

Abstract
Mutations in ATRX (alpha thalassemia/mental retardation syndrome X-linked), a chromatin-remodeling protein, are associated with the telomerase-independent ALT (alternative lengthening of telomeres) pathway of telomere maintenance in several types of cancer, including human gliomas. In telomerase-positive glioma cells, we found by immunofluorescence that ATRX localized not far from the chromosome ends but not exactly at the telomere termini. Chromatin immunoprecipitation (ChIP) experiments confirmed a subtelomeric localization for ATRX, yet short hairpin RNA (shRNA)-mediated genetic inactivation of ATRX failed to trigger the ALT pathway. Cohesin has been recently shown to be part of telomeric chromatin. Here, using ChIP, we showed that genetic inactivation of ATRX provoked diminution in the amount of cohesin in subtelomeric regions of telomerase-positive glioma cells. Inactivation of ATRX also led to diminution in the amount of TERRAs, noncoding RNAs resulting from transcription of telomeric DNA, as well as to a decrease in RNA polymerase II (RNAP II) levels at the telomeres. Our data suggest that ATRX might establish functional interactions with cohesin on telomeric chromatin in order to control TERRA levels and that one or the other or both of these events might be relevant to the triggering of the ALT pathway in cancer cells that exhibit genetic inactivation of ATRX.
INTRODUCTION
The linear chromosomes of eukaryotic organisms require particular protection at their extremities. Telomeres, which represent the ends of these chromosomes and contain repeated TG-rich sequences that do not code for proteins, as well as proteins of the shelterin complex, are essential for this protection (1). Telomeres protect chromosome ends from DNA repair activities that reseal chromosome internal DNA breaks occurring during DNA damage (2). Telomere sequences naturally erode with ongoing cell divisions, due to intrinsic mechanisms associated with the fixed 5′-to-3′ polarity of replication of the DNA of the genome. Below a certain threshold, shortened telomeres result in DNA damage-induced cell cycle arrest, which is the equivalent of replicative senescence in cultured cells.
By limiting the replicative potential of cells, telomere length acts as a biological clock, and telomere erosion serves as a barrier against tumorigenesis in healthy tissue. Paradoxically, telomere erosion or telomere dysfunction also induces chromosomal instability and favors the emergence of tumors (3). However, following cancer initiation, tumor cells must overcome the telomere-controlled replicative-senescence barrier, and all have an absolute necessity to maintain functional telomeres in order to sustain continuous and unlimited cell proliferation. In around 85 to 90% of cancer types, this occurs through upregulation of telomerase, a reverse transcriptase with a built-in RNA template specialized in telomeric DNA replication that is naturally repressed in most somatic tissues (4). In the remaining 10 to 15% of cancer types, an alternative pathway called the ALT (alternative lengthening of telomeres) pathway, functioning either by amplifying telomere sequences by homologous recombination or by sister chromatid exchange, is used (5–7). Although these numbers have been accepted for many years, a recent study performed on over 6,000 tumor tissues reported that only 3.7% of the tumors were ALT positive, using fluorescence in situ hybridization (FISH) to probe for ALT-specific promyelocytic leukemia (PML) bodies (8). Human gliomas are among the 5 to 15% of cancer types that can survive, owing to either telomerase or the ALT pathway (8, 9).
Recent clinical studies have highlighted the existence of a strong correlation between the occurrence of the ALT pathway of telomere maintenance in several types of ALT cancers and the presence of mutations in the ATRX (alpha thalassemia/mental retardation syndrome X-linked) gene (10, 11). ATRX is a DNA helicase/ATPase of the SWI2/SNF2 family that binds repeated sequences of DNA, particularly the G-rich ones, and has been implicated mainly in chromatin remodeling (12). In the present study, we have started to analyze possible alterations in some telomeric pathways in telomerase-positive human glioma cells in culture in response to short hairpin RNA (shRNA)-mediated genetic inactivation of ATRX. Among the principal results of the present study is the finding that partial ATRX inactivation led to a diminution in the level of transcription of telomeric DNA into the noncoding RNAs called TERRA. This occurred concomitantly with a decrease in the amounts of cohesin in subtelomeric regions. The present data provide clues to start to understand the tight association between the occurrence of a mutation in ATRX and that of the ALT pathway in both cultured cell lines and tumors.
MATERIALS AND METHODS
Cell lines, plasmids, cell culture, and transfection.
Glioma cancer cell lines, 8-MG-BA and Hs-683, were obtained from the American Type Culture Collection and the German Collection of Microorganisms and Cell Cultures and cultured in minimum Eagle's medium (MEM) and in Dulbecco's modified Eagle's medium (DMEM), respectively, supplemented with 10% fetal bovine serum (FBS) (PAA) in the presence of 5% CO2 in a 90% humidified incubator. Nontumoral human embryonic kidney (HEK-293; ATCC) cells were cultivated in DMEM plus 10% FBS. For the establishment of stable 8-MG-BA and Hs-683 cell lines, transfection was performed using FuGene HD reagent (Promega) and Lipofectamine 2000 reagent (Thermo Scientific Fisher, Invitrogen), respectively. Selection for plasmids with neomycin resistance was performed using G418 (800 μg/ml) for 15 days before isolation of the clones. The media were replaced every 72 h. To obtain ATRX shRNA clones, cells were transfected with the shATRX1 (sense) (5′-GATCCCCGAGGAAACCTTCAATTGTATTCAAGAGATACAATTGAAGGTTTCCTCTTTTTA-3′) and shATRX2 (sense) (5′GATCCCCGCAGAGAAATTCCTAAAGATTCAAGAGATCTTTAGGAATTTCTCTGCTTTTTA-3′) shRNA constructs that had been cloned in the pSuper.retro.neo plasmid, a generous gift from the Bérubé laboratory (13). The corresponding siATRX1 and siATRX2 synthetic small interfering RNA (siRNA) oligonucleotides, with exactly the same sequences (13), were obtained from Dharmacon-GE Healthcare. The IF-GFP-ATRX plasmid for ATRX overexpression was from Michael Dyer (plasmid 45444; Addgene, Cambridge, MA). Since this plasmid has been reported to be prone to IS10 insertion in exon 8, causing insertion of a premature stop codon, it was sequenced in that region (https://www.addgene.org/45444/). No mutation could be found, and we therefore assumed (having also visualized the expressed ATRX protein by Western blotting using anti-ATRX antibody) that ATRX was correctly expressed in these experiments (data not shown). Anti-ATRX mouse monoclonal antibody raised against amino acids 2193 to 2492 of human ATRX (Santa Cruz Biotechnology; sc-55584) was used for Western blot analysis. Anti-beta-actin chicken polyclonal antibody (Abcam; ab13822) was used as a loading control.
Q-RT-PCR and measurement of telomeric DNA transcription into TERRA.
Total RNA was isolated from cells using Nucleospin RNA II (Macherey-Nagel). Two micrograms of RNA was reverse transcribed using the Revertaid first-strand cDNA synthesis kit from Thermo Fisher Scientific-Fermentas, with gene-specific primers for ATRX, and using GAPDH expression levels as an internal control. The primers used for quantitative real-time PCR (Q-RT-PCR) were as follows: ATRX-F, 5′-TCCTTGCACACTCATCAGAAGAATC-3′, and ATRX-R, 5′-CGTGACGATCCTGAAGACTTGG-3′; GAPDH-F, 5′-GAGTCAACGGATTTGGTCGT-3′, and GAPDH-R, 5′-GACAAGCTTCCCGTTCTCAG-3′ (13). cDNAs were amplified in a Bio-Rad Mini-Opticon instrument using Mesa Green qPCR Mastermix Plus (Eurogentec), together with forward and reverse primers. Data were quantified using the Bio-Rad CFX Manager software. For PCR amplification of total TERRA, the following telomere-specific primers were used: Tel1b, 5′-CGGTTTGTTTGGGTTTGGGTTTGGGTTTGGGTTTGGGGT-3′, and Tel2b, 5′-GGCTTGCCTTACCCTTACCCTTACCCTTACCCTTACCCT-3′ (14, 15). The histograms in the figures represent the averages and standard deviations for three replicates. Student t tests were used for statistical analysis.
ChIP.
Chromatin immunoprecipitation (ChIP) was performed according to standard protocols. Samples were sonicated on ice until the size of chromatin fragments was around 500 bp, based on agarose gel electrophoresis. The purified DNA recovered by ChIP was denatured in 0.2 M NaOH by heating to 100°C for 10 min and used as a template for quantitative real-time PCR amplification. To measure binding at subtelomeric loci at positions ∼1.1 kb from the TTAGGG telomeric repeat, we used the following primers: 10q-2/-1122 5′ primer (5′-CAGAGACGAGTGGAACCTGAGTAAT-3′) and 10q-2/-1122 3′ primer (5′-TGGGCAAGCTGGTCCTGTAG-3′) (16). The different distinct genomic loci matching these primer sets computationally were 1q, 2q, 4q, 5q, 6q, 8p, 10q, 13q, 16q, 19p, 19q, 21q, 22q (Tel), and 2qfus (internal) (16). To measure binding at subtelomeric loci immediately adjacent to the TTAGGG repeat (bp 109 and 152), we used the XYq-1/-109 5′ primer (5′-CCCCTTGCCTTGGGAGAA-3′) and the XYq-1/-109 3′ primer (5′-GAAAGCAAAAGCCCCTCTGA-3′), matching the 9p, 19p, and XYq (Tel) genomic telomeric loci, as well as the 13q-152 5′ primer (5′-GCACTTGAACCCTGCAATACAG-3′) and the 13q-152 3′ primer (5′-CCTGCGCACCGAGATTCT-3′), matching the 1q, 2q, 4q, 5q, 6q, 10q, 13q, 16q, 21q, and 22q (Tel) genomic telomeric loci (16). The primers for analyzing binding at the nontelomeric locus GAPDH were GAPDH 5′ primer (5′-TGGGCTACACTGAGCACCAG-3′) and GAPDH 3′ primer (5′-GGGTGTCGCTGTTGAAGTCA-3′) (16). Student t tests were used for statistical analysis. The antibodies used for ChIP were anti-SMC1 mouse monoclonal (Bethyl Laboratories; A300-055A) and anti-RNA polymerase II rabbit polyclonal (Abcam 8WG16; ab817) antibodies, as well as the sc-55584 anti-ATRX mouse monoclonal antibody from Santa Cruz Biotechnology.
Micrococcal-nuclease digestion-based analysis of chromatin condensation.
Nuclei isolated from 107 cells were digested with 8 mU of micrococcal nuclease (MNase) (Sigma)/μg DNA at 37°C for the indicated times, as described previously (17). For dose-response experiments, 8 to 64 mU of MNase/μg DNA was added to chromatin before digestion for 5 min at 37°C. After digestion, the DNA was purified and separated onto a 1.5% agarose gel in the presence of ethidium bromide. Before transfer onto nylon membranes, the DNA was visualized under UV light for quantification of bulk chromatin digestion. Hybridization was performed with a 0.4-kb subtelomeric probe amplified from genomic DNA with the 10q-2/-1122 5′ primer (described above) and the subtelo-10q-R 3′ primer (5′-GCGCTCTGACTTTAAGTGGT-3′). To label the probe, purified PCR product was incubated with large Klenow fragment (NEB), random hexamers, and [α-32P]dCTP (PerkinElmer). After removal of unincorporated nucleotides, the probe was denatured and added to a prehybridized membrane in Ultrahyb solution (Ambion). Quantifications were performed with Image Gauge (GE Healthcare).
Measurements of ALT-specific C-circles.
The so-called C-circle assay was used for the detection of ALT activity. This assay, in which partially single-stranded telomeric (CCCTAA)n DNA circles (C-circles) are detected following rolling-circle amplification by the Phi29 polymerase, was performed exactly as described previously (18).
Immunofluorescence-FISH (IF-FISH) and detection of APBs.
Cells were permeabilized in Triton X-100 buffer (20 mM Tris-HCl, pH 8.0, 50 mM NaCl, 3 mM MgCl2, 0.5% Triton X-100, and 300 mM sucrose) for 5 min at room temperature and then fixed with 4% paraformaldehyde for 15 min at room temperature and permeabilized again in Triton X-100 buffer for 15 min at room temperature. The cells were then blocked in 2% bovine serum albumin (BSA) in phosphate-buffered saline (PBS) for 1 h at room temperature and then incubated for 1 h at room temperature with the 1st antibody diluted in 2% BSA. After a washing with 0.5% BSA in PBS, the cells were incubated for 1 h at room temperature in the 2nd antibody. When needed, FISH was next performed as follows. Briefly, the cells were fixed again with 4% paraformaldehyde for 15 min at room temperature, followed by washing in PBS and dehydration in a series of ethanol (70%, 2 min; 85%, 2 min; and 100%, 2 min). A telomeric Tel-Cy3 PNA probe from Panagene (Daejeon, South Korea) was prepared at 200 nM (final concentration) in 70% formamide, 0.5% NEN blocking reagent (PerkinElmer), 10 mM Tris-HCl, pH 7.4. The probe mixture was applied to the slides and denatured for 5 min at 75°C, followed by hybridization overnight at 37°C. The slides were then washed with 70% formamide, 10 mM Tris-HCl, pH 7.4, and 0.1% BSA, followed by washes in PBS and incubation with DAPI (4′,6-diamidino-2-phenylindole). Detection of ALT-associated promyelocytic leukemia nuclear bodies (APBs) using anti-PML antibody detection combined with telomere FISH (Tel-FISH) was conducted according to previously described protocols (9, 19). Mouse monoclonal anti-PML antibody, used for immunofluorescence analyses, was from Santa Cruz Biotechnology (sc-966). The anti-SMC1 and anti-ATRX antibodies used in immunofluorescence analyses were the same as those used for ChIP (see above).
Telomere length measurement by telomere restriction fragment (TRF) analysis.
To analyze telomere length, genomic DNAs were prepared, digested with RsaI and HinfI, and separated in a 0.9% agarose gel (in Tris-borate-EDTA [TBE]) run in TBE buffer overnight and, after denaturation, transferred and hybridized with a (TTAGGG)3 32P-labeled telomeric probe. The results were analyzed using a GE Storm phosphorimager and ImageGauge software.
RESULTS
ATRX localizes to subtelomeric regions of human glioma cells.
ATRX has recently been implicated in the telomerase-independent mechanism of telomere maintenance, the so-called ALT pathway. Thus, in several clinical studies, mutations in ATRX were found to be tightly associated with the ALT pathway in various types of tumors (10, 11, 20–23). This correlation has also been observed in cultured cells (24, 25). To understand how genetic inactivation of ATRX might interfere with telomeric pathways in tumor cells and, in particular, favor the ALT pathway, we used glioma cells in culture because gliomas have been reported to be among the 5 to 15% of tumors that exhibited ALT-maintained telomeres (9). Although several ALT-positive glioma cell lines were recently isolated (26–28), most of the glioma cell lines isolated to date utilize the telomerase pathway to maintain functional telomeres. With this in mind, the present objective was to analyze in telomerase-positive glioma cells possible alterations in telomeric pathways following genetic inactivation of ATRX, as described below. However, it was first necessary to try to determine the intracellular localization of ATRX in these cells to see if it was present near or at the telomeres, a localization that would be compatible with potential telomeric effects. Using a mouse monoclonal antibody raised against amino acids 2193 to 2492 of human ATRX, mapping near its C terminus, we analyzed the intracellular distribution of ATRX by immunofluorescence. In two human glioma cell lines, 8-MG-BA and Hs-683, as well as in the nontumoral HEK-293 human embryonic cell line, ATRX signals were present throughout the nuclei in the form of dense, sharp spots but were apparently absent from the cytoplasm, or at least present below the levels detectable by the antibody (Fig. 1). FISH using a telomeric (CCCTAA) PNA probe was performed in the same experiment in order to simultaneously localize the telomere termini at chromosome ends. Strikingly, a large number of ATRX nuclear spots localized very near a telomeric spot in both cell lines analyzed (Fig. 1). These experiments suggested that ATRX might be present in the subtelomeric regions of the chromosomes in these human glioma cell lines.
FIG 1.

ATRX localizes close to telomere ends of 8-MG-BA (top two rows) and Hs-683 (middle row) human glioma cells and of nontumoral HEK-293 cells (bottom row). Subcellular localization of ATRX (green) by immunofluorescence analysis using an anti-ATRX monoclonal antibody and simultaneous telomere FISH (red) indicated that ATRX frequently localized near—but not exactly at—the telomere ends.
To confirm this finding, we set out to detect possible physical interactions between ATRX and telomeric DNA. To do this, we used ChIP, a technique that has been widely used in studies on telomeric proteins. As shown in Fig. 2, in 8-MG-BA cells, ATRX was found to be enriched in a subtelomeric region located ∼1.1 kb from the junction between telomeric and subtelomeric sequences compared with a nontelomeric locus, GAPDH. To ascertain whether ATRX presence at subtelomeres might be meaningful, we next compared the levels at this subtelomeric location to those at another subtelomeric location, this time at a position close to the TTAGGG telomere repeat. Interestingly, ATRX was more abundant at the subtelomere at ∼1.1 kb from the terminal repeat than at the subtelomere-telomere junction; at the latter, ATRX enrichment was just slightly above that at the GAPDH locus (Fig. 2B). This finding was confirmed using another pair of primers to assess ATRX binding at positions close to the TTAGGG repeat other than those illustrated in Fig. 2B (see Materials and Methods) (Fig. 2C). Note that, in Fig. 2B, the ∼3-fold increase in ATRX detected at the subtelomeric position nucleotide (nt) −1122 compared to the subtelomeric position nt −109 cannot be simply due to the larger number of loci assessed for the subtelomere versus the subtelomere-telomere junction (see Materials and Methods). Indeed, in these experiments, the ChIP data were normalized to the DNA input in the PCRs.
FIG 2.

Enrichment of ATRX in subtelomeric regions by ChIP. (A) Schematic representation of the positions (in nucleotides) of the PCR primers used for ChIP analysis with respect to the telomeric terminal repeats, TTAGGG, together with the names of the telomeric genomic loci amplified by these primers (see Materials and Methods). (B and C) ATRX binding to subtelomeric regions of human 8-MG-BA glioma cells was measured relative to binding at the GAPDH locus, a nontelomeric locus, under the same conditions. The values obtained for GAPDH amplification were directly subtracted from those obtained for the subtelomeric loci. The bar graphs represent the average percentages of input for each ChIP (means and standard deviations [SD]). Each ChIP experiment was performed in duplicate.
Genetic inactivation of ATRX.
As illustrated above, telomerase-positive cultured cells, just like telomerase-positive tumors, exhibited nuclear ATRX, as seen by immunofluorescence, whereas ALT-positive tumors and cultured cells exhibited total loss of nuclear ATRX associated with mutations in ATRX (10, 24). This is also illustrated in Fig. 3A, in which we see that the 8-MG-BA and Hs-683 telomerase-positive glioma cells used in the present study, as well HeLa cells, contained nuclear ATRX, while the recently described ALT-positive TG20 glioma stem cells (26) and the widely used ALT-positive U2OS cells did not. We next used RNA interference (RNAi), both shRNA and siRNA, to deplete 8-MG-BA and Hs-683 glioma cells of ATRX RNA. Depending on the experiments and on the various clones generated following transfection of one or the other of two different ATRX shRNAs, we observed an extinction of ATRX RNA of between 60 and 75% as measured by Q-RT-PCR (Fig. 3B). In experiments using either one of two different siRNAs directed against ATRX sequences at targets identical to those defined for the two ATRX shRNAs, extinction of ATRX RNA was even more efficient, close to 75 to 80% (Fig. 3D). In these experiments with shRNAs and siRNAs against ATRX, we also evaluated the levels of ATRX protein after RNA extinction using Western blotting. The ATRX gene yields two major protein isoforms, a full-length 270- to 280-kDa protein and a truncated form of 170 to 180 kDa that colocalizes at pericentromeric heterochromatin (29). Note that the antibody used here could detect only the larger of the two isoforms, and the 170-kDa band thus represents a nonspecific band that we used as a loading control (Fig. 3C, E, and F). As shown in Fig. 3C and E, the 270- to 280-kDa ATRX isoform was barely detectable following RNAi application, if at all in some cases, in both cell lines compared with the negative controls transfected with an shRNA targeting a scrambled ATRX sequence or a control siRNA.
FIG 3.

Genetic inactivation of ATRX. (A) Cultured human cells, including both telomerase-positive cells (8-MG-BA and Hs-683 glioma cells and HeLa cells) and ALT-positive cells (TG20 glioma stem cells and U20S osteosarcoma cells), were processed for immunofluorescence microscopy and allowed to react with anti-ATRX monoclonal antibody. The green points indicate the presence of ATRX in the DAPI-stained nuclei (blue) of telomerase-positive cells, while ATRX was totally absent from the nuclei of ALT-positive cells. The photographs shown here are representative images. (B to F) 8-MG-BA and Hs-683 glioma cell lines stably expressing ATRX shRNA were established, and Q-RT-PCR (B) and Western blotting (C) were used to assess the degree of gene extinction in these cells, as indicated. ATRX RNA expression was plotted against that of GAPDH, which was chosen as an internal control because of its established invariable expression. Controls included cells transfected with scrambled shRNA (sh-SCR) and nontransfected cells (NT). The measurements shown here were performed on stable cell lines. In parallel, cells from the same lines were treated for 24, 48, or 72 h with ATRX siRNAs, and the amount of depletion was assessed by both Q-RT-PCR (D) and Western blotting (E). In panels C and E, the 170-kDa band is a nonspecific band (see the text) that we sometimes used as a loading control after performing simultaneous blotting with antiactin antibody to assess similar behavior (F). The images shown in panel E were taken from a single blot image. (G) Assessment of apoptosis and necrosis levels using the annexin V-FLUOS (fluorescein isothiocyanate [FITC])/propidium iodide assay to detect cell surface phosphatidylserine by flow cytometry in 8-MG-BA cells expressing ATRX (right) or scrambled (left) shRNA.
We repeatedly observed that ATRX-inhibited cells clearly started to lose adhesion to the cell culture substrate before reaching confluence in the culture dish (data not shown). We hypothesized that this might be related to apoptosis. Consequently, we used detection of cell surface phosphatidylserine (PS) with annexin V, a phospholipid-binding protein with high affinity for PS labeled in green with fluorescein (annexin-V-FLUOS staining kit; Roche) by flow cytometry as a marker for apoptosis. Cells were simultaneously stained with a DNA red-staining dye, propidium iodide, the exclusion of which, together with annexin V binding, indicated apoptotic cells. Interestingly, using this assay, we were able to determine that around 3% of atrx-minus cells were entering apoptosis, more than in cells transfected in parallel with the scrambled ATRX shRNA, in which only very few apoptotic events were present (Fig. 3G).
Next, we used Tel-FISH, in combination with γH2AX immunostaining, to detect possible telomeric DNA damage after ATRX had been genetically inactivated. The results of this analysis revealed that the numbers of telomere dysfunction-induced foci (TIF) per nucleus were similar in the glioma cells stably transfected with ATRX shRNA compared with controls transfected with scrambled ATRX shRNA (data not shown).
ATRX inactivation does not alter subtelomeric chromatin accessibility to micrococcal nuclease.
ATRX is a chromatin remodeler, and since we had shown (see above) that ATRX preferentially localized to subtelomeric regions, we next wished to test whether ATRX could impact higher-order chromatin structure at subtelomeres. To address this question, we performed MNase digestion assays on chromatin samples isolated from an 8-MG-BA cell line in which ATRX had been stably inactivated with ATRX shRNA and its control cell line expressing scrambled shRNA (Fig. 4). Time course experiments revealed very similar profiles of MNase digestion in both cell lines, suggesting that inactivation of ATRX does not impact higher-order subtelomeric chromatin structure (Fig. 4A). Similarly, when samples were digested for 5 min with various amounts of MNase, there was no dramatic difference in subtelomeric chromatin digestion between cells in which ATRX had been inactivated and control cells with wild-type ATRX (Fig. 4B). Based on these results, we conclude that the role of ATRX at the subtelomeres is unlikely to be related to massive remodeling of subtelomeric chromatin.
FIG 4.

shRNA-mediated inactivation of ATRX does not alter subtelomeric chromatin accessibility to MNase. (A) Chromatin isolated from 8-MG-BA glioma cells in which ATRX had been inactivated (shATRX) or not (shscrambled [shSCR]) was digested with MNase for the indicated times. (Left gel) Ethidium bromide (EtBr) staining of bulk chromatin. (Right gel) Southern blot with subtelomeric probe. (Far right) Quantification of the data. The signals obtained for mononucleosomes were normalized to the total signals measured for each time point (EtBr or Southern blot). (B) Chromatin samples from shATRX or shSCR 8-MG-BA cells were digested for 5 min with the indicated amounts of MNase (milliunits per microgram of DNA). (Far right) Quantification of the data.
Inactivation of ATRX results in decreased levels of TERRA and of RNAP II at subtelomeres.
Having shown that ATRX does not massively impact higher-order subtelomeric chromatin, and because of the previously established connection between ATRX and telomere transcription (30–32), we next wished to test the impact of ATRX depletion on TERRA levels in the glioma cell lines. Telomeric DNA is indeed transcribed into noncoding RNA, or TERRA (33). In mammalian cells, TERRA molecules have been proposed to regulate telomere structural maintenance and heterochromatin formation (31, 34). In mouse embryonic cells, upon inactivation of ATRX, TERRA levels were found to either increase (30) or remain unchanged (32), while in telomerase-positive human cells, inactivation of ATRX resulted in a diminution of TERRA (35). To further document this issue, we set out to measure TERRA levels in cultured glioma cells stably transfected with ATRX shRNA. Interestingly, we found that reducing ATRX expression led to a decrease in total TERRA levels in both the Hs-683 and the 8-MG-BA telomerase-positive glioma cell lines (Fig. 5A). Notably, this reduction was not due to changes in telomere length, as inactivation of ATRX did not lead to important changes in telomere length (Fig. 5B).
FIG 5.
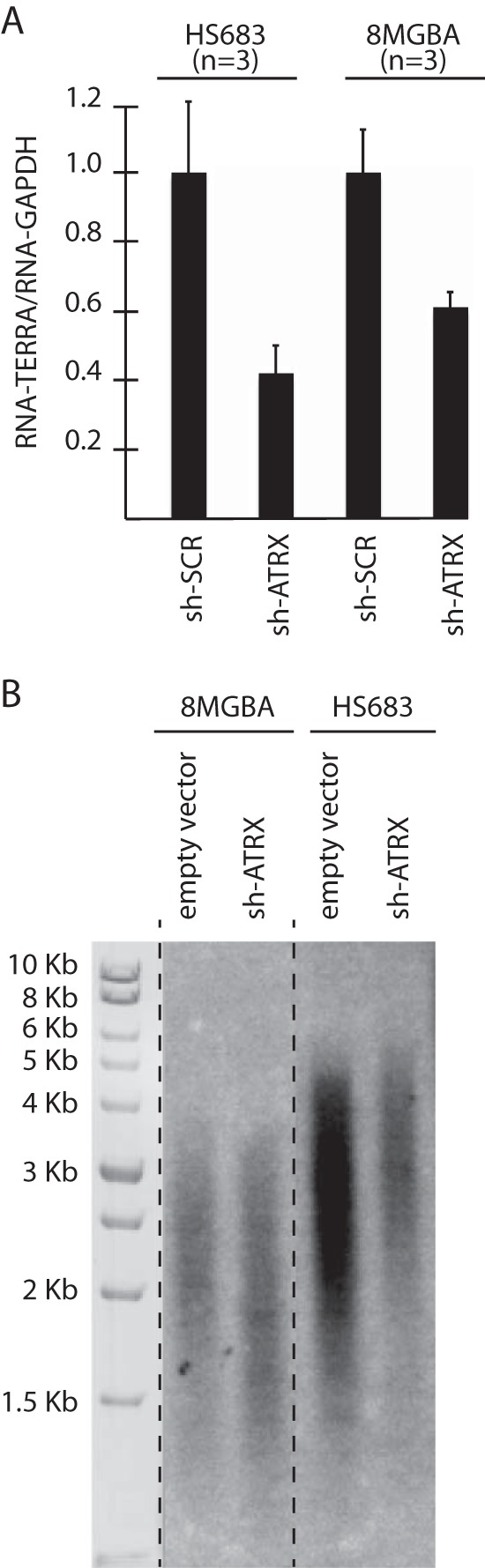
shRNA-mediated inactivation of ATRX provokes diminution of TERRA levels. (A) Q-RT-PCR analysis of total TERRA molecules in the indicated cell lines. The bar graph represents the average values of telomeric DNA transcription into TERRA relative to transcription of the GAPDH gene (means and SD) from the indicated number of independent experiments. Each experiment was performed in triplicate. (B) Telomere length measurements by TRF analysis in 8-MG-BA and Hs-683 cell lines following transfection with ATRX shRNA or empty vector after 30 population doublings. The size markers allowed us to evaluate the bulk size of telomeres in the cell populations, as the telomeres of all different sizes produced a smear, the middle of which represents the average size of the telomere population.
We next investigated whether the diminution of TERRA levels observed upon genetic inactivation of ATRX might result from perturbations in the recruitment of RNAP II. Accordingly, ChIP experiments revealed a drastic reduction in RNAP II recruitment at the subtelomeres of 8-MG-BA and Hs-683 cells expressing ATRX shRNA (Fig. 6A and B). Therefore, the present data suggest that ATRX promotes RNAP II recruitment at human subtelomeres.
FIG 6.
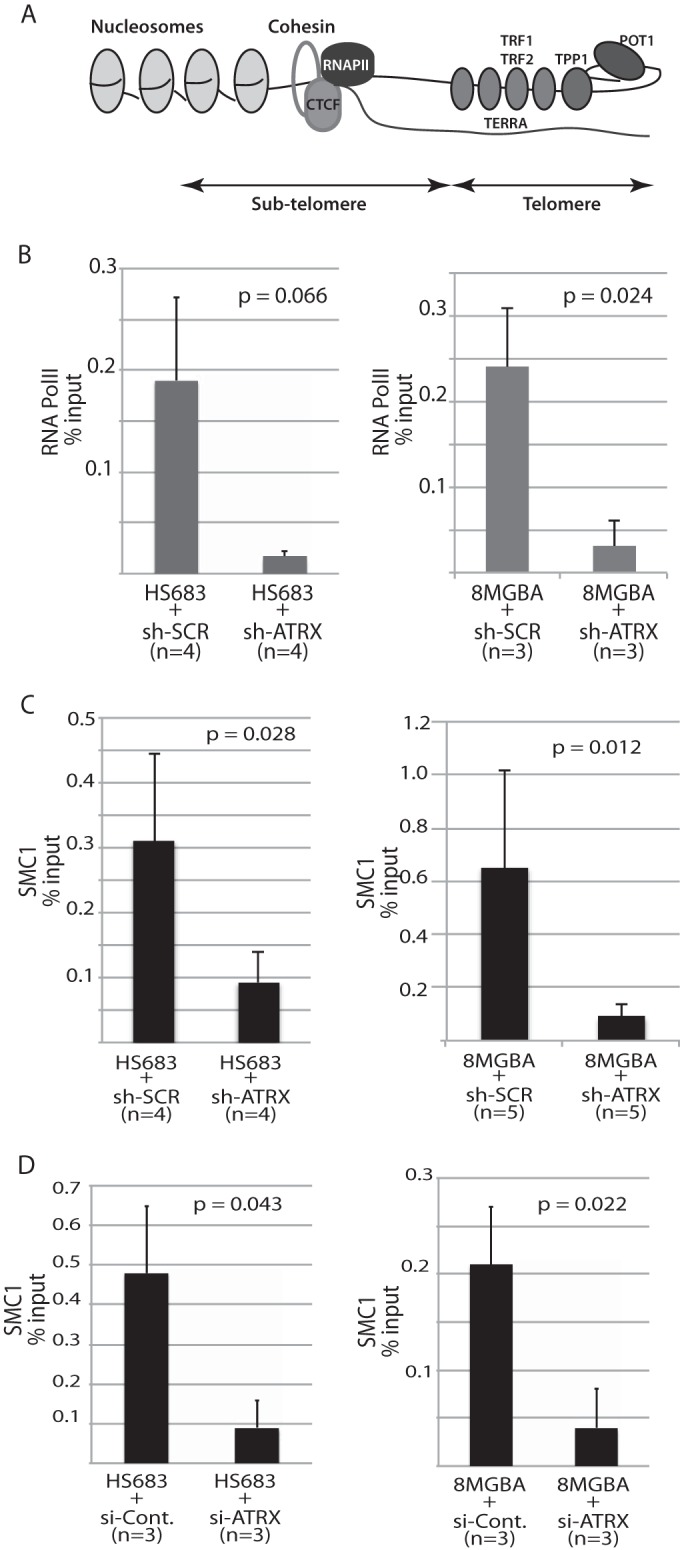
RNAi-mediated genetic inactivation of ATRX produces dramatic decreases in subtelomeric binding of cohesin/SMC1 and RNA polymerase II. (A) Schematic representation of the organization of the human subtelomere based on recent findings by Deng et al. (16) highlighting the emerging roles of cohesin and CTCF in telomeric DNA transcription into TERRA, partly through RNAP II access to the telomere. (B to D) ChIP experiments measuring by Q-RT-PCR the binding of RNA polymerase (Pol) II (B) and of cohesin/SMC1 (C and D) at the subtelomere using the 10q-2/-1122 primers (see Materials and Methods) relative to binding at the GAPDH locus, a nontelomeric locus, under the same conditions. The values obtained for GAPDH amplification were directly subtracted from those obtained for the subtelomeric loci. n represents the number of independent experiments for each condition. Each ChIP experiment was performed in triplicate, and means and SD are shown. In panel C, 8-MG-BA or Hs-683 cells were transfected with either ATRX shRNA or scrambled shRNA, while in panel D, the cells were transfected with ATRX siRNA or control siRNA (si-Cont.).
Downregulation of ATRX leads to decreased amounts of SMC1/cohesin at subtelomeres.
Our data indicate that ATRX depletion impairs RNAP II recruitment at subtelomeres. Recently, it was shown that in the postnatal mouse brain, ATRX was recruited concomitantly with cohesin at particular loci and that loss of ATRX impaired cohesin recruitment at these loci (36). In addition, recent studies have shown that cohesin is an integral component of human telomeric chromatin (16). We therefore set out to investigate whether inactivation of ATRX might affect the presence of cohesin at the telomeres by quantifying SMC1/cohesin abundance at subtelomeric loci located directly upstream of telomeric repeats (Fig. 6A). We used ChIP to analyze possible protein-DNA interactions between SMC1/cohesin and telomeric DNA. First, we could confirm the previous finding by Deng et al. (16) in HCT116 cells that cohesin is indeed also enriched at the subtelomeres of the two glioma cell lines analyzed here (Fig. 6C and D). We could further demonstrate that cells in which ATRX had been partially inactivated either by shRNA or by siRNA exhibited a dramatic decrease in the amount of SMC1/cohesin present at the subtelomere (Fig. 6C and D). Altogether, therefore, our data suggest that ATRX depletion impairs TERRA production through downregulation of SMC1/cohesin loading at the subtelomere.
Genetic inactivation of ATRX is not sufficient to trigger the ALT pathway of telomere maintenance.
Previous experiments showed that genetic inactivation of ATRX was not sufficient to trigger ALT in HeLa cells (25). We wished to confirm this result in a glioma cell line, because this type of cell, in tumors, has a predisposition to undergo ALT to maintain functional telomeres. We therefore used the so-called C-circle assay to measure the number of extrachromosomal telomeric DNA circles specific for the ALT pathway of telomere maintenance (18). In this assay, partially single-stranded telomeric (CCCTAA)n DNA circles (C-circles), which presumably represent the result of the exchange between repeated telomeric sequences during the ALT processes, are detected following rolling-circle amplification by the Phi29 polymerase, which can amplify up to 70 kb of (TTAGGG)n DNA sequences in the absence of dCTP. Production of these extrachromosomal telomeric DNA molecules that have been excised from the recombining telomeres, the C-circles, accompanies all types of ALT cancer cells studied to date (18). The experiments performed here indicated that genetic inactivation of ATRX was not sufficient to trigger the appearance of the C-circles in these telomerase-positive glioma cells (Fig. 7A). This was assessed at early passages, 3 and 6, following transfection with shRNAs/siRNAs (data not shown), as well as later, after 30 passages (Fig. 7A). We also assessed the presence of the ALT-associated so-called APBs (9, 19) in 8-MG-BA cells depleted of ATRX or not by RNAi. Consistent with the finding described above that the ALT-associated C-circles were not induced by ATRX inactivation, no APBs could be detected following efficient depletion of ATRX (Fig. 7B). On the other hand, as expected and as shown previously (26), cells from the TG20 glioma stem cell line exhibited clearly visible APBs in the form of PML bodies perfectly colocalizing with telomere termini labeled by FISH (Fig. 7B). Conversely, an ALT-positive U2OS cell line overexpressing ATRX in a stable manner did not result, after up to 2 months of selection, in the inhibition of the number of ALT-associated C-circles in these cells compared with cells expressing an empty vector (data not shown). It should be noted, however, that ATRX isoform 1 expressed from the IF-GFP-ATRX plasmid is actually not completely wild type because it lacks exon 6. Although perhaps unlikely, the possibility that the missing sequences have an effect on the function of the protein and the inability of the ATRX construct to reverse the ALT pathway in ALT-positive cells cannot be completely ruled out.
FIG 7.

Genetic inactivation of ATRX is not sufficient to trigger the ALT pathway. (A) Detection of ALT pathway activity in telomerase-positive glioma cells transfected with ATRX shRNA and in ALT-positive U2OS cells, using the C-circle assay. At the top is a schematic illustration of the main successive steps of the reaction, as described previously (18). At the bottom right, the actual measurements performed in cultured cells by Southern blotting with a radiolabeled telomeric probe are shown. ATRX shRNA-transfected cells were harvested after 30 passages. Thirty nanograms of DNA was loaded for each reaction in the dot blot apparatus. (B) Detection by immunofluorescence of the PML bodies in the indicated cells using simultaneous labeling of the PML protein (green) and of telomeric DNA at chromosome ends using a PNA FISH probe (red). The presence of yellow signals upon merging the green and red signals indicated the localization of the ALT-specific PMLs, the APBs, in the TG20 cells at the telomeres but their absence in the 8-MG-BA and Hs-683 cells transfected with ATRX shRNA. The images are representative of observations of approximately 100 cells for each condition.
DISCUSSION
In the present paper, we report that genetic inactivation of the chromatin-remodeling protein ATRX in human telomerase-positive cancer cells impinges both on the presence of cohesin in subtelomeric regions of the chromosomes and on the level of transcription of telomeric DNA into TERRA. The latter possibly resulted from impaired transcription, as RNAP II levels were also severely depressed following ATRX inactivation. Previous findings, mainly from clinical studies, have pointed to the simultaneous occurrence of a mutation in ATRX and of the telomerase-independent ALT pathway in several types of tumors (10, 11, 20–23). This list is not exhaustive, as other, more recent studies have confirmed these early findings. However, these studies failed to identify possible targets of ATRX. The experiments performed here indicated that genetic inactivation of ATRX was not sufficient to trigger the appearance of the ALT-associated C-circles in telomerase-positive glioma cells, thus confirming a recent study using the same approach in HeLa cells (25). The present data provide important clues to start to understand the relationship between genetic inactivation of ATRX and the ALT pathway.
In the present study, we found by both IF-FISH and ChIP that ATRX localized at the subtelomeres but not at the very ends of the telomeres of glioma cells. ATRX enrichment has been previously reported at subtelomeres and telomeres of human primary cells and mouse embryonic stem cells (37, 38). More recently, Atrx-null mouse embryonic brain cells were shown to exhibit telomeric damage that could be worsened by treatment with the G-quadruplex ligand telomestatin, suggesting a role for ATRX in the replication of telomeric G4-DNA structures (39). In mouse embryonic stem cells, ATRX and H3.3 colocalized within PML bodies containing telomeric DNA (40). Interestingly, such PML bodies likely correspond to the APBs found in ALT-positive cancer cells (9, 19). These mouse embryonic stem cells have not been reported to experience any ALT-mediated pathway of telomere maintenance, and obviously, the role of ATRX in these cells is different from that in cancer cells, as the latter have lost ATRX function. It is tempting to speculate that, in both humans and mice, ATRX's prominent role is in the maintenance of telomeric chromatin and that, for some as yet unknown reasons, loss of ATRX function in human tumors leads to the triggering of the ALT pathway, while in mouse embryonic stem cells, ATRX helps in telomeric DNA replication. It is also tempting to speculate that, in telomerase-positive human cancer cells, ATRX also helps in telomeric DNA replication and that upon fortuitous loss of ATRX function, perturbation in telomere replication leads to recombination-based ALT.
Reducing ATRX expression led here to a decrease in total TERRA levels in both the Hs-683 and the 8-MG-BA telomerase-positive glioma cell lines. This was in contrast to recent findings in mouse embryonic stem cells, in which inactivation of ATRX led to upregulation of TERRA (30), and in mouse embryonic fibroblasts, in which TERRA levels remained unchanged upon ATRX inactivation (32), but was in agreement with another recent study in which loss of ATRX function led to a reduction in TERRA levels in a human telomerase-positive cell line (35). Overall, this suggests that regulation of telomere transcription is different in mice and humans. In addition, based on previous findings (16), the present data suggest that the diminution in cohesin levels at subtelomeres in cells with ATRX inactivated might be responsible for the diminution of TERRA levels observed here. Indeed, in that study, inactivation of RAD21/cohesin resulted in severe impairment of TERRA transcription (16). Moreover, the present data tend to suggest that ATRX may, possibly through an effect on cohesin recruitment and/or maintenance at telomeric chromatin, facilitate TERRA transcription by increasing RNAP II binding at the telomeres. Alternatively, the present, as well as the recent (35), findings that ATRX inactivation diminished TERRA levels might be explained by the occurrence of a concomitant loss of control of histone/DNA methylation at the subtelomeres. Indeed, in mouse embryonic stem cells, disruption of ATRX/H3.3 binding led to a loss of control of the telomeric histone methylation pattern (39), and in human cells, TERRA abundance is negatively regulated by methylation of TERRA promoter CpG islands (41). Of note, it was recently reported that genetic inactivation of ATRX led to persistence of both TERRA and RPA foci at telomeres of HeLa cells in G2/M, suggesting a role for ATRX in cell cycle-dependent regulation of TERRA abundance at telomeres (42). It was proposed that the hnRPA1-mediated RPA-displacing activity normally inhibited by TERRA only during early S phase (43) was now abnormally inhibited at G2/M due to genetic inactivation of ATRX and that the resulting elevation of telomeric RPA levels led to recruitment of ATR and potentially to the recombination events characteristic of ALT (42).
How the global reduction of TERRA levels upon ATRX knockdown and the impact of ATRX on TERRA cell cycle regulation are connected is still unknown. Recent data have shown that the NuRD deacetylase complex was recruited at ALT telomeres through orphan receptor binding and induced hypoacetylation of telomeric chromatin, potentially causing a diminution in the markers associated with open -state chromatin (44). The telomeres of ALT cells are decompacted compared to those of telomerase-positive cells (35), and Conomos et al. (44) have proposed that NuRD recruitment might partly compensate for this decompaction of telomeric chromatin. One hypothesis is that this dramatic decondensation of telomeric chromatin in ALT cells needs to be partially compensated for in order to avoid excessive activation of the DNA damage response at telomeres and chromosomal instability. We now propose that ATRX loss of function might act in synergy with recruitment of NuRD as part of a putative feedback mechanism aimed at partly reconstituting functional telomeric heterochromatin compatible with survival (a model is shown in Fig. 8). In addition, as discussed above, inactivation of ATRX might play an important role in the TERRA-induced RPA-to-POT1 switch that potentially results in inappropriate presence of ATR at the telomeres and triggering of ALT (42, 43). Further experimentation will be required to test these models. In addition, how telomeric chromatin relaxation, TERRA upregulation, and ATRX loss contribute to the ALT mechanism remains to be established.
FIG 8.
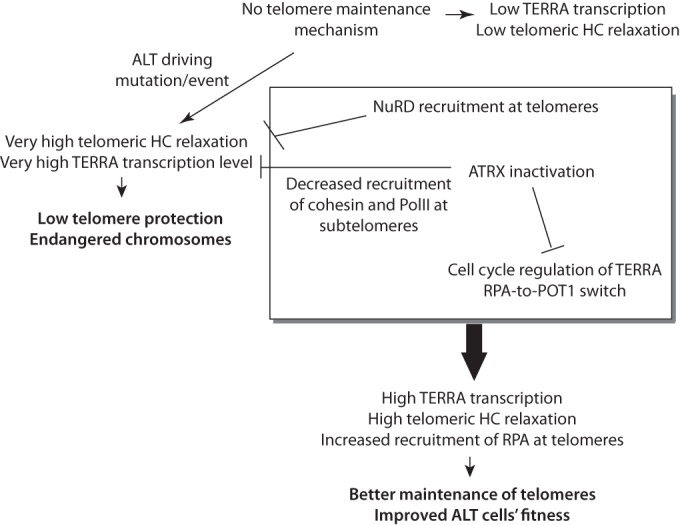
Model of possible roles of ATRX in ALT induction (see the text) (HC, heterochromatin).
It is now clear that repression of ATRX cannot, on its own, activate ALT, although, on the other hand, several pieces of data suggest that mutation of ATRX is necessary for ALT induction. Alternatively, ATRX could act as a suppressor of ALT in telomerase-positive cells, which could also explain the association between an ATRX mutation and the ALT status. In this hypothesis, reestablishing ATRX expression in ALT cells should inhibit the ALT mechanism. However, under our experimental conditions, at least, overexpression of ATRX did not seem to alter the level of ALT-specific C-circles. Therefore, if ATRX is an inhibitor of ALT, other conditions, such as cooverexpression of one or several other proteins, must be fulfilled in order to observe the result of this action.
In the budding yeast Saccharomyces cerevisiae, type II telomeric recombination generates highly heterogeneous terminal restriction fragments, which can attain 20 kb or more after telomere elongation, resembling those present in human ALT cells and probably mechanistically similar (45). On the other hand, in telomerase-negative yeast cells, type I recombination amplifies the subtelomeric sequences. It is notable that, in human cells, the absence of ATRX, which we find to be mainly localized at the subtelomeres, is associated with ALT recombination (at least in all clinical samples analyzed), an event that presumably takes place only at the level of telomeric sequences, mainly involving TTAGGG repeats (46). Strikingly, ATRX is a homolog of S. cerevisiae Rad54 (47; http://www.ensembl.org) and Rad54 is essential for operating type I recombination at S. cerevisiae subtelomeres (48, 49). In yeast, Rad54 is required for subtelomeric recombination but not for telomeric recombination, while human ATRX appears to have to be inactivated before telomeric recombination can take place. Telomerase-negative S. cerevisiae cells lacking the RAD54 gene survive by using ALT-like type II recombination (48, 49). It is tempting to speculate that in humans, telomerase-negative tumor cells with a fortuitous mutation in ATRX cannot perform subtelomeric recombination and that they amplify only their telomeric sequences, just like S. cerevisiae rad54 mutants that can perform only ALT-like type II recombination. In this hypothesis, telomerase-negative tumors with wild-type ATRX amplifying their subtelomeric sequences may potentially exist. Notably, there are some types of human cancer in which a substantial subset of the tumors do not have evidence either of ALT or of telomerase activity (50, 51). Perhaps some of these tumors survive owing to subtelomeric recombination. It should also be noted that the presence of TGAGGG, TCAGGG, and TTGGGG variant repeats, found in large quantities in ALT cells, implies subtelomeric recombination during the process of acquisition of ALT (46). We speculate that some subtelomeric factors, such as cohesin (the subtelomeric localization of which was found here to depend on ATRX), might be involved in subtelomeric recombination, for instance, owing to its role in maintaining sister chromatids in close proximity, thereby favoring recombination.
In summary, our findings reveal cohesin, as part of telomeric chromatin, to be a potentially important actor in the mechanisms of the telomerase-independent ALT pathways that accompany all known cases of tumors with a mutation in the chromatin remodeler ATRX. Together with those of Episkopou et al. and Flynn et al. (35, 42), our data also suggest that the potential control of TERRA by ATRX represents a key event in the functioning of the ALT pathway (Fig. 8). Our present finding that ATRX controls telomeric cohesin, together with the recent finding by Deng et al. (16) that telomeric cohesin regulates TERRA transcription, dictates a necessary orientation of future research toward understanding these relationships. It might also be pertinent to try to find functional interactions between ASF1 and ATRX (and to know whether cohesin might play a role in such interactions), as ASF1, a major regulator of chromatin organization, was recently found to negatively control the ALT pathway (52). Finally, recent data showing, in mice, that ATRX regulates the expression of genes in intragenic G-rich regions and allows optimal RNAP II function at these sites (32) leave open the possibility that loss of ATRX function in ALT tumors correlates with the acquisition of a particular transcriptional program uniquely compatible with the survival of cells that have undergone telomeric recombination.
ACKNOWLEDGMENTS
We are very grateful to Nathalie Bérubé for providing the ATRX shRNA constructs, Michael Dyer for the gift of the IF-GFP-ATRX plasmid, Valérie Gouilleux and François Boussin for the gift of cell lines, Jérome Bourgeais for help with the ChIP experiments, Nicole Ishac for help with the experiments on apoptosis, and Laetitia Corset for technical assistance. We also thank the Département Génomique PPF ASB facility at University François Rabelais of Tours for access to the Storm phosphorimager.
This work was supported by grants from the LIGUE Grand-Ouest contre le Cancer (Comités Charente, Eure-et-Loir, Ille-et-Vilaine, Indre-et-Loire, Morbihan, Sarthe, Vendée, Vienne) and the Fondation de France to M.C. and by grants from the Fonds National de la Recherche Scientifique (Belgium) to H.E. and A.D.
REFERENCES
- 1.Palm W, de Lange T. 2008. How shelterin protects mammalian telomeres. Annu Rev Genet 42:301–334. doi: 10.1146/annurev.genet.41.110306.130350. [DOI] [PubMed] [Google Scholar]
- 2.de Lange T. 2009. How telomeres solve the end-protection problem. Science 326:948–952. doi: 10.1126/science.1170633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Artandi SE, DePinho RA. 2010. Telomeres and telomerase in cancer. Carcinogenesis 31:9–18. doi: 10.1093/carcin/bgp268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nandakumar J, Cech TR. 2013. Finding the end: recruitment of telomerase to telomeres. Nat Rev Mol Cell Biol 14:69–82. doi: 10.1038/nrm3505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bryan TM, Englezou A, Dalla-Pozza L, Dunham MA, Reddel RR. 1997. Evidence for an alternative mechanism for maintaining telomere length in human tumors and tumor-derived cell lines. Nat Med 3:1271–1274. doi: 10.1038/nm1197-1271. [DOI] [PubMed] [Google Scholar]
- 6.Cesare AJ, Reddel RR. 2010. Alternative lengthening of telomeres: models, mechanisms and implications. Nat Rev Genet 11:319–330. doi: 10.1038/nrg2763. [DOI] [PubMed] [Google Scholar]
- 7.Durant ST. 2012. Telomerase-independent paths to immortality in predictable cancer sub-types. J Cancer 3:67–82. doi: 10.7150/jca.3965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Heaphy CM, Subhawong AP, Hong SM, Goggins MG, Montgomery EA, Gabrielson E, Netto GJ, Epstein JI, Lotan TL, Westra WH, Shih IM, Iacobuzio-Donahue CA, Maitra A, Li QK, Eberhart CG, Taube JM, Rakheja D, Kurman RJ, Wu TC, Roden RB, Argani P, De Marzo AM, Terracciano L, Torbenson M, Meeker AK. 2011. Prevalence of the alternative lengthening of telomeres telomere maintenance mechanism in human cancer subtypes. Am J Pathol 179:1608–1615. doi: 10.1016/j.ajpath.2011.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Henson JD, Hannay JA, McCarthy SW, Royds JA, Yeager TR, Robinson RA, Wharton SB, Jellinek DA, Arbuckle SM, Yoo J, Robinson BG, Learoyd DL, Stalley PD, Bonar SF, Yu D, Pollock RE, Reddel RR. 2005. A robust assay for alternative lengthening of telomeres in tumors shows the significance of alternative lengthening of telomeres in sarcomas and astrocytomas. Clin Cancer Res 11:217–225. [PubMed] [Google Scholar]
- 10.Heaphy CM, de Wilde RF, Jiao Y, Klein AP, Edil BH, Shi C, Bettegowda C, Rodriguez FJ, Eberhart CG, Hebbar S, Offerhaus GJ, McLendon R, Rasheed BA, He Y, Yan H, Bigner DD, Oba-Shinjo SM, Marie SK, Riggins GJ, Kinzler KW, Vogelstein B, Hruban RH, Maitra A, Papadopoulos N, Meeker AK. 2011. Altered telomeres in tumors with ATRX and DAXX mutations. Science 333:425. doi: 10.1126/science.1207313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schwartzentruber J, Korshunov A, Liu XY, Jones DTW, Pfaff E, Jacob K, Sturm D, Fontebasso AM, Quang DK, Tönjes M, Hovestadt V, Albrecht S, Kool M, Nantel A, Konermann C, Lindroth A, Jäger N, Rausch T, Ryzhova M, Korbel JO, Hielscher T, Hauser P, Garami M, Klekner A, Bognar L, Ebinger M, Schuhmann MU, Scheurlen W, Pekrun A, Frühwald MC, Roggendorf W, Kramm C, Dürken M, Atkinson J, Lepage P, Montpetit A, Zakrzewska M, Zakrzewski K, Liberski PP, Dong Z, Siegel P, Kulozik AE, Zapatka M, Guha A, Malkin D, Felsberg J, Reifenberger G, von Deimling A, Ichimura K, Collins VP, Witt H, Milde T, Witt O, Zhang C, Castelo-Branco P, Lichter P, Faury D, Tabori U, Plass C, Majewski J, Pfister SM, Jabado N. 2012. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 482:226–231. doi: 10.1038/nature10833. [DOI] [PubMed] [Google Scholar]
- 12.Bérubé NG. 2011. ATRX in chromatin assembly and genome architecture during development and disease. Biochem Cell Biol 89:435–444. doi: 10.1139/o11-038. [DOI] [PubMed] [Google Scholar]
- 13.Ritchie K, Seah C, Moulin J, Isaac C, Dick F, Bérubé NG. 2008. Loss of ATRX leads to chromosome cohesion and congression defects. J Cell Biol 180:315–324. doi: 10.1083/jcb.200706083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sampl S, Pramhas S, Stern C, Preusser M, Marosi C, Holzmann K. 2012. Expression of telomeres in astrocytoma WHO grade 2 to 4: TERRA level correlates with telomere length, telomerase activity, and advanced clinical grade. Transl Oncol 5:56–65. doi: 10.1593/tlo.11202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.O'Callaghan N, Dhillon V, Thomas P, Fenech M. 2008. A quantitative real-time PCR method for absolute telomere length. Biotechniques 44:807–809. doi: 10.2144/000112761. [DOI] [PubMed] [Google Scholar]
- 16.Deng Z, Wang Z, Stong N, Plasschaert R, Moczan A, Chen HS, Hu S, Wikramasinghe P, Davuluri RV, Bartolomei MS, Riethman H, Lieberman PM. 2012. A role for CTCF and cohesin in subtelomere chromatin organization, TERRA transcription, and telomere end protection. EMBO J 31:4165–4178. doi: 10.1038/emboj.2012.266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Episkopou H, Kyrtopoulos SA, Sfikakis PP, Fousteri M, Dimopoulos MA, Mullenders LHF, Souliotis VL. 2009. Association between transcriptional activity, local chromatin structure, and the efficiencies of both subpathways of nucleotide excision repair of melphalan adducts. Cancer Res 69:4424–4433. doi: 10.1158/0008-5472.CAN-08-3489. [DOI] [PubMed] [Google Scholar]
- 18.Henson JD, Cao Y, Huschtscha LI, Chang AC, Au AYM, Pickett HA, Reddel RR. 2009. DNA C-circles are specific and quantifiable markers of alternative-lengthening-of-telomeres activity. Nat Biotechnol 27:1181–1186. doi: 10.1038/nbt.1587. [DOI] [PubMed] [Google Scholar]
- 19.Yeager T, Neumann A, Englezou A, Huschtscha L, Noble J, Reddel R. 1999. Telomerase-negative immortalized human cells contain a novel type of promyelocytic leukemia (PML) body. Cancer Res 59:4175–4179. [PubMed] [Google Scholar]
- 20.Jiao Y, Killela PJ, Reitman ZJ, Rasheed AB, Heaphy CM, de Wilde RF, Rodriguez FJ, Rosemberg S, Oba-Shinjo SM, Nagahashi Marie SK, Bettegowda C, Agrawal N, Lipp E, Pirozzi C, Lopez G, He Y, Friedman H, Friedman AH, Riggins GJ, Holdhoff M, Burger P, McLendon R, Bigner DD, Vogelstein B, Meeker AK, Kinzler KW, Papadopoulos N, Diaz LA, Yan H. 2012. Frequent ATRX, CIC, FUBP1 and IDH1 mutations refine the classification of malignant gliomas. Oncotarget 3:709–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kannan K, Inagaki A, Silber J, Gorovets D, Zhang J, Kastenhuber ER, Heguy A, Petrini JH, Chan TA, Huse JT. 2012. Whole exome sequencing identifies ATRX mutation as a key molecular determinant in lower-grade glioma. Oncotarget 3:1194–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu XY, Gerges N, Korshunov A, Sabha N, Khuong-Quang DA, Fontebasso AM, Fleming A, Hadjadj D, Schwartzentruber J, Majewski J, Dong Z, Siegel P, Albrecht S, Croul S, Jones DT, Kool M, Tonjes M, Reifenberger G, Faury D, Zadeh G, Pfister S, Jabado N. 2012. Frequent ATRX mutations and loss of expression in adult diffuse astrocytic tumors carrying IDH1/IDH2 and TP53 mutations. Acta Neuropathol 124:615–625. doi: 10.1007/s00401-012-1031-3. [DOI] [PubMed] [Google Scholar]
- 23.Nguyen DN, Heaphy CM, de Wilde RF, Orr BA, Odia Y, Eberhart CG, Meeker AK, Rodriguez FJ. 2013. Molecular and morphologic correlates of the alternative lengthening of telomeres phenotype in high-grade astrocytomas. Brain Pathol 23:237–243. doi: 10.1111/j.1750-3639.2012.00630.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bower K, Napier CE, Cole SL, Dagg RA, Lau LMS, Duncan EM, Moy EL, Reddel RR. 2012. Loss of wild-type ATRX expression in somatic cell hybrids segregates with activation of alternative lengthening of telomeres. PLoS One 7:e50062. doi: 10.1371/journal.pone.0050062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lovejoy CA, Li W, Reisenweber S, Thongthip S, Bruno J, de Lange T, De S, Petrini JHJ, Sung PA, Jasin M, Rosenbluh J, Zwang Y, Weir BA, Hatton C, Ivanova E, Macconaill L, Hanna M, Hahn WC, Lue NF, Reddel RR, Jiao Y, Kinzler K, Vogelstein B, Papadopoulos N, Meeker AK, ALT Starr Cancer Consortium . 2012. Loss of ATRX, genome instability, and an altered DNA damage response are hallmarks of the alternative lengthening of telomeres pathway. PLoS Genet 8:e1002772. doi: 10.1371/journal.pgen.1002772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Silvestre DC, Pineda Marti JR, Hoffschir F, Studler J-M, Mouthon M-A, Pflumio F, Junier M-P, Chneiweisse H, Boussin FD. 2011. Alternative lengthening of telomeres in human glioma stem cells. Stem Cells 29:440–451. doi: 10.1002/stem.600. [DOI] [PubMed] [Google Scholar]
- 27.Heaphy CM, Schreck KC, Raabe E, Mao XG, An P, Chu Q, Poh W, Jiao Y, Rodriguez FJ, Odia Y, Meeker AK, Eberhart CG. 2013. A glioblastoma neurosphere line with alternative lengthening of telomeres. Acta Neuropathol 126:607–608. doi: 10.1007/s00401-013-1174-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Borodovsky A, Meeker AK, Kirkness EF, Zhao Q, Eberhart CG, Gallia GL, Riggins GJ. 2015. A model of a patient-derived IDH1 mutant anaplastic astrocytoma with alternative lengthening of telomeres. J Neurooncol 121:479–487. doi: 10.1007/s11060-014-1672-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Garrick D, Samara V, McDowell TL, Smith AJ, Dobbie L, Higgs DR, Gibbons RJ. 2004. A conserved truncated isoform of the ATR-X syndrome protein lacking the SWI/SNF-homology domain. Gene 326:23–34. doi: 10.1016/j.gene.2003.10.026. [DOI] [PubMed] [Google Scholar]
- 30.Goldberg AD, Banaszynski LA, Noh KM, Lewis PW, Elsaesser SJ, Stadler S, Dewell S, Law M, Guo X, Li X, Wen D, Chapgier A, DeKelver RC, Miller JC, Lee YL, Boydston EA, Holmes MC, Gregory PD, Greally JM, Rafii S, Yang C, Scambler PJ, Garrick D, Gibbons RJ, Higgs DR, Cristea IM, Urnov FD, Zheng D, Allis CD. 2010. Distinct factors control histone variant H3.3 localization at specific genomic regions. Cell 140:678–691. doi: 10.1016/j.cell.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Arnoult N, Van Beneden A, Decottignies A. 2012. Telomere length regulates TERRA levels through increased trimethylation of telomeric H3K9 and HP1α. Nat Struct Mol Biol 19:948–956. doi: 10.1038/nsmb.2364. [DOI] [PubMed] [Google Scholar]
- 32.Levy MA, Kernohan KD, Jiang Y, Bérubé NG. 2015. ATRX promotes gene expression by facilitating transcriptional elongation through guanine-rich coding regions. Hum Mol Genet 24:1824–1835. doi: 10.1093/hmg/ddu596. [DOI] [PubMed] [Google Scholar]
- 33.Azzalin CM, Reichenbach P, Khoriauli L, Giulotto E, Lingner J. 2007. Telomeric repeat containing RNA and RNA surveillance factors at mammalian chromosome ends. Science 318:798–801. doi: 10.1126/science.1147182. [DOI] [PubMed] [Google Scholar]
- 34.Deng Z, Norseen J, Wiedmer A, Riethman H, Lieberman PM. 2009. TERRA RNA binding to TRF2 facilitates heterochromatin formation and ORC recruitment at telomeres. Mol Cell 35:403–413. doi: 10.1016/j.molcel.2009.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Episkopou H, Draskovic I, Van Beneden A, Tilman G, Mattiussi M, Gobin M, Arnoult N, Londono-Vallejo A, Decottignies A. 2014. Alternative lengthening of telomeres is characterized by reduced compaction of telomeric chromatin. Nucleic Acids Res 42:4391–4405. doi: 10.1093/nar/gku114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kernohan KD, Jiang Y, Tremblay DC, Bonivissuto AC, Eubanks JH, Mann MRW, Bérubé NG. 2010. ATRX partners with cohesin and MeCP2 and contributes to developmental silencing of imprinted genes in the brain. Dev Cell 18:191–202. doi: 10.1016/j.devcel.2009.12.017. [DOI] [PubMed] [Google Scholar]
- 37.Law MJ, Lower KM, Voon HP, Hughes JR, Garrick D, Viprakasit V, Mitson M, De Gobbi M, Marra M, Morris A, Abbott A, Wilder SP, Taylor S, Santos GM, Cross J, Ayyub H, Jones S, Ragoussis J, Rhodes D, Dunham I, Higgs DR, Gibbons RJ. 2010. ATR-X syndrome protein targets tandem repeats and influences allele-specific expression in a size-dependent manner. Cell 143:367–378. doi: 10.1016/j.cell.2010.09.023. [DOI] [PubMed] [Google Scholar]
- 38.Wong LH, McGhie JD, Sim M, Anderson MA, Ahn S, Hannan RD, George AJ, Morgan KA, Mann JR, Choo KHA. 2010. ATRX interacts with H3.3 in maintaining telomere structural integrity in pluripotent embryonic stem cells. Genome Res 20:351–360. doi: 10.1101/gr.101477.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Watson LA, Solomon LA, Li JR, Jiang Y, Edwards M, Shin-ya K, Beier F, Bérubé NG. 2013. Atrx deficiency induces telomere dysfunction, endocrine defects, and reduced life span. J Clin Invest 123:2049–2063. doi: 10.1172/JCI65634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chang FTM, McGhie JD, Chan FL, Tang MC, Anderson MA, Mann JR, Choo KHA, Wong LH. 2013. PML bodies provide an important platform for the maintenance of telomeric chromatin integrity in embryonic stem cells. Nucleic Acids Res 41:4447–4458. doi: 10.1093/nar/gkt114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nergadze SG, Farnung BO, Wischnewski H, Khoriauli L, Vitelli V, Chawla R, Giulotto E, Azzalin CM. 2009. CpG-island promoters drive transcription of human telomeres. RNA 15:2186–2194. doi: 10.1261/rna.1748309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Flynn RL, Cox KE, Jeitany M, Wakimoto H, Bryll AR, Ganem NJ, Bersani F, Pineda JR, Suvà ML, Benes CH, Haber DA, Boussin FD, Zou L. 2015. Alternative lengthening of telomeres renders cancer cells hypersensitive to ATR inhibitors. Science 347:273–277. doi: 10.1126/science.1257216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Flynn RL, Centore RC, O'Sullivan RJ, Rai R, Tse A, Songyang Z, Chang S, Karlseder J, Zou L. 2011. TERRA and hnRNPA1 orchestrate an RPA-to-POT1 switch on telomeric single-stranded DNA. Nature 471:532–536. doi: 10.1038/nature09772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Conomos D, Reddel RR, Pickett HA. 2014. NuRD-ZNF827 recruitment to telomeres creates a molecular scaffold for homologous recombination. Nat Struct Mol Biol 21:760–770. doi: 10.1038/nsmb.2877. [DOI] [PubMed] [Google Scholar]
- 45.Bhattacharyya MK, Lustig AJ. 2006. Telomere dynamics in genome stability. Trends Biochem Sci 31:114–122. doi: 10.1016/j.tibs.2005.12.001. [DOI] [PubMed] [Google Scholar]
- 46.Conomos D, Pickett HA, Reddel RR. 2013. Alternative lengthening of telomeres: remodeling the telomere architecture. Front Oncol 3:27. doi: 10.3389/fonc.2013.00027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Picketts DJ, Higgs DR, Bachoo S, Blake DJ, Quarrell OW, Gibbons RJ. 1996. ATRX encodes a novel member of the SNF2 family of proteins: mutations point to a common mechanism underlying the ATR-X syndrome. Hum Mol Genet 5:1899–1907. doi: 10.1093/hmg/5.12.1899. [DOI] [PubMed] [Google Scholar]
- 48.Le S, Moore JK, Haber JE, Greider CW. 1999. RAD50 and RAD51 define two pathways that collaborate to maintain telomeres in the absence of telomerase. Genetics 152:143–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Teng SC, Zakian VA. 1999. Telomere-telomere recombination is an efficient bypass pathway for telomere maintenance in Saccharomyces cerevisiae. Mol Cell Biol 19:8083–8093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hakin-Smith V, Jellinek DA, Levy D, Carroll T, Teo M, Timperley WR, McKay MJ, Reddel RR, Royds JA. 2003. Alternative lengthening of telomeres and survival in patients with glioblastoma multiforme. Lancet 361:836–838. doi: 10.1016/S0140-6736(03)12681-5. [DOI] [PubMed] [Google Scholar]
- 51.Ulaner GA, Huang HY, Otero J, Zhao Z, Ben-Porat L, Satagopan JM, Gorlick R, Meyers P, Healey JH, Huvos AG, Hoffman AR, Ladanyi M. 2003. Absence of a telomere maintenance mechanism as a favorable prognostic factor in patients with osteosarcoma. Cancer Res 63:1759–1763. [PubMed] [Google Scholar]
- 52.O'Sullivan RJ, Arnoult N, Lackner DH, Oganesian L, Haggblom C, Corpet A, Almouzni G, Karlseder J. 2014. Rapid induction of alternative lengthening of telomeres by depletion of the histone chaperone ASF1. Nat Struct Mol Biol 21:167–174. doi: 10.1038/nsmb.2754. [DOI] [PMC free article] [PubMed] [Google Scholar]