparticles-based multi-drug delivery system for overcoming multi-drug resistance. Biomaterials. 2011;32:7711–7720. doi: 10.1016/j.biomaterials.2011.06.066. [DOI] [PubMed] [Google Scholar]
- 19.Van SM, Mellaerts R, Thi TD, Martens JA, Van Humbeeck J, Annaert P, et al. Preventing release in the acidic environment of the stomach via occlusion in ordered mesoporous silica enhances the absorption of poorly soluble weakly acidic drugs. J Pharm Sci. 2011;100:4864–4876. doi: 10.1002/jps.22703. [DOI] [PubMed] [Google Scholar]
- 20.Ganesh M, Hemalatha P, Mei Mei P, Palanichamy M, Ubaidulla U, Jang HT. Synthesis and characterization of pharmaceutical surfactant templated mesoporous silica: its application to controlled delivery of duloxetine. Mater Res Bull. 2014;51:228–235. doi: 10.1016/j.materresbull.2013.12.037. [DOI] [Google Scholar]
- 21.Roggers R, Kanvinde S, Boonsith S, Oupick D. The practicality of mesoporous silica nanoparticles as drug delivery devices and progress toward this goal. AAPS PharmSciTech. 2014;15:1163–1171. doi: 10.1208/s12249-014-0142-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ganesh M, Lee SG. Synthesis, characterization and drug release capability of new cost effective mesoporous silica nano particles for ibuprofen drug delivery. Int J Cont Auto. 2013;6:207–216. doi: 10.14257/ijca.2013.6.5.20. [DOI] [Google Scholar]
- 23.Salonen J, Laitinen L, Kaukonen AM, Tuura J, Bjorkqvist M, Heikkila T, et al. Mesoporous silicon microparticles for oral drug delivery: loading and release of five model drugs. J Contol Release. 2005;108:362–374. doi: 10.1016/j.jconrel.2005.08.017. [DOI] [PubMed] [Google Scholar]
- 24.Popovici RF, Seftel EM, Mihai GD, Popovici E, Voicu AV. Controlled drug delivery system based on ordered mesoporous silica matrices of captopril as angiotensin-converting enzyme inhibitor drug. J Pharm Sci. 2011;100:704–714. doi: 10.1002/jps.22308. [DOI] [PubMed] [Google Scholar]
- 25.Ng JBS, Kamali-Zare P, Brismar H, Bergstrom L. Release and molecular transport of cationic and anionic fluorescent molecules in mesoporous silica spheres. Langmuir. 2008;24:11096–11102. doi: 10.1021/la801179v. [DOI] [PubMed] [Google Scholar]
- 26.Vinita V, Nagesh U, Mittapalli N, Kumar P, DR. Reddy’s Laboratories, assignee. Pharmaceutical formulations comprising duloxetine, United States patent, 20090226517A1, 2009
- 27.Zhao R-K, Cheng G, Tang J, Song J, Peng W-X. Pharmacokinetics of duloxetine hydrochloride enteric-coated tablets in healthy Chinese volunteers: a randomized, open-label, single- and multiple-dose study. Clin Ther. 2009;31:1022–1036. doi: 10.1016/j.clinthera.2009.05.005. [DOI] [PubMed] [Google Scholar]
- 28.Gopferich A, Tessmar J. Polyanhydride degradation and erosion. Adv Drug Deliv Rev. 2002;54:911–931. doi: 10.1016/S0169-409X(02)00051-0. [DOI] [PubMed] [Google Scholar]
- 29.Patel K, Padhye S, Nagarsenker M. Duloxetine HCl lipid nanoparticles: preparation, characterization, and dosage form design. AAPS PharmSciTech. 2012;13:125–133. doi: 10.1208/s12249-011-9727-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ganesh M, Hemalatha P, Mei Mei P, Rajasekar K, Jang HT. A new fluoride mediated synthesis of mesoporous silica and their usefulness in controlled delivery of duloxetine hydrochloride a serotonin re-uptake inhibitor. J Ind Eng Chem. 2012;18:684–689. doi: 10.1016/j.jiec.2011.11.121. [DOI] [Google Scholar]
- 31.Yunoos M, Sankar DG, Kumar BP, Hameed S, Hussain A. Simple UV spectrophotometric determination of duloxetine hydrochloride in bulk and in pharmaceutical formulations. E-J Chem. 2010;7:785–788. doi: 10.1155/2010/709176. [DOI] [Google Scholar]
- 32.Ritger PL, Peppas NA. A simple equation for description of solute release. I. Fickian and non-Fickian release from non-swellable devices in the form of slabs, spheres, cylinders or discs. J Control Release. 1987;5:23–36. doi: 10.1016/0168-3659(87)90034-4. [DOI] [PubMed] [Google Scholar]
- 33.Peppas NA. Analysis of Fickian and non-Fickian drug release from polymers. Pharm Acta Helv. 1985;60:110–111. [PubMed] [Google Scholar]
- 34.Marjo CE, Bhadbhade M, Hook JM, Rich AM. Polymorphism and a metastable Solvate of duloxetine hydrochloride. Mol Pharm. 2011;8:2454–2464. doi: 10.1021/mp200455u. [DOI] [PubMed] [Google Scholar]
- 35.Newa M, Bhandari KH, Oh DH, Kim YR, Sung JH, Kim JO, et al. Enhanced dissolution of ibuprofen using solid dispersion with poloxamer. 407. Arch Pharm Res. 2008;31:1497–1507. doi: 10.1007/s12272-001-2136-8. [DOI] [PubMed] [Google Scholar]
- 36.Labudzinska A, Gorczynska K. The UV difference spectra as a characteristic feature of phenols and aromatic amines. J Mol Str. 1995;349:469–472. doi: 10.1016/0022-2860(95)08810-I. [DOI] [Google Scholar]
- 37.Sugimoto Y, Nishimura S, Imoto E. The ultraviolet spectra of the thiophene derivatives. Opera 1959;71–81
- 38.Landau MV, Varkey SP, Herskowitz M, Regev O, Pevzner S, Sen T, et al. Wetting stability of Si-MCM-41 mesoporous material in neutral, acidic and basic aqueous solutions. Micro Meso Mater. 1999;33:149–163. doi: 10.1016/S1387-1811(99)00133-X. [DOI] [Google Scholar]
- 39.Higuchi T. Rate of release of medicaments from ointment bases containing drugs in suspension. J Pharm Sci. 1961;50:874–875. doi: 10.1002/jps.2600501018. [DOI] [PubMed] [Google Scholar]
- 40.Kuang Y, Zhao L, Zhang S, Zhang F, Dong M, Xu S. Morphologies, preparations and applications of layered double hydroxide micro-/nanostructures. Materials. 2010;3:5220–5235. doi: 10.3390/ma3125220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wei M, Pu M, Guo J, Han J, Li F, He J, et al. Intercalation of l-dopa into layered double hydroxides: enhancement of both chemical and stereochemical stabilities of a drug through host-guest interactions. Chem Mater. 2008;20:5169–5180. doi: 10.1021/cm800035k. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(DOC 7874 kb)