

Abstract
Inhibition of adipocyte lipolysis by insulin is important for whole-body energy homeostasis; its disruption has been implicated as contributing to the development of insulin resistance and type 2 diabetes mellitus. The main target of the antilipolytic action of insulin is believed to be phosphodiesterase 3B (PDE3B), whose phosphorylation by Akt leads to accelerated degradation of the prolipolytic second messenger cyclic AMP (cAMP). To test this hypothesis genetically, brown adipocytes lacking PDE3B were examined for their regulation of lipolysis. In Pde3b knockout (KO) adipocytes, insulin was unable to suppress β-adrenergic receptor-stimulated glycerol release. Reexpressing wild-type PDE3B in KO adipocytes fully rescued the action of insulin against lipolysis. Surprisingly, a mutant form of PDE3B that ablates the major Akt phosphorylation site, murine S273, also restored the ability of insulin to suppress lipolysis. Taken together, these data suggest that phosphorylation of PDE3B by Akt is not required for insulin to suppress adipocyte lipolysis.
INTRODUCTION
Adipose tissue serves the highly specialized function of storing excess energy in the form of triglycerides (TG) until a time of caloric need. These lipid stores are then hydrolyzed into glycerol and free fatty acids (FFAs), a process termed lipolysis, and released into circulation to provide energy to other tissues. The intricate balance maintained by adipose tissue in response to nutritional status is essential for whole-body energy homeostasis. The importance of the control of lipid storage is illustrated by the consequences of an excess of dietary lipids, which leads to inappropriate deposition of neutral lipid in nonadipose cell types and insulin (Ins) resistance (1). While much is known about the regulatory mechanisms that govern lipid metabolism, there are questions that remain unresolved, such as how lipolysis is suppressed following nutrient intake.
Adipocytes are specifically poised to respond to lipolytic stimulation in a dynamic fashion. During fasting, catecholamines initiate the canonical β-adrenergic receptor (β-AR) signaling cascade, leading to the generation of the second messenger cyclic AMP (cAMP) and subsequent activation of protein kinase A (PKA). Two key protein targets of PKA, perilipin 1 (PLIN1) and hormone-sensitive lipase (HSL), help facilitate the robust lipolytic response resulting in the sequential hydrolysis of TG first to diacylglycerol (DG), then to monoacylglycerol (MG), and finally to glycerol and FFA (2). PLIN1 associates with and protects the neutral lipid droplet from lipases in the unstimulated (basal) state to maintain lipid storage (3). However, phosphorylation of PLIN1 by PKA leads to the release of comparative gene identification-58 (CGI-58), allowing it to associate with and activate adipose triglyceride lipase (ATGL), the first enzyme in the lipolytic cascade (4, 5). In addition, upon PKA phosphorylation, HSL translocates from the cytosol to the surface of the lipid droplet, where it hydrolyzes DG to MG (6, 7). The final hydrolysis step, by monoglyceride lipase (MGL), converts MG into glycerol and free fatty acid.
Conversely, following a meal, insulin signals to the adipocyte to take up glucose from the circulation for de novo synthesis of lipids and esterification of fatty acids. Insulin binds to its receptor, tyrosine kinase, leading to phosphorylation of insulin receptor substrate (IRS) and subsequent activation of phosphatidylinositol 3-kinase (PI3K). The phosphatidylinositol (3,4,5)-trisphosphate (PIP3) generated at the membrane recruits Akt, which then is activated by two upstream kinases, PDK1 and mTORC2. Akt, the major node of insulin action, promotes the translocation of the facilitated glucose transporter GLUT4 to the membrane, where it catalyzes increased glucose uptake (8). Akt is also believed to mediate the antilipolytic effect of insulin via phosphorylation of phosphodiesterase 3B (PDE3B), resulting in enhanced hydrolysis of its substrate, cAMP (9–11). The reduction in cAMP levels in the adipocyte allows phosphatases to return lipolytic signaling to the basal state. The most convincing evidence for a role for PDE3B derives from the observation that insulin does not suppress lipolysis in primary adipocytes from mice lacking an intact Pde3b gene (12). While the requirement for PDE3B in the regulation of lipolysis is generally accepted, the mechanism by which it is regulated by insulin has been called into question, since the antilipolytic effects of insulin do not correlate with decreases in PKA activity at maximal levels of adrenergic stimulation of lipolysis (13), and Akt has been suggested to be dispensable for this process (14, 15). To address this issue, we used a genetic approach to examine the requirement of PDE3B and its phosphorylation for insulin-mediated antilipolysis in cultured brown adipocytes. We found that while PDE3B is important for the control of lipolytic activity, the phosphorylation of its major Akt site is not required for insulin to suppress lipolysis.
MATERIALS AND METHODS
Materials.
Akt inhibitor VIII (AktiVIII) and cilostamide were obtained from EMD Chemicals (Gibbstown, NJ); MK-2206 dihydrochloride was obtained from Selleck Chemicals (Houston, TX); rolipram, CL 316,243, and 3-isobutyl-1-methylxanthine (IBMX) were obtained from Sigma-Aldrich (St. Louis, MO). [2,8-3H]cyclic AMP, ammonium salt, was obtained from American Radiolabeled Chemicals, Inc. (St. Louis, MO).
Buffers.
Modified radioimmunoprecipitation assay (RIPA) buffer contained 50 mM Tris (pH 7.4), 150 mM NaCl, 0.5% sodium deoxycholate, 1% Triton X-100, 1× Complete protease inhibitor cocktail (Roche Diagnostics, Mannheim, Germany), and 1× phosphatase inhibitor cocktails 2 and 3 (Sigma, St. Louis, MO). Hypotonic lysis medium (HLM) contained 50 mM HEPES (pH 7.5), 50 mM sucrose, 1 mM EDTA, 100 mM NaCl, 5 mM NaF, and 1× protease/phosphatase inhibitors. Krebs-Ringer phosphate (KRP) buffer contained 136 mM NaCl, 4.7 mM KCl, 10 mM NaPO4 (pH 7.4), 0.9 mM MgSO4, and 0.9 mM CaCl2.
Western blot analysis.
Cell lysates were loaded and separated on 10% SDS-PAGE gels and were then transferred to nitrocellulose membranes. Membranes were probed with primary antibodies overnight at 4°C. The primary antibodies against pan-Akt (4691P), pAkt Ser473 (4060S), ATGL, fatty acid binding protein 4 (FABP4), HSL, pHSL Ser660, PRAS40, HSP90, and PLIN1 (9349S) were obtained from Cell Signaling Technology (Beverly, MA); anti-PRAS40 [pThr246] was obtained from Invitrogen (Grand Island, NY); the anti-p-perilipin 1 antibody, raised against a peptide near phosphorylated Ser522 of human perilipin 1, was obtained from Vala Sciences (San Diego, CA); an antitubulin primary antibody was obtained from Sigma (St. Louis, MO); and primary antibodies against peroxisome proliferator-activated receptor gamma (PPARγ) and PDE3B (SC-11838) were obtained from Santa Cruz Biotechnology, Inc. (Dallas, TX). All antibodies were used at dilutions suggested by the manufacturer. Infrared-labeled secondary antibodies were obtained from Rockland, Inc. (Gilbertsville, PA). We used an Odyssey infrared imaging system (Li-Cor Biosciences, Lincoln, NE) for all immunoblotting.
Cell culture.
Brown preadipocytes were isolated from the stromal vascular fractions of the interscapular fat pads of wild-type (WT) (JAX 129/SvJ) and Pde3b knockout (Pde3b-KO) mice, immortalized, and differentiated as described previously (16, 17). Experiments were performed on days 6 to 8 postdifferentiation. 3T3-L1 preadipocytes were maintained and differentiated as described previously (18), except that the differentiation medium contained 10 μg/ml insulin (Sigma). Experiments were performed with 3T3-L1 adipocytes 7 to 10 days postdifferentiation.
Glycerol release.
Adipocytes were serum starved in low-glucose Dulbecco's modified Eagle medium (DMEM) containing 0.2% bovine serum albumin (BSA) for 2 to 3 h. Experiments were conducted in KRP containing 4% fatty-acid-free BSA (Roche) with agonists and inhibitors for 1 h at 37°C. Reactions were stopped on ice, and media were collected and assayed for glycerol content by using free glycerol reagent (Sigma) according to the manufacturer's instructions. Protein was harvested from each well and was used for normalization. Data were calculated as a rate of release (in nanograms of glycerol per microgram of protein per minute).
Membrane and lipid droplet isolation.
Adipocytes in 10-cm dishes were scraped in HLM with protease inhibitors and were lysed using a Dounce homogenizer (12 strokes). Lysates were centrifuged at low speed (200 × g) for 5 min. For membrane isolation, the supernatant was transferred to a new tube and was centrifuged at 100,000 × g for 30 min. Membrane pellets were resuspended in HLM alone (for the PDE activity assay) or in HLM with 1% Triton X-100 (for Western blotting). Lipid droplets were isolated using a modified version of the sucrose density gradient centrifugation method (19). Briefly, the supernatant was mixed with a 60% sucrose solution (3:1 ratio by volume). Five milliliters of a 5% sucrose solution was layered on top, followed by 3.5 ml of HLM. Samples were centrifuged at 15,000 rpm for 30 min at 4°C using an SW41 Ti swinging bucket rotor without a brake. The fat layer was scraped off the top and was mixed with HLM.
Cyclic AMP-phosphodiesterase activity.
PDE3 activity, the portion of total PDE activity inhibited by 1 μM cilostamide, a selective PDE3 inhibitor (PDE3i), was measured by a modification of our published method using 0.1 μM [3H]cAMP (45,000 cpm) as the substrate (20).
Triglyceride content.
Intracellular triglycerides were measured by hydrolyzing triglyceride in the whole-cell lysate into free glycerol using triglyceride reagent (Sigma) for 1 h at 37°C. The free glycerol was then measured using free glycerol reagent (Sigma). Glycerol for triglyceride is calculated by subtracting the glycerol in the lysate from the total glycerol from the hydrolysis reaction. The glycerol was normalized to total protein in the lysate.
Hexose uptake.
The uptake of hexose into brown adipocytes was measured as described previously (21). Briefly, serum-starved adipocytes were treated with 100 nM insulin with or without Akt inhibitors for 15 min at 37°C. The assay was initiated by adding 0.5 μCi 2-[3H]deoxyglucose and 0.1 mM 2-deoxyglucose. The reaction was stopped by immediately washing the plates in ice-cold phosphate-buffered saline (PBS) 8 times after 15 min. Samples were collected, protein measured, and radioactivity counted. Uptake was also measured in the presence of 10 μM cytochalasin B and was subtracted from all values.
Intracellular cAMP measurement.
The intracellular cAMP concentration was measured by using a cAMP Biotrak enzyme immunoassay (EIA) kit (nonacetylation protocol) according to the manufacturer's instructions.
Generation of PDE3B retrovirus.
Human PDE3B cDNA (OriGene Technologies, Inc.) was initially cloned by PCR into the pENTR/dTOPO vector. Single amino acid mutations were introduced via QuikChange II XL site-directed mutagenesis (Agilent Technologies). Upon sequence verification, cDNA was subcloned into the pMIGR1 retroviral vector (22), and high-titer retrovirus was generated using BOSC23 cells as described previously (23).
Statistical analysis.
Experiments were analyzed using a two-tailed t test or one-way analysis of variance (ANOVA) with Tukey's posttest. Statistics were calculated using Prism, version 5 (GraphPad Software, Inc.).
RESULTS
Insulin suppresses glycerol release in 3T3-L1 and brown adipocytes independently of Akt activity.
Immortalized brown fat cells present an excellent in vitro model for studying the mechanisms of adipocyte function. Although they serve the distinct physiological function of thermogenesis instead of fat storage, brown adipocytes possess the same lipolytic machinery as white adipocytes (24, 25). In fact, we have shown previously that the regulation of lipolysis in primary brown adipocytes is comparable to its regulation in vivo (26). In order to be certain that immortalized brown adipocytes behave in a manner similar to the better-established model of white adipocyte biology, 3T3-L1 cells, we compared the regulation of lipolysis by insulin in the two cell lines. Stimulation of lipolysis by a submaximal concentration (0.3 nM) of isoproterenol (Iso), a β1, β2-AR agonist, increased glycerol release, which was inhibited by 25 nM insulin (Ins), in both 3T3-L1 and brown adipocytes (Fig. 1A). Furthermore, insulin suppressed glycerol release even when Akt phosphorylation was inhibited by 10 μM AktiVIII or 10 μM MK-2206 (Fig. 1A and B). Under the same conditions, insulin-stimulated glucose transport was completely blocked by either of these inhibitors (Fig. 1C). Of note, whereas MK-2206 increased Iso-dependent glycerol release, presumably by an off-target effect, lipolysis was still inhibited by insulin, albeit to a lesser degree, in brown adipocytes. These data support our previous observations for 3T3-L1 adipocytes that under conditions in which Akt is inhibited to levels that completely prevent the stimulation of glucose uptake by insulin, suppression of lipolysis is maintained (15).
FIG 1.

Insulin suppresses glycerol release in both 3T3-L1 and brown adipocytes independently of Akt activity. (A) Glycerol release was measured at the basal state and upon stimulation with 0.3 nM isoproterenol (Iso) in the presence or absence of 25 nM insulin (Ins), 10 μM AktiVIII, or 10 μM MK-2206 for 1 h in both 3T3-L1 and brown adipocytes. Data from 3T3-L1 (n = 3) and brown adipocytes (n = 6) were normalized to those with Iso alone. Symbols indicate significant differences (P < 0.05) for insulin suppression (*) and for Iso alone versus Iso plus MK-2206 (#). (B) Cell lysates from the experiment for which results are shown in panel A were assessed for Akt phosphorylation by Western blotting. (C) The uptake of 2-deoxyglucose into brown adipocytes was measured upon stimulation with 100 nM insulin in the presence or absence of AktiVIII or MK-2206 at the same concentrations used in the experiment for which results are shown in panel A. Values represent the results of 3 experiments. (D) Glycerol release from WT brown adipocytes upon stimulation with 0.3 nM Iso in the presence or absence of 25 nM insulin, 10 μM rolipram (PDE4i), 10 μM cilostamide (PDE3i), or a combination of PDE3i and PDE4i. Data were normalized to those for Iso with no inhibitor. Values represent the results of 3 to 7 experiments. Symbols indicate significant differences (P < 0.05) for insulin suppression (*) and between Iso with a PDE inhibitor and Iso alone (#). All data are presented as means ± standard errors of the means. Statistical analysis was performed using one-way ANOVA and Tukey's posttest.
The prevalent model for insulin-mediated antilipolysis involves Akt-mediated modulation of PDE3B activity. In light of our findings suggesting that brown adipocytes have an Akt-independent mechanism by which insulin acts to suppress lipolysis, we measured glycerol release upon inhibition of PDE3 and PDE4, the major cAMP-PDEs in adipocytes (27, 28), to determine the requirement for PDEs in this process in brown adipocytes. A 10 μM concentration of rolipram, a PDE4-specific inhibitor (PDE4i), had no significant effect on Iso stimulation or insulin suppression of glycerol release (Fig. 1D). However, treatment of cells with 10 μM cilostamide, a PDE3-specific inhibitor (PDE3i), resulted in an 80% increase in Iso-stimulated glycerol release over that with Iso alone. Furthermore, insulin could not suppress glycerol release in the presence of the PDE3 inhibitor, in agreement with previous reports for white adipocytes (29). Upon inhibition of both PDE3 and PDE4 by a combination of cilostamide and rolipram, we observed a dramatic increase in basal and Iso-stimulated glycerol release that could not be suppressed by insulin (Fig. 1D). These data support the notion that the PDE3 isoform is the major cAMP-degrading enzyme in both white and brown adipocytes and is therefore important for the regulation of lipolysis.
Characterization of WT and Pde3b-KO brown adipocytes.
In order to address the requirement for PDE3B and its modulation by insulin in the regulation of lipolysis in a genetic model, we isolated brown preadipocytes from the interscapular brown fat pads of Pde3b-KO mice and their wild-type (WT) littermates and established immortalized cell lines (12). Immunoblotting for PDE3B protein in membranes from differentiated WT and Pde3b-KO brown adipocytes revealed expression in WT but not KO adipocytes (Fig. 2A). As a more sensitive assay for PDE3B in Pde3b-KO brown adipocytes, we measured its activity by assessing the hydrolysis of [3H]cAMP. In membrane lysates from wild-type brown adipocytes, PDE3 accounted for ∼80% of total PDE activity, while less than 10% was attributed to other IBMX-sensitive cAMP-PDEs, in agreement with previous reports (30). In membrane lysates from Pde3b-null brown adipocytes, cAMP-PDE activity was undetectable, in agreement with the idea that this isoform accounts for most of the membrane cAMP hydrolytic activity in fat cells (Fig. 2B). Of note, there was no compensatory induction of cAMP-PDE activity in membranes from Pde3b-null adipocytes.
FIG 2.
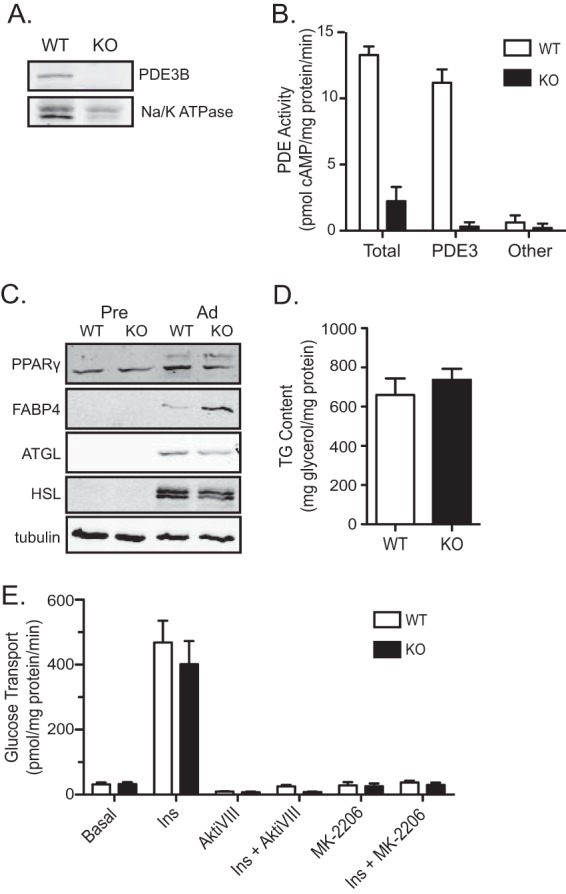
Characterization of WT and Pde3b-KO brown adipocytes. (A) Immunoblot for PDE3B protein in crude membrane lysates harvested from WT and Pde3b-KO brown adipocytes. Na/K ATPase, a membrane-bound protein, was used as a loading control. (B) A radioactive assay for PDE activity was performed on membrane lysates. For the PDE3-specific activity, 10 μM cilostamide was added to the reaction mixture for both WT and Pde3b-KO adipocytes, and the activity was subtracted from total activity. Other cAMP-PDE activity was calculated based on inhibition after the addition of 100 μM IBMX. (C) The protein expression of peroxisome proliferator-activated receptor gamma (PPARγ), fatty acid binding protein 4 (FABP4), adipose triglyceride lipase (ATGL), and hormone-sensitive lipase (HSL) for the two genotypes was compared via Western blot analysis. (D and E) Triglyceride (TG) content (n = 3) (D) and glucose transport (n = 3) (E) were measured in WT and Pde3b-KO adipocytes. All data are presented as means ± standard errors of the means.
To test whether deletion of PDE3B and subsequent disruption of normal cAMP regulation had an effect on adipocyte conversion, whole-cell lysates from WT and KO preadipocytes and differentiated adipocytes were immunoblotted for peroxisome proliferator-activated receptor gamma (PPARγ), the master regulator of adipogenesis, fatty acid binding protein 4 (FABP4), and ATGL and HSL, key markers of mature adipocytes (31). The expression of these proteins in KO cells was unchanged from that in wild-type adipocytes (Fig. 2C). Pde3b-KO brown adipocytes also stored amounts of triglyceride comparable to those in WT cells (Fig. 2D). Furthermore, ablation of PDE3B protein had no effect on insulin-stimulated glucose uptake or its blockade by Akt inhibitors, in agreement with data from primary adipocytes isolated from epididymal white fat pads of Pde3b-KO mice (Fig. 2E) (12). Thus, there were no marked differences in the fat cell phenotype between WT and Pde3b-KO brown adipocytes.
Lack of PDE3B in brown adipocytes impairs the action of insulin on lipolysis.
To assess lipolysis in Pde3b-KO adipocytes, we measured glycerol release after treatment of WT and Pde3b-KO brown adipocytes with increasing doses of Iso and CL 316,243 (a β3-AR agonist) with or without 25 nM insulin for 1 h. We observed a dose-dependent increase in glycerol release in response to both adrenergic agents in WT and KO adipocytes. Surprisingly, there was no statistically significant enhancement in glycerol release in KO adipocytes. As expected, insulin suppressed glycerol release from WT brown adipocytes by ∼80% after treatment of adipocytes with a 0.3 nM dose of Iso or CL 316,243 and by 60% after treatment with a 3 nM dose of Iso or CL 316,243 (Fig. 3A and B). However, Pde3b-KO adipocytes showed a marked impairment in the suppression of glycerol release by insulin (Fig. 3A and B).
FIG 3.

Pde3b-KO brown adipocytes have impaired insulin-mediated suppression of lipolysis. (A and B) Glycerol release from WT (solid lines) and Pde3b-KO (dashed lines) brown adipocytes without stimulation or upon stimulation with 0.3 nM or 3 nM isoproterenol (Iso) (A) or CL 316,243 (B) in the presence (×) or absence (○) of 25 nM insulin. Data were normalized to a 0.3 nM dose in Pde3b-KO adipocytes. Values represent results for 3 to 7 experiments. Asterisks indicate significant differences (P < 0.05) for insulin suppression in WT cells. (C) Representative immunoblots for tubulin and phosphorylated and total perilipin 1 (PLIN1), HSL, and Akt in whole-cell lysates harvested at the termination of glycerol release assays. (D) Intracellular cAMP measurement in WT and Pde3b-KO adipocytes upon stimulation with 1 μM forskolin (Fsk) with or without the addition of 25 nM insulin. Data were normalized to basal values of WT cells. Values represent results for 5 experiments. All data are presented as means ± standard errors of the means. Statistical analysis was performed using one-way ANOVA with Tukey's posttest.
We next probed the phosphorylation states of two PKA substrates important in the lipolytic pathway, HSL and PLIN1, under the same conditions as those of the glycerol release assay. Iso increased the phosphorylation of Ser517 of perilipin 1 and Ser651 of HSL, the sites thought most important for hormone-stimulated lipolysis (32, 33), with a dose dependency that correlated well with glycerol release in WT brown adipocytes (Fig. 3C). Insulin substantially blocked Iso-stimulated PLIN1 and HSL phosphorylation in WT adipocytes. Interestingly, there was a smaller but consistent effect of insulin on the phosphorylation of these PKA substrates in KO brown adipocytes stimulated with 0.3 nM Iso (Fig. 3C). Taken together, these data confirm that phosphorylation of the lipolytic substrates PLIN1 and HSL correlates well with glycerol release in WT cells, but they also suggest that insulin can antagonize PKA-dependent phosphorylation in the absence of PDE3B.
In view of the effect of insulin on the phosphorylation of PKA substrates in Pde3b-KO cells, we measured cAMP in both WT and Pde3b-KO brown adipocytes. Upon stimulation with 1 μM forskolin (Fsk) with or without 25 nM Ins for 10 min, WT and KO adipocytes accumulated intracellular cAMP at levels that were 3-fold above basal levels and were not affected by insulin in either cell line (Fig. 3D).
Phosphorylation of PDE3B at its canonical Akt site is not required for the antilipolytic effect of insulin.
The inability of insulin to suppress lipolysis in Pde3b-KO adipocytes confirms a requirement for the enzyme and its associated basal activity; however, the data do not resolve whether the modulation of PDE3B activity by hormones is essential for this process. In order to test the role of phosphorylation and subsequent enhanced activity of PDE3B in the suppression of lipolysis, we reexpressed WT PDE3B or PDE3B phosphorylation site mutants in Pde3b-KO brown adipocytes. Pde3b-KO adipocytes were infected with a retrovirus containing the cDNA encoding human WT PDE3B, cDNA with an S295A or S318A mutation (corresponding to murine Ser273 or Ser296, respectively), or cDNA with both S295A and S318A (2SA), by using green fluorescent protein (GFP) as a vector control. For ease of discussion, we will refer to the S273/S295 site as the Akt site and to the S296/S318 site as the PKA site, as described in prior publications (34–36). Upon differentiation, adipocyte membranes were isolated and assayed for PDE3B expression and activity. Exogenous WT PDE3B and mutant PDE3B were expressed at equivalent levels that were at least 5-fold greater than those of endogenous PDE3B (Fig. 4A). In the infected cells, the activities of all overexpressed PDE3B variants were approximately 6 times endogenous PDE3 activity (Fig. 2B and 4B). Both Iso and insulin increased PDE3B activity in Pde3b-KO adipocytes overexpressing wild-type PDE3B. Cells expressing PDE3B with the Akt site, the PKA site, or both sites ablated did not respond to Iso or insulin with increases in PDE3B activity (Fig. 4B). These data confirm that these residues are indeed the main targets of catecholamine- and insulin-dependent phosphorylation and mediate the regulation of enzyme activity.
FIG 4.

Phosphorylation of PDE3B at putative Akt and PKA sites is not required for the antilipolytic effect of insulin in brown adipocytes. (A) Immunoblot for PDE3B protein in crude membrane lysates harvested from WT and Pde3b-KO brown adipocytes and from KO adipocytes overexpressing the indicated human PDE3B proteins. Na/K ATPase was used as a membrane loading control. (B) PDE3 activity in membrane fractions. Values represent results for 2 to 3 independent experiments. *, P < 0.001. (C) Representative immunoblot of the lipid droplet fraction and whole-cell lysate (WCL) for PDE3B using perilipin 1 and HSP90 as lipid droplet and cytosolic markers, respectively. For “+WT+Ins” samples, KO plus WT adipocytes were treated with 100 nM insulin for 15 minutes prior to fractionation. (D and E) Glycerol release was measured without Iso or upon the addition of 0.3 or 3 nM Iso (D) or 3 nM Iso (E) in the presence or absence of 25 nM Ins. Data are expressed as percentages of the response of WT (D) or Pde3b-KO (E) adipocytes upon stimulation with 3 nM Iso. Vec, vector; NT, no treatment (basal). Values represent results for 3 experiments. Symbols indicate significant differences (P < 0.05) for insulin suppression (*), for comparison with the WT group with 3 nM Iso (#), for comparison with the KO-plus-S318A group with 3 nM Iso (^), or for comparison with the KO-plus-2SA group with 3 nM Iso (+). (F) Representative immunoblots for phosphorylated perilipin 1 (PLIN1) and HSL in whole-cell lysates harvested at the end of the glycerol release assays. All data are presented as means ± standard errors of the means. Statistical analysis was performed using one-way ANOVA and Tukey's posttest.
We next assessed the subcellular distribution of PDE3B by isolating lipid droplets using density gradient centrifugation. Endogenous and overexpressed PDE3B localized to the lipid droplet fraction. The association of overexpressed WT PDE3B with the lipid droplet with the lipid droplet was unchanged upon treatment with insulin (Fig. 4C). As expected based on the significant overexpression, WT PDE3B in KO adipocytes displayed lower maximal Iso-stimulated glycerol release than parental KO cells. In spite of this, WT PDE3B-expressing null cells responded robustly to insulin in the suppression of lipolysis (Fig. 4D). All the phosphorylation site mutants of PDE3B, including one in which the putative Akt site was ablated, also restored the ability of insulin to antagonize Iso-stimulated glycerol release to Pde3b-KO adipocytes comparably. Mutation of the PKA site alone or in combination with the Akt site (2SA) enhanced Iso-stimulated lipolysis over that for the WT protein (Fig. 4D). These data are consistent with previous studies indicating that PKA phosphorylation of PDE3B increases its activity, functioning as a negative-feedback signal (34, 36, 37). To assess whether other residues are required for the regulation of lipolysis by insulin, we mutated one, two, or three other conserved serines to alanine (human S296 [murine S274], S442 [murine S421], and S495 [murine S474]), in addition to the Akt and PKA sites; we refer to these as the 3SA, 4SA, and 5SA mutants, respectively. When these proteins were overexpressed in Pde3b-KO adipocytes, insulin still suppressed Iso-stimulated glycerol release to an extent similar to that in cells overexpressing the WT enzyme (Fig. 4E). Phosphorylation of PLIN1 and HSL paralleled glycerol release in the retrovirus-infected cells, with restoration of insulin action by all PDE3B proteins and increases in phosphorylation when the PKA site serine was mutated to alanine (Fig. 4F). Taken together, these data suggest that phosphorylation of PDE3B at its major Akt site is required for the activation of PDE3B by insulin but not for the suppression of lipolysis in brown adipocytes.
DISCUSSION
Hormone regulation of lipolysis has garnered attention recently as support has grown for the idea that elevated circulating free fatty acid levels can lead to ectopic deposition of lipid in multiple tissues, potentially leading to insulin resistance and cellular dysfunction. Moreover, the recent recognition of “selective insulin resistance” in liver and adipose tissue has brought to the fore the concept that insulin uses multiple intracellular signaling pathways to carry out its biological actions (38–40). Thus, it seemed timely to reassess the molecular mechanism by which lipolysis is regulated in fat cells, and in particular how insulin suppresses fatty acid release. Insulin stimulates glucose transport, generating glycerol-3-phosphate, which promotes reesterification and thus reduces FFA secretion. However, insulin also inhibits the cleavage of triglyceride into glycerol and fatty acids, and it is the nature of the signaling pathway controlling this process that has proven controversial. In this report, we use a genetic approach to test one of the tenets of the hypothesis that PDE3B phosphorylation is required for insulin-dependent antilipolysis and find instead that the major Akt phosphorylation site in PDE3B is dispensable for the action of insulin.
A persistent question dating back almost to the discovery of cAMP has been whether insulin carries out many of its actions by reducing the intracellular levels of this second messenger. Almost 30 years ago, Londos and colleagues carefully examined the effects of insulin on lipolysis and PKA activity, concluding that, depending on the degree of adrenergic simulation, the suppression of lipolysis by insulin could not always be fully accounted for by the decrease in PKA activity (13, 41). Nonetheless, although these data questioned the relationship between changes in cAMP and insulin-dependent antilipolysis, a generally accepted model has emerged in which Akt, whose activity is elevated by insulin, phosphorylates and stimulates PDE3B, thus reducing cAMP levels and PKA activity (9–11, 42). Considerable data support this hypothesis, which is made all the more compelling by analogy to the well-established mediation of insulin-regulated glucose metabolism by Akt, as well as the absence of the antilipolytic effects of insulin in Pde3b-KO adipocytes (12). Pharmacological inhibition of PI3K, Akt, and PDE3B blocks the ability of insulin to suppress lipolysis (43–46). Additionally, the predominant Akt phosphorylation site in murine PDE3B has been mapped to murine Ser273 and has been shown to be necessary for in vitro and in vivo phosphorylation and activation of the enzyme (34, 35, 47). On the other hand, conflicting studies have provided evidence that Akt and cAMP degradation are not required for insulin to inhibit lipolysis (14, 15, 48, 49). Most recently, we found that under conditions of submaximal β-adrenergic stimulation, the ability of insulin to suppress lipolysis did not require Akt2, and possibly did not require any Akt activity (15). One explanation for this discrepancy in the literature is that both Akt-dependent and Akt-independent pathways exist in the adipocyte, and depending on the experimental conditions, one pathway will predominate. In this study, we assessed the requirement for PDE3B phosphorylation in insulin-mediated antilipolysis in an in vitro model system, differentiated immortalized brown adipocytes. While confirming the observation that primary mouse adipocytes deficient in PDE3B do increase lipolysis in response to β-adrenergic stimulation but no longer respond to insulin, we found that reexpression of a mutant version of PDE3B lacking the major Akt and PKA phosphorylation sites reestablished insulin responsiveness in a manner indistinguishable from that of wild-type PDE3B. By showing that the enhancement of PDE3B activity via phosphorylation is not required for antilipolysis, we provide additional support for an Akt-independent mechanism for insulin-dependent lipolysis.
Several aspects of these data give us confidence in the physiological relevance of the results in spite of the artificial nature of the experimental system. Even though the retrovirus-encoded cDNAs were expressed at levels considerably higher than that of endogenous PDE3B, and thus, the absolute level of isoproterenol-stimulated lipolysis was reduced, insulin still inhibited glycerol release and PKA substrate phosphorylation as in WT adipocytes. Importantly, mutation of candidate phosphorylation sites in PDE3B completely abrogated the ability of Iso or insulin to activate the enzyme, even though the mutagenesis had no effect on insulin-regulated lipolysis. In contrast to the lack of effect of ablation of the Akt phosphorylation site of PDE3B, mutation of the PKA phosphorylation site led to an increase in lipolysis, which was nonetheless inhibited by insulin. PKA phosphorylation of PDE3B is activating, which serves as a negative-feedback loop; interruption of this circuit would be expected to potentiate isoproterenol-stimulated glycerol release, as found in our experiments (34, 36). Importantly, this provides strong support for the notion that, had activation of PDE3B by Akt been important for antilipolysis, the experimental system would have had the sensitivity to detect this, since the magnitudes of PDE3B activation by Akt and PKA are similar (34, 35, 50, 51). Furthermore, we have shown that no other residue previously identified as a potential target of phosphorylation is important in this process. Taken as a whole, these results suggest strongly that PDE3B phosphorylation by Akt and PKA is not required for insulin to suppress lipolysis, in agreement with our recently published findings with Akt2-deficient mice (26).
Although the data in this paper argue against an obligate role for insulin-mediated phosphorylation and subsequent activation of PDE3B in antilipolysis in adipocytes, treatment with insulin was nonetheless associated with a marked decrease in the phosphorylation of canonical PKA substrates in KO adipocytes. Londos and colleagues noted this phenomenon, suggesting that it was due to the activation of serine/threonine protein phosphatases by insulin (13, 52). We advance the alternative hypothesis that insulin works by inhibiting PKA activity locally on the lipid droplet. Moreover, although our data exclude an obligate role for PDE3B activation via phosphorylation in antilipolysis, insulin could be promoting the assembly of a protein complex that includes PDE3B or other negative regulators of PKA signaling on the lipid droplet. The loss of insulin action in the Pde3b-null adipocytes is consistent with this idea. Localized regulation of PKA activity by the assembly of signaling complexes containing components of the cAMP/PKA pathway is a recurrent theme in cellular physiology, and such a mechanism seems plausible for the regulation of lipolysis at the lipid droplet (53–55). In support of this hypothesis, in primary and 3T3-L1 adipocytes, PDE3B associates with caveolin-1, a scaffolding protein within plasma membrane caveolae, which localizes to the lipid droplet upon β-adrenergic stimulation (56–60). Additionally, in the current study, we show that endogenous PDE3B localizes to the lipid droplet fraction of brown adipocytes, suggesting that this compartment might be a site of PDE3B action. Furthermore, treatment of 3T3-L1 adipocytes with insulin has been reported to induce the formation of large macromolecular complexes (“signalosomes”) isolated by gel filtration (Superose 6 columns) of solubilized microsomal membranes. These signalosomes contain phosphorylated/activated PDE3B and signaling molecules, e.g., IRS, PI3K, Akt, caveolin-1, PP2A, and 14-3-3, which coimmunoprecipitate with PDE3B and are potentially involved in its regulation (60, 61). In our studies, perilipin phosphorylation was the best correlate of glycerol release in WT cells, in agreement with the idea that compartmentalized lipid-droplet-associated PKA activity and/or substrate phosphorylation is the ultimate molecular target for insulin signaling, whether or not this process involves PDE3B.
In conclusion, these data confirm that PDE3B is essential for the antilipolytic effect of insulin and that insulin modulates PDE3B activity via phosphorylation, but they also show that phosphorylation of PDE3B is not required for insulin-dependent antilipolysis. Furthermore, these results are consistent with the hypothesis that Akt is dispensable for this antilipolysis, at least under some conditions. Identification of the presently unknown pathway(s) as well as the role of PDE3B in the antilipolytic action of insulin will provide a better understanding of lipid metabolism and might uncover potential targets of therapeutic intervention for insulin-resistant syndromes.
ACKNOWLEDGMENTS
We thank Russell A. Miller for generating the pENTR-PDE3B plasmid and Paul M. Titchenell and William J. Quinn for critical reading of the manuscript.
This work was supported by NIH grants R01DK093959 to M.J.B. and F32DK884443 to L.M.D. Vincent Manganiello and Faiyaz Ahmad are supported by the NHLBI Intramural Research Program.
REFERENCES
- 1.Savage DB, Petersen KF, Shulman GI. 2007. Disordered lipid metabolism and the pathogenesis of insulin resistance. Physiol Rev 87:507–520. doi: 10.1152/physrev.00024.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jaworski K, Sarkadi-Nagy E, Duncan RE, Ahmadian M, Sul HS. 2007. Regulation of triglyceride metabolism. IV. Hormonal regulation of lipolysis in adipose tissue. Am J Physiol Gastrointest Liver Physiol 293:G1–G4. doi: 10.1152/ajpgi.00554.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brasaemle DL, Rubin B, Harten IA, Gruia-Gray J, Kimmel AR, Londos C. 2000. Perilipin A increases triacylglycerol storage by decreasing the rate of triacylglycerol hydrolysis. J Biol Chem 275:38486–38493. doi: 10.1074/jbc.M007322200. [DOI] [PubMed] [Google Scholar]
- 4.Granneman JG, Moore HP, Krishnamoorthy R, Rathod M. 2009. Perilipin controls lipolysis by regulating the interactions of AB-hydrolase containing 5 (Abhd5) and adipose triglyceride lipase (Atgl). J Biol Chem 284:34538–34544. doi: 10.1074/jbc.M109.068478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Greenberg AS, Egan JJ, Wek SA, Garty NB, Blanchette-Mackie EJ, Londos C. 1991. Perilipin, a major hormonally regulated adipocyte-specific phosphoprotein associated with the periphery of lipid storage droplets. J Biol Chem 266:11341–11346. [PubMed] [Google Scholar]
- 6.Egan JJ, Greenberg AS, Chang MK, Wek SA, Moos MC, Londos C. 1992. Mechanism of hormone-stimulated lipolysis in adipocytes: translocation of hormone-sensitive lipase to the lipid storage droplet. Proc Natl Acad Sci U S A 89:8537–8541. doi: 10.1073/pnas.89.18.8537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sztalryd C, Xu G, Dorward H, Tansey JT, Contreras JA, Kimmel AR, Londos C. 2003. Perilipin A is essential for the translocation of hormone-sensitive lipase during lipolytic activation. J Cell Biol 161:1093–1103. doi: 10.1083/jcb.200210169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Taniguchi CM, Emanuelli B, Kahn CR. 2006. Critical nodes in signalling pathways: insights into insulin action. Nat Rev Mol Cell Biol 7:85–96. doi: 10.1038/nrm1837. [DOI] [PubMed] [Google Scholar]
- 9.Czech MP, Tencerova M, Pedersen DJ, Aouadi M. 2013. Insulin signalling mechanisms for triacylglycerol storage. Diabetologia 56:949–964. doi: 10.1007/s00125-013-2869-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Arner P, Langin D. 2014. Lipolysis in lipid turnover, cancer cachexia, and obesity-induced insulin resistance. Trends Endocrinol Metab 25:255–262. doi: 10.1016/j.tem.2014.03.002. [DOI] [PubMed] [Google Scholar]
- 11.Frühbeck G, Méndez-Giménez L, Fernández-Formoso J-A, Fernández S, Rodríguez A. 2014. Regulation of adipocyte lipolysis. Nutr Res Rev 27:63–93. doi: 10.1017/S095442241400002X. [DOI] [PubMed] [Google Scholar]
- 12.Choi YH, Park S, Hockman S, Zmuda-Trzebiatowska E, Svennelid F, Haluzik M, Gavrilova O, Ahmad F, Pepin L, Napolitano M, Taira M, Sundler F, Stenson Holst L, Degerman E, Manganiello VC. 2006. Alterations in regulation of energy homeostasis in cyclic nucleotide phosphodiesterase 3B-null mice. J Clin Invest 116:3240–3251. doi: 10.1172/JCI24867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Londos C, Honnor RC, Dhillon GS. 1985. cAMP-dependent protein kinase and lipolysis in rat adipocytes. III. Multiple modes of insulin regulation of lipolysis and regulation of insulin responses by adenylate cyclase regulators. J Biol Chem 260:15139–15145. [PubMed] [Google Scholar]
- 14.Smith U, Carvalho E, Mosialou E, Beguinot F, Formisano P, Rondinone C. 2000. PKB inhibition prevents the stimulatory effect of insulin on glucose transport and protein translocation but not the antilipolytic effect in rat adipocytes. Biochem Biophys Res Commun 268:315–320. doi: 10.1006/bbrc.2000.2145. [DOI] [PubMed] [Google Scholar]
- 15.Choi SM, Tucker DF, Gross DN, Easton RM, DiPilato LM, Dean AS, Monks BR, Birnbaum MJ. 2010. Insulin regulates adipocyte lipolysis via an Akt-independent signaling pathway. Mol Cell Biol 30:5009–5020. doi: 10.1128/MCB.00797-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Klein J, Fasshauer M, Ito M, Lowell BB, Benito M, Kahn CR. 1999. β3-Adrenergic stimulation differentially inhibits insulin signaling and decreases insulin-induced glucose uptake in brown adipocytes. J Biol Chem 274:34795–34802. doi: 10.1074/jbc.274.49.34795. [DOI] [PubMed] [Google Scholar]
- 17.Tseng YH, Kriauciunas KM, Kokkotou E, Kahn CR. 2004. Differential roles of insulin receptor substrates in brown adipocyte differentiation. Mol Cell Biol 24:1918–1929. doi: 10.1128/MCB.24.5.1918-1929.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yin W, Mu J, Birnbaum MJ. 2003. Role of AMP-activated protein kinase in cyclic AMP-dependent lipolysis in 3T3-L1 adipocytes. J Biol Chem 278:43074–43080. doi: 10.1074/jbc.M308484200. [DOI] [PubMed] [Google Scholar]
- 19.Brasaemle DL, Wolins NE. 2006. Isolation of lipid droplets from cells by density gradient centrifugation. Curr Protoc Cell Biol Chapter 3:Unit 3.15. doi: 10.1002/0471143030.cb0315s29. [DOI] [PubMed] [Google Scholar]
- 20.Manganiello V, Vaughan M. 1973. An effect of insulin on cyclic adenosine 3′:5′-monophosphate phosphodiesterase activity in fat cells. J Biol Chem 248:7164–7170. [PubMed] [Google Scholar]
- 21.Fingar DC, Hausdorff SF, Blenis J, Birnbaum MJ. 1993. Dissociation of pp70 ribosomal protein S6 kinase from insulin-stimulated glucose transport in 3T3-L1 adipocytes. J Biol Chem 268:3005–3008. [PubMed] [Google Scholar]
- 22.Pear WS, Miller JP, Xu L, Pui JC, Soffer B, Quackenbush RC, Pendergast AM, Bronson R, Aster JC, Scott ML, Baltimore D. 1998. Efficient and rapid induction of a chronic myelogenous leukemia-like myeloproliferative disease in mice receiving P210 bcr/abl-transduced bone marrow. Blood 92:3780–3792. [PubMed] [Google Scholar]
- 23.Pear WS, Nolan GP, Scott ML, Baltimore D. 1993. Production of high-titer helper-free retroviruses by transient transfection. Proc Natl Acad Sci U S A 90:8392–8396. doi: 10.1073/pnas.90.18.8392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lafontan M, Berlan M. 1993. Fat cell adrenergic receptors and the control of white and brown fat cell function. J Lipid Res 34:1057–1091. [PubMed] [Google Scholar]
- 25.Cannon B, Nedergaard J. 2004. Brown adipose tissue: function and physiological significance. Physiol Rev 84:277–359. doi: 10.1152/physrev.00015.2003. [DOI] [PubMed] [Google Scholar]
- 26.Koren S, DiPilato LM, Emmett MJ, Shearin AL, Chu Q, Monks B, Birnbaum MJ. 2015. The role of mouse Akt2 in insulin-dependent suppression of adipocyte lipolysis in vivo. Diabetologia 58:1063–1070. doi: 10.1007/s00125-015-3532-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schmitz-Peiffer C, Reeves ML, Denton RM. 1992. Characterization of the cyclic nucleotide phosphodiesterase isoenzymes present in rat epididymal fat cells. Cell Signal 4:37–49. doi: 10.1016/0898-6568(92)90006-T. [DOI] [PubMed] [Google Scholar]
- 28.Coudray C, Charon C, Komas N, Mory G, Diot-Dupuy F, Manganiello V, Ferre P, Bazin R. 1999. Evidence for the presence of several phosphodiesterase isoforms in brown adipose tissue of Zucker rats: modulation of PDE2 by the fa gene expression. FEBS Lett 456:207–210. doi: 10.1016/S0014-5793(99)00934-5. [DOI] [PubMed] [Google Scholar]
- 29.Snyder PB, Esselstyn JM, Loughney K, Wolda SL, Florio VA. 2005. The role of cyclic nucleotide phosphodiesterases in the regulation of adipocyte lipolysis. J Lipid Res 46:494–503. doi: 10.1194/jlr.M400362-JLR200. [DOI] [PubMed] [Google Scholar]
- 30.Zhang X, Carey GB. 2004. Plasma membrane-bound cyclic AMP phosphodiesterase activity in 3T3-L1 adipocytes. Comp Biochem Physiol B Biochem Mol Biol 137:309–316. doi: 10.1016/j.cbpc.2003.12.005. [DOI] [PubMed] [Google Scholar]
- 31.Cristancho AG, Lazar MA. 2011. Forming functional fat: a growing understanding of adipocyte differentiation. Nat Rev Mol Cell Biol 12:722–734. doi: 10.1038/nrm3198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Miyoshi H, Perfield JW, Souza SC, Shen WJ, Zhang HH, Stancheva ZS, Kraemer FB, Obin MS, Greenberg AS. 2007. Control of adipose triglyceride lipase action by serine 517 of perilipin A globally regulates protein kinase A-stimulated lipolysis in adipocytes. J Biol Chem 282:996–1002. doi: 10.1074/jbc.M605770200. [DOI] [PubMed] [Google Scholar]
- 33.Anthonsen MW, Rönnstrand L, Wernstedt C, Degerman E, Holm C. 1998. Identification of novel phosphorylation sites in hormone-sensitive lipase that are phosphorylated in response to isoproterenol and govern activation properties in vitro. J Biol Chem 273:215–221. doi: 10.1074/jbc.273.1.215. [DOI] [PubMed] [Google Scholar]
- 34.Kitamura T, Kitamura Y, Kuroda S, Hino Y, Ando M, Kotani K, Konishi H, Matsuzaki H, Kikkawa U, Ogawa W, Kasuga M. 1999. Insulin-induced phosphorylation and activation of cyclic nucleotide phosphodiesterase 3B by the serine-threonine kinase Akt. Mol Cell Biol 19:6286–6296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lindh R, Ahmad F, Resjö S, James P, Yang JS, Fales HM, Manganiello V, Degerman E. 2007. Multisite phosphorylation of adipocyte and hepatocyte phosphodiesterase 3B. Biochim Biophys Acta 1773:584–592. doi: 10.1016/j.bbamcr.2007.01.010. [DOI] [PubMed] [Google Scholar]
- 36.Palmer D, Jimmo SL, Raymond DR, Wilson LS, Carter RL, Maurice DH. 2007. Protein kinase A phosphorylation of human phosphodiesterase 3B promotes 14-3-3 protein binding and inhibits phosphatase-catalyzed inactivation. J Biol Chem 282:9411–9419. doi: 10.1074/jbc.M606936200. [DOI] [PubMed] [Google Scholar]
- 37.Smith CJ, Vasta V, Degerman E, Belfrage P, Manganiello VC. 1991. Hormone-sensitive cyclic GMP-inhibited cyclic AMP phosphodiesterase in rat adipocytes. Regulation of insulin- and cAMP-dependent activation by phosphorylation. J Biol Chem 266:13385–13390. [PubMed] [Google Scholar]
- 38.Brown MS, Goldstein JL. 2008. Selective versus total insulin resistance: a pathogenic paradox. Cell Metab 7:95–96. doi: 10.1016/j.cmet.2007.12.009. [DOI] [PubMed] [Google Scholar]
- 39.Gonzalez E, Flier E, Molle D, Accili D, McGraw TE. 2011. Hyperinsulinemia leads to uncoupled insulin regulation of the GLUT4 glucose transporter and the FoxO1 transcription factor. Proc Natl Acad Sci U S A 108:10162–10167. doi: 10.1073/pnas.1019268108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Perry RJ, Camporez J-PG, Kursawe R, Titchenell PM, Zhang D, Perry CJ, Jurczak MJ, Abudukadier A, Han MS, Zhang X-M, Ruan H-B, Yang X, Caprio S, Kaech SM, Sul HS, Birnbaum MJ, Davis RJ, Cline GW, Petersen KF, Shulman GI. 2015. Hepatic acetyl CoA links adipose tissue inflammation to hepatic insulin resistance and type 2 diabetes. Cell 160:745–758. doi: 10.1016/j.cell.2015.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Honnor RC, Dhillon GS, Londos C. 1985. cAMP-dependent protein kinase and lipolysis in rat adipocytes. II. Definition of steady-state relationship with lipolytic and antilipolytic modulators. J Biol Chem 260:15130–15138. [PubMed] [Google Scholar]
- 42.Duncan RE, Ahmadian M, Jaworski K, Sarkadi-Nagy E, Sul HS. 2007. Regulation of lipolysis in adipocytes. Annu Rev Nutr 27:79–101. doi: 10.1146/annurev.nutr.27.061406.093734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rahn T, Ridderstråle M, Tornqvist H, Manganiello V, Fredrikson G, Belfrage P, Degerman E. 1994. Essential role of phosphatidylinositol 3-kinase in insulin-induced activation and phosphorylation of the cGMP-inhibited cAMP phosphodiesterase in rat adipocytes. Studies using the selective inhibitor wortmannin. FEBS Lett 350:314–318. [DOI] [PubMed] [Google Scholar]
- 44.Berggreen C, Gormand A, Omar B, Degerman E, Göransson O. 2009. Protein kinase B activity is required for the effects of insulin on lipid metabolism in adipocytes. Am J Physiol Endocrinol Metab 296:E635–E646. doi: 10.1152/ajpendo.90596.2008. [DOI] [PubMed] [Google Scholar]
- 45.Eriksson H, Ridderstråle M, Degerman E, Ekholm D, Smith CJ, Manganiello VC, Belfrage P, Tornqvist H. 1995. Evidence for the key role of the adipocyte cGMP-inhibited cAMP phosphodiesterase in the antilipolytic action of insulin. Biochim Biophys Acta 1266:101–107. doi: 10.1016/0167-4889(94)00237-9. [DOI] [PubMed] [Google Scholar]
- 46.Tan S-X, Fisher-Wellman K, Fazakerley D, Ng Y, Pant H, Li J, Meoli C, Coster ACF, Stöckli J, James DE. 2015. Selective insulin resistance in adipocytes. J Biol Chem 290:11337–11348. doi: 10.1074/jbc.M114.623686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Onuma H, Osawa H, Yamada K, Ogura T, Tanabe F, Granner DK, Makino H. 2002. Identification of the insulin-regulated interaction of phosphodiesterase 3B with 14-3-3 β protein. Diabetes 51:3362–3367. doi: 10.2337/diabetes.51.12.3362. [DOI] [PubMed] [Google Scholar]
- 48.Gabbay RA, Lardy HA. 1985. The antilipolytic effect of insulin does not require adenylate cyclase or phosphodiesterase action. FEBS Lett 179:7–11. doi: 10.1016/0014-5793(85)80179-4. [DOI] [PubMed] [Google Scholar]
- 49.Nishino N, Tamori Y, Kasuga M. 2007. Insulin efficiently stores triglycerides in adipocytes by inhibiting lipolysis and repressing PGC-1α induction. Kobe J Med Sci 53:99–106. [PubMed] [Google Scholar]
- 50.Degerman E, Smith CJ, Tornqvist H, Vasta V, Belfrage P, Manganiello VC. 1990. Evidence that insulin and isoprenaline activate the cGMP-inhibited low-Km cAMP phosphodiesterase in rat fat cells by phosphorylation. Proc Natl Acad Sci U S A 87:533–537. doi: 10.1073/pnas.87.2.533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Omatsu-Kanbe M, Cushman SW, Manganiello VC, Taira M. 1995. Insulin stimulates hormone-sensitive cyclic GMP-inhibited cyclic nucleotide phosphodiesterase in rat brown adipose cells. FEBS Lett 374:187–191. doi: 10.1016/0014-5793(95)01112-R. [DOI] [PubMed] [Google Scholar]
- 52.Egan JJ, Greenberg AS, Chang MK, Londos C. 1990. Control of endogenous phosphorylation of the major cAMP-dependent protein kinase substrate in adipocytes by insulin and beta-adrenergic stimulation. J Biol Chem 265:18769–18775. [PubMed] [Google Scholar]
- 53.Marx SO, Kurokawa J, Reiken S, Motoike H, D'Armiento J, Marks AR, Kass RS. 2002. Requirement of a macromolecular signaling complex for beta adrenergic receptor modulation of the KCNQ1-KCNE1 potassium channel. Science 295:496–499. doi: 10.1126/science.1066843. [DOI] [PubMed] [Google Scholar]
- 54.Dodge-Kafka KL, Soughayer J, Pare GC, Carlisle Michel JJ, Langeberg LK, Kapiloff MS, Scott JD. 2005. The protein kinase A anchoring protein mAKAP coordinates two integrated cAMP effector pathways. Nature 437:574–578. doi: 10.1038/nature03966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hinke SA, Navedo MF, Ulman A, Whiting JL, Nygren PJ, Tian G, Jimenez-Caliani AJ, Langeberg LK, Cirulli V, Tengholm A, Dell'Acqua ML, Santana LF, Scott JD. 2012. Anchored phosphatases modulate glucose homeostasis. EMBO J 31:3991–4004. doi: 10.1038/emboj.2012.244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nilsson R, Ahmad F, Swärd K, Andersson U, Weston M, Manganiello V, Degerman E. 2006. Plasma membrane cyclic nucleotide phosphodiesterase 3B (PDE3B) is associated with caveolae in primary adipocytes. Cell Signal 18:1713–1721. doi: 10.1016/j.cellsig.2006.01.010. [DOI] [PubMed] [Google Scholar]
- 57.Berger K, Lindh R, Wierup N, Zmuda-Trzebiatowska E, Lindqvist A, Manganiello VC, Degerman E. 2009. Phosphodiesterase 3B is localized in caveolae and smooth ER in mouse hepatocytes and is important in the regulation of glucose and lipid metabolism. PLoS One 4:e4671. doi: 10.1371/journal.pone.0004671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Brasaemle DL, Dolios G, Shapiro L, Wang R. 2004. Proteomic analysis of proteins associated with lipid droplets of basal and lipolytically stimulated 3T3-L1 adipocytes. J Biol Chem 279:46835–46842. doi: 10.1074/jbc.M409340200. [DOI] [PubMed] [Google Scholar]
- 59.Cohen AW, Razani B, Schubert W, Williams TM, Wang XB, Iyengar P, Brasaemle DL, Scherer PE, Lisanti MP. 2004. Role of caveolin-1 in the modulation of lipolysis and lipid droplet formation. Diabetes 53:1261–1270. doi: 10.2337/diabetes.53.5.1261. [DOI] [PubMed] [Google Scholar]
- 60.Ahmad F, Lindh R, Tang Y, Ruishalme I, Öst A, Sahachartsiri B, Strålfors P, Degerman E, Manganiello VC. 2009. Differential regulation of adipocyte PDE3B in distinct membrane compartments by insulin and the β3-adrenergic receptor agonist CL316243: effects of caveolin-1 knockdown on formation/maintenance of macromolecular signalling complexes. Biochem J 424:399–410. doi: 10.1042/BJ20090842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ahmad F, Lindh R, Tang Y, Weston M, Degerman E, Manganiello VC. 2007. Insulin-induced formation of macromolecular complexes involved in activation of cyclic nucleotide phosphodiesterase 3B (PDE3B) and its interaction with PKB. Biochem J 404:257–268. doi: 10.1042/BJ20060960. [DOI] [PMC free article] [PubMed] [Google Scholar]