

Abstract
Serum retinol-binding protein 4 (RBP4) is the sole specific transport protein for retinol in the blood, but it is also an adipokine with retinol-independent, proinflammatory activity associated with obesity, insulin resistance, type 2 diabetes, and cardiovascular disease. Moreover, two separate studies reported that patients with proliferative diabetic retinopathy have increased serum RBP4 levels compared to patients with mild or no retinopathy, yet the effect of increased levels of RBP4 on the retina has not been studied. Here we show that transgenic mice overexpressing RBP4 (RBP4-Tg mice) develop progressive retinal degeneration, characterized by photoreceptor ribbon synapse deficiency and subsequent bipolar cell loss. Ocular retinoid and bisretinoid levels are normal in RBP4-Tg mice, demonstrating that a retinoid-independent mechanism underlies retinal degeneration. Increased expression of pro-interleukin-18 (pro-IL-18) mRNA and activated IL-18 protein and early-onset microglia activation in the retina suggest that retinal degeneration is driven by a proinflammatory mechanism. Neither chronic systemic metabolic disease nor other retinal insults are required for RBP4 elevation to promote retinal neurodegeneration, since RBP4-Tg mice do not have coincident retinal vascular pathology, obesity, dyslipidemia, or hyperglycemia. These findings suggest that elevation of serum RBP4 levels could be a risk factor for retinal damage and vision loss in nondiabetic as well as diabetic patients.
INTRODUCTION
Serum retinol-binding protein 4 (RBP4) is the sole specific transport protein for vitamin A (retinol) in the blood (1–4). The eye is the organ most dependent on the RBP4-mediated delivery of retinol to maintain optimal function, as RBP4 loss of function in either mice or humans impairs retinal function, leading to visual impairment, while other organ systems remain intact (5–7). However, in the past decade numerous clinical studies have linked increased serum levels of RBP4 to disease, including obesity (8, 9), insulin resistance (8–13), type 2 diabetes (9, 13), and cardiovascular disease (hypertension, atherosclerosis, stroke) (14–18). This appears to be more than a spurious correlation, since recent studies have demonstrated that RBP4 is an adipokine (adipose-derived cytokine) with retinoid-independent, proinflammatory activity that contributes to the development of insulin resistance (13, 19–21). Moreover, a human single nucleotide polymorphism that increases RBP4 promoter activity confers a 2-fold increase for the risk of type 2 diabetes (22, 23).
Several studies have provided mechanistic insights into RBP4-induced insulin resistance. Mice with genetic or pharmacologic elevation of RBP4 levels develop insulin resistance (13), whereas lowering of RBP4 levels improves insulin sensitivity in mice (13, 24). RBP4 inhibits insulin signaling in adipocytes indirectly by activating proinflammatory cytokine production in macrophages through retinol-independent and Toll-like receptor 4 (TLR4)- and c-Jun N-terminal kinase (JNK)-dependent pathways (19). Transgenic mice overexpressing RBP4 (RBP4-Tg mice) have increased adipose infiltration of macrophages and CD4 T cells through a JNK-dependent pathway that does not involve the RBP4 receptor STRA6 (stimulated by retinoic acid) (20). However, RBP4 can also directly inhibit insulin signaling in adipocytes through the RBP4 receptor STRA6, activating JAK2/STAT5-mediated expression of SOCS3 (suppressor of cytokine signaling), a cytosolic inhibitor of insulin receptor signaling (25, 26). In addition, we have previously shown that RBP4 induces a proinflammatory phenotype in human endothelial cells through a retinol- and STRA6-independent mechanism (27), which could contribute to cardiovascular disease as well as insulin resistance.
The incidences of obesity, insulin resistance, type 2 diabetes, and cardiovascular disease continue to increase, and these diseases are not mutually exclusive, often yielding comorbid conditions. For instance, obesity is a significant risk factor for developing both type 2 diabetes and cardiovascular disease (28–32), and patients with type 2 diabetes have a 2- to 4-fold increased risk for cardiovascular disease (33–35). Moreover, type 2 diabetes is associated with deranged function in several tissues, which can cause debilitating neuropathy, nephropathy, and retinopathy (36–38). In fact, even with good control of blood glucose levels, nearly all type 1 diabetics and over 60% of type 2 diabetics will develop clinically significant retinal disease (diabetic macular edema or diabetic retinopathy) and vision loss within 20 years of diabetes onset (38–40). This statistic highlights the role of additional factors, besides hyperglycemia, that continue to promote tissue dysfunction and damage during diabetes. In nondiabetic patients, hypertension can lead to retinopathy (41–43), while obesity increases the risk for the development and progression of age-related macular (retinal) degeneration (44, 45). Since RBP4 is a proinflammatory adipokine that is often elevated in patients with obesity, cardiovascular disease, and type 2 diabetes, we sought to determine whether the elevation of RBP4 levels could promote retinal dysfunction and/or degeneration.
RBP4−/− mice have retinal dysfunction due to insufficient retinol delivery to the eye (5). RBP4-Tg mice were previously generated and bred with RBP4−/− mice as a means to restore retinol delivery and rescue retinal function (5, 46). However, visual function was not directly measured in RBP4-Tg mice on a wild-type background. Here we show that RBP4-Tg mice develop progressive retinal dysfunction and degeneration, characterized by photoreceptor ribbon synapse deficiency and subsequent bipolar cell loss. Importantly, retinoid and bisretinoid levels are normal in the eyes of RBP4-Tg mice, demonstrating that a retinoid-independent mechanism underlies retinal dysfunction and degeneration. Increased levels of expression of pro-interleukin-18 (pro-IL-18) mRNA and activated (cleaved) IL-18 protein and early-onset microglia activation in the retina suggest that retinal degeneration is driven by a proinflammatory mechanism. Notably, RBP4-Tg mice maintain normal body mass and normal blood glucose, triglyceride, and insulin levels, demonstrating that systemic metabolic dysregulation is not necessary to promote retinal degeneration. These studies reveal that RBP4-Tg mice develop retinal neuroinflammation and neurodegeneration in the absence of retinal vascular pathology, obesity, dyslipidemia, and hyperglycemia. This suggests that elevation of the serum RBP4 level could be a risk factor for retinal damage and vision loss in nondiabetic as well as diabetic patients.
MATERIALS AND METHODS
Mice.
RBP4-Tg mice, which overexpress human RBP4 under the control of the mouse muscle creatine kinase (MCK) promoter on a C57BL/6J background, have been described previously (46) and were kindly provided by Loredana Quadro. All animal studies were approved by the Institutional Animal Care and Use Committee of the University of Oklahoma Health Sciences Center (OUHSC). Mice were fed a standard chow diet (Labdiet 5001) ad libitum. This diet contains a sufficient amount of vitamin A (15 IU g−1). All studies were performed on age-, sex-, and strain-matched wild-type controls. Both sexes were used in all studies, and no sex-dependent differences in phenotype were observed. Importantly, RBP4-Tg mice were confirmed to be free of the retinal degeneration rd8 mutation that is known to be present in some C57BL/6 colonies (data not shown).
Measurements of body mass and blood glucose levels and insulin tolerance tests.
Mice were fasted for 6 h before measurements were taken. A OneTouch Ultra 2 glucose monitor was used to measure blood glucose levels. To assess insulin tolerance, mice were intraperitoneally (i.p.) injected with 0.75 unit/kg of body weight of insulin (Humulin R; Eli Lilly & Company, Indianapolis, IN), and the blood glucose level was measured at 0, 15, 30, 60, 90, and 120 min after injection, as previously described (47).
Serum measurements.
Mouse serum was collected by superficial temporal vein puncture from live mice that had fasted for 6 to 16 h, depending on the endpoint analysis. Enzyme-linked immunosorbent assay (ELISA) kits for mouse adiponectin (catalog no. MRP300; R&D Systems, Minneapolis, MN), mouse insulin (catalog no. 90080; Crystal Chem, Downers Grove, IL), mouse transthyretin (TTR; catalog no. IRKTAH1161; Innovative Research, Novi, MI), human RBP4 (catalog no. DRB400; R&D Systems, Minneapolis, MN), mouse RBP4 (catalog no. MRB400; R&D Systems, Minneapolis, MN), and mouse IL-18 (catalog no. 7625; MBL International Corp., Woburn, MA) were used to quantify the target molecules in serum samples. Serum triglycerides were quantified using a commercial kit (catalog no. ab65336; Abcam, Cambridge, MA).
To quantify the serum retinol level, 100 μl of mouse serum was diluted in 1 ml of 0.025 M KOH, retinoids were extracted in hexane, and the solvent was evaporated under argon gas. Dried retinoid samples were resuspended in 200 μl of mobile-phase buffer (11.2% ethyl acetate, 2.0% dioxane, 1.4% octanol, 85.4% hexane) and injected into the high-performance liquid chromatography (HPLC) machine (515 HPLC pump; Waters Corp., Milford, MA) at an isocratic flow rate of 1 ml/min for separation using a normal-phase 5-μm column (4.6 mm [inner diameter] by 250 mm [length]; Lichosphere SI-60; Alltech, Deerfield, IL). The all-trans-retinol absorbance was monitored at 320 nm. The serum retinol level was quantified from the area of the all-trans-retinol peak using synthetic purified all-trans-retinol as a standard for calibration (Empower software; Waters Corp.).
Quantification of RBP4 and albumin in retina tissue.
Mice were sacrificed by CO2 asphyxiation and perfused with phosphate-buffered saline (PBS) at a flow rate of 8 ml/min for 3 min to remove blood cells. The retinas were rapidly collected using Winkler's method, snap-frozen, and stored at −80°C. The retinas were suspended in lysis buffer [50 mM Tris-HCl, pH 7.8, 0.1 M NaCl, 5 mM EDTA, 0.1% SDS, 1 mM 4-(2-aminoethyl)benzenesulfonyl fluoride hydrochloride, 0.1% Triton X-100, 2.5% glycerol] and sonicated on ice at a 20% intensity for 2 rounds of 5 s each. Insoluble material was removed by centrifugation, and the protein concentration was determined by the Bradford colorimetric assay. ELISA kits for human RBP4 (catalog no. DRB400; R&D Systems, Minneapolis, MN), mouse RBP4 (catalog no. MRB400; R&D Systems, Minneapolis, MN), and mouse albumin (catalog no. 80630; Crystal Chem, Downers Grove, IL) were used to quantify RBP4 and albumin levels in retinal lysates. RBP4 and albumin levels were normalized according to the total retinal protein content in each sample.
OKT analyses.
Visual function was measured using a virtual optokinetic tracking (OKT) system (OptoMotry; CerebralMechanics Inc.) designed for rapid, quantifiable behavioral measurements of spatial vision in a virtual environment (48). All optokinetic tracking experiments were performed using an OptoMotry device designed for rodent use (CerebralMechanics Inc.). The animals were placed inside an enclosed box that was placed on a platform surrounded by four computer monitors forming a square. The monitors display continuous vertical sine wave gratings rotating across the monitors at 12 degrees/s. These appear to the animal to be a virtual three-dimensional rotating cylinder. The animal's ability to visualize the sine wave was monitored via a video camera positioned directly above the animal to display an image perpendicular to the animal's field of vision. The rotation of the virtual cylinder was constantly centered at the animal's viewing position to ensure a consistent viewing distance. Tracking movements were identified as slow, steady head movements in the direction of the rotating grating. For measurements of spatial frequency threshold, the mice were tested at spatial frequencies ranging from 0.034 to 0.664 cycle per degree. The OptoMotry device employs a proprietary algorithm to accept the input from the masked observer and automatically adjusts the testing stimuli on the basis of whether the animal exhibits the correct or incorrect tracking reflex. All measurements of contrast threshold were performed at a spatial frequency threshold of 0.064 cycle per degree. The contrast sensitivity was calculated as a reciprocal of the Michelson contrast from the screen's luminance: (maximum – minimum)/(maximum + minimum).
ERG.
Mice were weighed on the day prior to the electroretinography (ERG) test and dark adapted overnight. At 30 min prior to the test, 1 drop of 1% cyclopentolate hydrochloride ophthalmic solution (Cyclogyl) was delivered to each cornea for pupil dilation. The mice were anesthetized with an intraperitoneal injection of a cocktail of ketamine and xylazine at 85 and 14 mg/kg, respectively. Body temperature was maintained at 37°C using a heating pad. Pupils were dilated a second time with phenylephrine hydrochloride ophthalmic solution (10%) 5 min prior to performing the ERG procedure. Excess fluid was wicked from eyes, whiskers were trimmed, and hypromellose ophthalmic demulcent solution (Gonak; 2.5%) was applied to each cornea. A stainless steel needle electrode was gently hooked into the right cheek to serve as a reference. A stainless steel needle electrode was placed just under the skin of the tail to serve as the ground. Platinum wire electrodes were placed on the corneal surface of each eye, and a drop of saline was added after the baseline was established. A seven-step protocol was run using an Espion E2 system with a Ganzfeld ColorDome system (Diagnosys LLC, Lowell, MA). The first five steps were performed under scotopic conditions and consisted of 5 single flashes of 10-fold increasing flash intensity, starting at a light intensity of 0.004 (S)cd · s/m2 [where (S) refers to scotopic units]. After light adaptation, two separate photopic readings were taken at light intensities of 3 (S)cd · s/m2 and 10 (S)cd · s/m2 using a 10-(S)cd · m−2 background light; for each photopic light intensity reading, the readings from 15 flashes were averaged.
Quantitative histological analyses of retina.
A flamed needle was used to scorch the superior side of the cornea to demarcate the vertical meridian, and the eyes were enucleated with part of the optic nerve still attached. The eyes were fixed in 4% paraformaldehyde for 24 h, subsequently transferred into PBS, and then dehydrated and paraffin embedded for sectioning along the superior-inferior retinal axis. Sections were deparaffinized and stained with hematoxylin and eosin, and images were scanned at ×40 magnification on a Ventana Coreo Au slide scanner (Tissue Sciences Facility, University of Nebraska Medical Center). Scanned images underwent quantitative analysis using Ventana image viewer software (Ventana Medical Systems Inc., Tucson, AZ). Beginning at the optic nerve head and extending into the retinal periphery in 250-μm increments, the thickness of the total retina, outer nuclear layer (ONL), and inner nuclear layer (INL) was measured. In the case of the ONL and INL, the number of cell nuclei spanning each layer was counted to alleviate differences that may arise simply due to fixation or histological processing.
Quantification of visual cycle retinoids and bisretinoids A2E and iso-A2E in mouse eyes by HPLC.
For quantification of visual cycle retinoids, mice were dark adapted overnight for 16 h and sacrificed in the dark under dim red light, and the eyes were enucleated and stored at −80°C with protection from light. All further sample processing and HPLC were performed in the dark under a dim red light. The whole eyes were homogenized with a glass grinder in lysis buffer [10 mM NH2OH, 50% ethanol, 50% 2-(N-morpholino)ethanesulfonic acid, pH 6.5], and retinoids were extracted with hexane. Solvent was evaporated under argon gas, and dried retinoid samples were resuspended in 200 μl of mobile phase (11.2% ethyl acetate, 2.0% dioxane, 1.4% octanol, 85.4% hexane) and injected into the HPLC machine (515 HPLC pump; Waters Corp., Milford, MA) at an isocratic flow rate of 1 ml/min for separation using a normal-phase 5-μm column (Lichosphere SI-60; 4.6 mm [inner diameter] by 250 mm [length]; Alltech, Deerfield, IL). Each retinoid isomer was quantified from the area of its corresponding peak, determined by using synthetic purified retinoid standards for calibration (Empower software; Waters Corp.).
For quantification of the bisretinoids N-retinylidene-N-retinylethanolamine (A2E) and iso-A2E, mice were reared under normal cyclic light and fed standard chow ad libitum. At 9 months of age, the mice were sacrificed, their eyes were enucleated, and the eyecup (retinal pigment epithelium [RPE], sclera, and choroid) was dissected out. A total of 4 eyecups (from 2 mice of the same genotype and sex) were pooled to generate a single sample for HPLC measurement. The eyecups were stored at −80°C until analysis. Samples were homogenized in a glass grinder in PBS, and bisretinoids were extracted three times using chloroform-methanol (2:1; vol/vol). The pooled organic phases were dried under a stream of argon gas and resuspended in 100 μl of methanol. A2E and iso-A2E were separated using a mobile-phase gradient of methanol in water (85 to 96% methanol and 0.1% trifluoroacetic acid) through a C18 reverse-phase column (particle size, 3.5 μm; 4.6 by 150 mm; Waters, Milford, MA) at a flow rate of 1 ml/min. A Waters 2996 photodiode array detector was used to monitor the absorbance of A2E and iso-A2E at 430 nm. A2E and iso-A2E levels were quantified from the area of each peak using purified A2E and iso-A2E standards for calibration (Empower software; Waters Corp.).
Immunohistochemistry.
Mouse eyes were enucleated, and posterior eyecups were dissected and fixed in 4% paraformaldehyde for 2 h. The eyecups were cryoprotected in graded sucrose solutions (10 to 30% in PBS), embedded in optimal cutting temperature compound, and snap-frozen in liquid nitrogen. Cryosections of the retina were cut to a thickness of 8 μm and processed for immunohistochemistry as previously described (49). Briefly, retinal cryosections were coated in blocking solutions (10% goat or donkey serum, 5% bovine serum albumin, 1% fish gelatin, and 0.5% Triton X-100 in PBS) for 1 h before incubation overnight at 4°C in primary antibodies diluted in blocking buffer. After the sections were washed in PBS, they were incubated with appropriate fluorophore-conjugated secondary antibodies for 2 h at room temperature. The sections were then rinsed with PBS, Vectashield mounting medium (Vector Laboratories, Burlingame, CA) and DAPI (4′,6-diamidino-2-phenylindole) were applied, a cover slip was placed over the sections, and the sections were imaged with a Leica SP2 MP confocal microscope (Leica Microsystems, Buffalo Grove, IL).
For labeling of the retina and RPE whole mounts, posterior eyecups were dissected and fixed in 4% paraformaldehyde for 30 min. The entire neural retina was carefully peeled off from the RPE-choroid and sclera. Retina and RPE whole mounts were permeabilized separately in 1% Triton X-100–PBS overnight at 4°C and blocked in blocking buffer (5% goat serum and 0.5% Triton X-100 in PBS) overnight at 4°C. The retina and RPE whole mounts were then incubated for 2 days on a shaker at 4°C in primary antibodies dissolved in 0.1% Triton X-100 with 1% goat serum in PBS. After they were washed in PBS, the retina and RPE whole mounts were incubated with fluorophore-conjugated secondary antibodies for 2 h at room temperature. The whole mounts were then washed in PBS, coated in Vectashield mounting medium, flat mounted, and viewed and imaged using a Leica SP2 MP confocal microscope.
The following antibodies were used: from Enzo Life Sciences, bassoon (catalog no. SAP7F407); from Cedarlane Labs, ribeye (catalog no. 192103); from EMD Millipore, calbindin (catalog no. ab1778), NeuN (catalog no. MAB377), cone arrestin (catalog no. AB15282), S-opsin (catalog no. ab5407), M-opsin (catalog no. ab5405), rhodopsin (catalog no. MAB5356), VGLUT1 (catalog no. ab5905), glutamine synthetase (catalog no. mab302), glial fibrillary acidic protein (GFAP; catalog no. mab3402), GLYT1 (catalog no. AB1770), and ZO-1 (catalog no. AB2272); from Abcam, beta-III tubulin (catalog no. ab18207); from Santa Cruz Biotechnology, protein kinase C alpha (PKC-alpha; catalog no. sc-208); from Biovendor, secretagogin (catalog no. RD 184120100); from Life Technologies, calretinin (catalog no. 18-0291); from Leico Technologies, CD11b (catalog no. C227); from Wako, Iba-1 (catalog no. 019-19741); from the Developmental Studies Hybridoma Bank at the University of Iowa, CD31 (catalog no. 2H8-c); from Exalpha, Chx10 (catalog no. X1179P); and from the Zebrafish International Resource Center, ZNP-1 (catalog no. ZDB-ATB-081002-25).
Funduscopy and fluorescein angiography.
Mice were sedated by i.p. administration of ketamine and xylazine at 85 and 14 mg/kg, respectively. Following sedation, the pupils were dilated by topical administration of cyclopentolate and phenylephrine ophthalmic solution (Cyclomydril). The fundus of the sedated animals was observed and recorded using a Micron III small-animal funduscope (Phoenix Research). Standard color fundus photos were first captured for each eye without any exciter/barrier filters. The mice were then administered 10% fluorescein sodium at 1 μl/g of body weight via i.p. injection. Within 5 min of fluorescein administration, exciter/barrier filters for a target wavelength of 488 nm were employed to capture fluorescence-driven fundus photographs.
Retinal leukostasis assays.
Retinal leukostasis was measured by labeling the retinal vasculature and adherent leukocytes with fluorescein isothiocyanate-conjugated concanavalin A (FITC-ConA; Vector Laboratories, Burlingame, CA) according to a published method (50). The animals were deeply anesthetized by intraperitoneal injection of the cocktail of ketamine (85 mg/kg) and xylazine (14 mg/kg). Then, the chest cavity was opened and the descending aorta was clamped. A 25-gauge cannula was inserted into the left ventricle, and drainage was achieved by opening the right atrium. The animals were perfused with PBS containing 0.1 mg/ml heparin at a constant flow rate of 8 ml/min for 2 min to wash out erythrocytes and nonadherent leukocytes. Next, the animals were perfused with FITC-ConA at 500 μg/ml in PBS at 4 ml/min for 2 min to label the adherent leukocytes and vascular endothelial cells. Residual unbound FITC-ConA was removed by perfusion with PBS containing 0.1 mg/ml heparin at a constant flow rate of 8 ml/min for 2 min. The retinas were carefully dissected, fixed with 4% paraformaldehyde, and flat mounted. The retinas were then visualized using an Olympus BX-60 fluorescence microscope (Olympus, Tokyo, Japan), and the total number of adherent leukocytes per retina was determined.
Confocal and 3D imaging.
A Leica SP2 MP confocal microscope (Leica Microsystems, Wetzlar, Germany) was used for imaging of the retinal cryosections and whole mounts. Cryosection images were acquired using a 63× oil immersion objective, and each z-series was taken at 0.5-μm intervals. Image stacks were processed using Leica Application Suite advanced fluorescence 3 (LAS AF 3) software. Retina and RPE whole-mount multiplane z-series were collected using a 40× or 60× oil immersion objective. Each z-series spanned 50 μm in depth and comprised 100 optical sections taken at 0.5-μm intervals. Microglial cell numbers in retinal whole mounts stained with CD11b or Iba-1 antibodies were counted, using z-series stacks that encompassed the inner plexus versus the outer plexus separately and that were collected using a 40× oil immersion objective. A single image (magnification, ×40; corresponding surface area, 0.14 mm2) from the midperiphery of each retinal whole mount was randomly selected and used for counting 3 to 5 independent retinas from each age-genotype group.
Image stacks were processed with an image analysis software package (Volocity; PerkinElmer, Waltham, MA) to generate a 3-dimensional (3D) rendering of the in vivo spatial distribution and activation of microglia. The z-stack images were processed for 3D data visualization using the same image analysis software package.
Quantitative reverse transcription-PCR (qRT-PCR).
Retinas were dissected and immediately submerged in the TRIzol reagent (Life Technologies, Carlsbad, CA) for storage at −80°C. Thawed samples were homogenized with a motorized pestle, and RNA was extracted using an Ambion PureLink RNA minikit (Life Technologies). RNA was reverse transcribed to generate cDNA. Quantitative real-time PCR was performed in triplicate for each cDNA sample using SYBR green (Invitrogen) as previously described (47). The primer sequences are as follows: (i) for IL-18, forward (Fwd) primer 5′-GTCTGTGATGCCCTTAGATG-3′ and reverse (Rev) primer 5′-AGCTTATGACCCGCACTTAC-3′; (ii) for IL-1β, Fwd primer 5′-TGGGCCTCAAAGGAAAGA-3′ and Rev primer 5′-GGTGCTGATGTACCAGTT-3′; (iii) for Nlrp3, Fwd primer 5′-ATTACCCGCCCGAGAAAGG-3′ and Rev primer 5′-TCGCAGCAAAGATCCACACAG-3′; (iv) for GAPDH (glyceraldehyde-3-phosphate dehydrogenase), Fwd primer 5′-ATTGTCAGCAATGCATCCTG-3′ and Rev primer 5′-ATGGACTGTGGTCATGAGCC-3′; (v) for retinoic acid receptor beta (RARβ), Fwd primer 5′-GGACCTTGAGGAACCAACAA-3′ and Rev primer 5′-TGGCATTGATCCAGGAATTT-3′; and (vi) for hypoxanthine phosphoribosyltransferase (HPRT), Fwd primer 5′-GCAAACTTTGCTTTCCCTGGTT-3′ and Rev primer 5′-CAAGGGCATATCCAACAACA-3′. The relative RNA amount was calculated with the 2-ΔΔCT threshold cycle (CT) method and normalized to the amount of GAPDH or HPRT as the internal control. At least 3 distinct biological samples from mice of each genotype were examined.
Western blotting.
Retinal lysate and serum protein concentrations were determined by Bradford analysis, and equal amounts of protein lysates were loaded in each lane of SDS-polyacrylamide gels for Western blotting. Immunoblotting for actin (retina lysates) or albumin (serum) was performed on each Western blot to confirm equal protein loading. The following antibodies were used: from R&D Systems Inc., IL-1β (catalog no. AF-401-NA); from Sigma-Aldrich, actin-horseradish peroxidase (catalog no. A3854); and from BioVision, IL-18 (catalog no. 5180R).
Caspase-1 activity assay on retina lysates.
Caspase-1 activity was measured according to the manufacturer's instructions (catalog no. AB39470; Abcam). Briefly, each retina was homogenized in the lysis buffer provided in the kit, sonicated on ice at a 20% intensity for two 5-s bursts, and centrifuged at 10,000 × g for 1 min at 4°C. The supernatant was collected, and its protein concentration was determined using the Bradford assay. Fifty microliters of supernatant from each sample was transferred to a clear-bottom 96-well plate, 50 μl of reaction buffer (containing 10 mM dithiothreitol) and 5 μl of 4 mM YVAD–p-nitroanilide (YVAD-pNA) substrate were added, and the mixture was incubated at 37°C for 1 h. Active human caspase-1 (catalog no. ab39901; Abcam) was used as a positive control and to set the standard curve for caspase-1 activity units. Caspase-1 activity was measured by reading the plate at 405 nm. Absorbance values were converted to caspase-1 activity units and adjusted for the total amount of protein per sample.
RESULTS
RBP4-Tg mice are a model for studying the effects of increased serum RBP4 levels in the context of normal serum glucose, insulin, and lipid levels.
Total serum RBP4 levels in RBP4-Tg mice were initially 10-fold higher than those in the wild-type controls, although serum RBP4 levels decreased with age in RBP4-Tg mice, likely due to transgene silencing; nonetheless, RBP4-Tg mice maintained a ∼6-fold increase in serum total RBP4 levels by 9 months of age (Fig. 1A). Serum all-trans-retinol levels increased 3.8-fold compared to those in the wild type, and the majority of RBP4 likely circulates as holo-RBP4 (retinol bound), on the basis of a serum RBP4/retinol molar ratio of 0.79 (Table 1). Serum transthyretin (TTR) levels were significantly increased, albeit by a modest amount (25%) (Fig. 1B), and TTR levels followed the same trend as serum RBP4 levels by decreasing with aging, but unlike RBP4 levels, serum TTR levels were nonsignificantly increased by 6 months of age (data not shown). RBP4-Tg mice that were fed standard chow ad libitum maintained a normal body mass and normal fasting levels of serum glucose (Fig. 1C to F), as well as normal levels of insulin, triglyceride, and adiponectin (Fig. 1G to I). These results are similar to those presented in a previous report (20). In contrast to previous reports, insulin tolerance tests showed no change in insulin sensitivity in RBP4-Tg mice (Fig. 1J and K). Thus, RBP4-Tg mice represent a model for studying the physiologic consequences of specifically increasing serum RBP4 levels in the absence of systemic insulin resistance, hyperglycemia, hyperinsulinemia, or hyperlipidemia and their confounding comorbidities.
FIG 1.

RBP4-Tg mice have 10- to 6-fold increases in serum RBP4 levels and maintain a normal body mass and fasting blood glucose, serum insulin, and triglyceride levels. (A) Total (mouse and human transgene) RBP4 levels in mouse serum (n ≥ 4 mice per age-genotype combination; WT, wild type). (B) TTR levels in mouse serum at 2 months of age (n ≥ 5 mice per genotype; data were analyzed by Student's t test). (C and D) Blood glucose levels in mice that had fasted for 6 h (n ≥ 6 mice per measurement). (E and F) Body mass measurements in mice that had fasted for 6 h (n ≥ 6 mice per measurement). (G) Insulin levels in the sera of 9-month-old mice that had fasted for 6 h (no significant difference between genotypes was detected on the basis of Student's t test). (H) Triglyceride levels in serum from 4-month-old mice that had fasted for 6 h (no significant difference between genotypes was detected on the basis of Student's t test). (I) Adiponectin levels in serum from 4-month-old mice that had fasted for 6 h (no significant difference between genotypes was detected on the basis of Student's t test). (J and K) Results of insulin tolerance tests in which male mice aged 17 to 21 weeks (n = 7 to 8 mice per genotype) were fasted for 6 h and then given a single i.p. injection of insulin (0.75 unit/kg) at time zero, followed by blood glucose measurements at 0, 15, 30, 60, 90, and 120 min postinjection. All graph values are means ± SEMs. **, P < 0.01 by Student's t test; ***, P < 0.001 by Student's t test.
TABLE 1.
Mouse serum levels of retinol and RBP4a
| Mouse | Retinol concn (μM) | RBP4 concn (μg/ml) |
Total RBP4 concn (μM) | RBP4/retinol molar ratio | ||
|---|---|---|---|---|---|---|
| Mouse | Human | Total | ||||
| Wild type | 1.87 ± 0.35 | 17.9 ± 0.8 | 0 | 17.9 ± 0.8 | 0.85 ± 0.04 | 0.46 |
| RBP4-Tg | 7.11 ± 0.36 | 18.2 ± 1.6 | 100.4 ± 8.9 | 118.6 ± 10.5 | 5.65 ± 0.47 | 0.79 |
Serum from the same set of male mice aged 10 weeks was used to measure retinol and RBP4 levels by HPLC and ELISA, respectively. Values are means ± standard deviations for ≥5 mice per genotype.
Visual acuity and retinal function progressively decline with age in RBP4-Tg mice.
To understand whether RBP4 may have direct or indirect effects on the retina, we investigated whether RBP4-Tg mice have increased levels of RBP4 in retinal tissue. PBS perfusion removed most blood from retinal tissue, as determined by an ∼6-fold reduction in the albumin concentration in perfused versus nonperfused retinas (data not shown), and albumin levels were similar in perfused retinas from wild-type and RBP4-Tg mice (Fig. 2B). We found that RBP4-Tg mice had significant increases in total RBP4 levels and the RBP4/albumin ratio in the retina compared to those in the retinas of the wild-type controls (Fig. 2A to C), suggesting that RBP4 may directly target retinal tissue.
FIG 2.

Visual acuity is significantly reduced in RBP4-Tg mice. (A to C) RBP4 and albumin levels in perfused retinas from mice (WT, wild type) aged 6 months (n ≥ 5 mice per genotype); (D and E) OKT response to changes in spatial frequency; (F and G) OKT response to changes in grating contrast. All OKT analyses were performed on ≥6 mice per genotype. Values are means ± SEMs. n.s., not significant by Student's t test; *, P < 0.05 by Student's t test; **, P < 0.01 by Student's t test; ***, P < 0.001 by Student's t test.
To assess visual acuity and contrast sensitivity, we performed optokinetic tracking (OKT) experiments, which clearly showed that RBP4-Tg mice have a significant reduction in both visual contrast and spatial frequency thresholds as early as 1 month of age (Fig. 2D and F). The OKT response was more severely reduced by 6 months of age (Fig. 2E and G). Since reductions in visual acuity and contrast vision can result from dysfunction in a number of different visual system pathways (i.e., cataracts, brain perception, retina), we performed electroretinography (ERG) analyses to directly measure retinal function in response to light stimuli. Under scotopic (dark-adapted) conditions, RBP4-Tg mice showed a significant reduction in the ERG inner retinal response (b wave), which began by 1 month of age and progressively worsened with aging (Fig. 3B and D). Likewise, under photopic (light-adapted) conditions, RBP4-Tg mice showed a significant reduction in the ERG inner retinal response (b wave) by 1 month of age, which continued to decline with aging (Fig. 3C). The scotopic photoreceptor response (a wave) was not significantly reduced until 6 months of age but declined with aging thereafter (Fig. 3A). These data suggest that RBP4-Tg mice develop an early dominant dysfunction in inner retinal signaling leading to secondary photoreceptor dysfunction.
FIG 3.

RBP4-Tg mice have dominant and progressive inner retinal dysfunction leading to secondary photoreceptor dysfunction. (A and B) Scotopic ERG a-wave and b-wave amplitudes in mice aged 1 to 9 months (WT, wild type). (C) Photopic ERG b-wave amplitudes in mice aged 1 to 9 months. Values are means ± SEMs for ≥6 mice per age-genotype combination. n.s., not significant by one-way analysis of variance (ANOVA) with Tukey's post hoc test; *, P < 0.05 by one-way ANOVA with Tukey's post hoc test; **, P < 0.01 by one-way ANOVA with Tukey's post hoc test; ***, P < 0.001 by one-way ANOVA with Tukey's post hoc test. (D) Representative scotopic ERG responses to increasing light intensities (0.004 to 40 cd · s/m2) in mice aged 6 months. Note the pronounced b-wave decline compared to the more subtle a-wave reduction.
Retinal degeneration coincides with the progressive loss of retinal function in RBP4-Tg mice.
Quantitative histological analyses were performed to measure retinal thickness. RBP4-Tg mice had normal retinal development and thickness at 1 month of age (Fig. 4A) but significant thinning of both the outer and inner nuclear layers of the retina, which are comprised of photoreceptor and second-order neuron cell bodies, respectively, by 6 months of age (Fig. 4B). Second-order neurons of the inner nuclear layer are responsible for generating and modulating the ERG b-wave response. In RBP4-Tg mice, thinning of the retinal inner nuclear layer was the most significant and coincided with the predominant ERG b-wave deficit (Fig. 3). Immunohistochemical analyses of retinal cryosections demonstrated that the expression of key rod and cone phototransduction proteins appeared to be normal in RBP4-Tg mice (Fig. 4C to F), even at 6 months of age. Taken together, these data suggest that photoreceptor cell integrity is largely intact and that the second-order neurons of the INL preferentially degenerate in RBP4-Tg mice.
FIG 4.

RBP4-Tg mice have age-dependent inner retinal degeneration. (A and B) Quantitative histological analyses of the total retina, INL, and ONL thickness for retinas from mice aged 1 month (A) or 6 months (B) (WT, wild-type; ONH, optic nerve head). Values are means ± SEMs for ≥8 mice for each data point. *, P < 0.05 by one-way analysis of variance (ANOVA) with Tukey's post hoc test; **, P < 0.01 by one-way ANOVA with Tukey's post hoc test; ***, P < 0.001 by one-way ANOVA with Tukey's post hoc test. (C to E) Representative confocal images of retinal paraffin sections from 9-month-old mice immunofluorescently labeled for rhodopsin (C), cone S-opsin (D), cone M-opsin (E). (F) Representative confocal images of retinal cryosections from 6-month-old mice immunofluorescently labeled for cone arrestin. DAPI counterstaining is shown in blue. Confocal images were acquired under a 63× magnification objective, but rhodopsin images are shown with an additional digital zoom of ×2. All images shown represent the maximum stack of z-plane acquisition. All images were acquired from central retina sections and are representative of those from 4 to 6 independent samples from mice of each genotype. Bars, 10 μm (C to E) and 20 µm (F).
Retinas of RBP4-Tg mice have normal visual cycle retinoids and bisretinoid A2E levels.
Since RBP4 is primarily recognized for its role in transporting and delivering retinoids to target tissues (1–4), including its specific importance for the delivery of optimal levels of retinol to the retinal pigment epithelium for metabolism by the retinoid visual cycle (5–7), we measured the levels of each major retinoid isomer of the visual cycle in the retina. Retinal levels of 11-cis-retinal, all-trans-retinal, and retinyl ester were not significantly different between RBP4-Tg and wild-type mice, with the exception of a slight but significant increase in the level of the visual chromophore 11-cis-retinal in RBP4-Tg mouse retinas at 2 months of age (Fig. 5A to C). A modest increase in the level of a visual chromophore such as this has not been associated with any form of retinal degeneration and does not explain the retinal degeneration observed in RBP4-Tg mice.
FIG 5.

RBP4-Tg mice have normal levels of retinal visual cycle retinoids and bisretinoids A2E and iso-A2E. (A to C) HPLC-based quantification of visual cycle retinoid isomers in eyes from 2- to 11-month-old mice dark adapted for 16 h prior to euthanization. Values are means ± SEMs for 4 mice per genotype-age combination. ns, not significant. P values were determined by Student's t test. (D) HPLC-based quantification of bisretinoid A2E and iso-A2E levels in eyecups from 9-month-old mice reared in cyclic room light. Values are means ± SEMs for 4 samples (4 eyecups per sample from 8 mice total) per genotype; n.s., not significant by Student's t test. (E) Quantitative RT-PCR was performed to measure RARβ mRNA levels in retinas from mice aged 3 months. Values are means ± SEMs for 3 samples per group. ns, not significant by Student's t test.
Retinoid visual cycle activity is known to contribute to the generation of by-products that are nondegradable and toxic to cells (51, 52). These retinoid-derived by-products slowly accumulate within the subretinal space and retinal pigment epithelium (RPE) during normal aging but can accumulate more rapidly in some types of retinal degeneration, in which toxic retinoid derivatives have been shown to promote retinal degeneration (51–55). Despite no change in the levels of retinoid isomers of the visual cycle itself, we reasoned that it was possible that RBP4-Tg mice have increased retinol uptake that promotes excessive visual cycle activity leading to the increased accumulation of toxic retinoid by-products of the visual cycle, such as the bisretinoid N-retinylidene-N-retinylethanolamine (A2E), the most abundant toxic retinoid derivative associated with retinal degeneration (51, 52). Therefore, we measured A2E levels in mouse eyecups (RPE, sclera, choroid). There was no significant increase in the level of A2E or its isomer, iso-A2E, in the eyecups of RBP4-Tg mice compared to that in the eyecups of the wild-type controls at 9 months of age (Fig. 5D). Moreover, RBP4-Tg mice had no observable deposits or RPE atrophy upon retinal fundus exam (Fig. 6A), and the RPE pigmentation (Fig. 6B and C) and tight junctions (Fig. 6D) remained intact.
FIG 6.
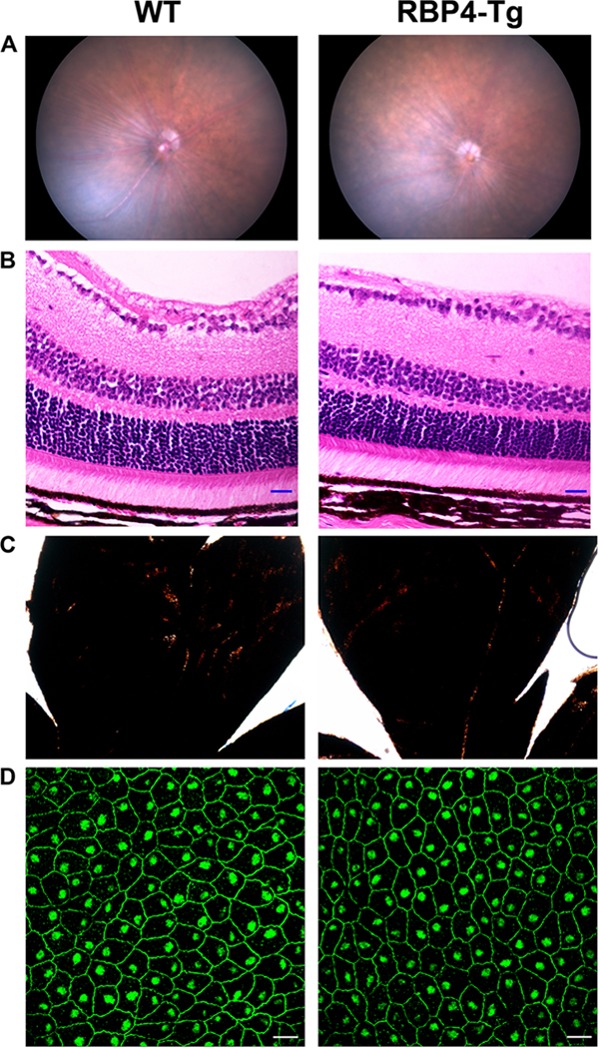
RBP4-Tg mice have normal retinal stratification and RPE pigmentation and integrity. (A) Representative retinal fundus images of wild-type (WT) and RBP4-Tg mice aged 6 months. (B) Hematoxylin- and eosin-stained retinal sections from mice aged 6 months. Magnifications, ×40; bars, 20 μm. (C) Bright-field images of retinal whole mounts showing normal RPE pigmentation in 6-month-old mice. Magnifications, ×4. (D) Representative confocal images of RPE whole mounts from 6-month-old mice immunofluorescently labeled for ZO-1 to demarcate tight junctions. Confocal images were acquired under a 63× magnification objective and represent the maximum stack of z-plane acquisition. Bars, 20 μm. The images in panels B and D were acquired from central retina sections. All images are representative of those from 4 to 6 independent samples from mice of each genotype.
To address the possibility that RBP4-Tg mouse retinas have increased levels of retinoic acid, we measured the level of expression of mRNA for the retinoic acid receptor beta (RARβ) gene, which is itself a retinoic acid target gene whose mRNA shows increased expression in response to retinoic acid treatment (56–60). There was no significant difference in RARβ mRNA levels between wild-type and RBP4-Tg mouse retinas (Fig. 5E), suggesting that retinoic acid signaling is unchanged in RBP4-Tg mouse retinas. These data indicate that retinoid metabolism is largely unchanged in RBP4-Tg mice and that retinal degeneration is retinoid independent.
Loss of retinal synaptic connectivity and reduced bipolar cell density in RBP4-Tg mice.
Since RBP4-Tg mice have predominant INL thinning, we performed immunohistochemistry on retinal cryosections to evaluate the integrity of each major cell type of the INL. At 6 months of age, there was significant bipolar cell loss, and both rod and cone bipolar cell types were affected (Fig. 7A to C). All other INL cell subtypes, including amacrine, horizontal, and Müller cells, appeared normal (Fig. 7D to G). Retinal whole-mount staining also showed no loss of retinal ganglion cells or their axons (Fig. 7H).
FIG 7.

RBP4-Tg mice have an age-related loss of rod and cone bipolar cells. (A to G) Representative confocal images of retinal cryosections from 6-month-old mice immunofluorescently labeled for distinct INL cell types, as follows: all bipolar cells (green, Chx10) and rod bipolar cells (red, PKC-α) (A), rod bipolar cells (PKC-α) (B), cone bipolar cells (secretagogin) (C), calcium-binding amacrine and ganglion cells (calretinin) (D), glycinergic amacrine cells (GLYT1) (E), horizontal cells (calbindin) (F), and Müller cells (glutamine synthetase [GS]) (G). (H) Representative confocal images of retinal whole mounts immunolabeled with NeuN (green) to stain retinal ganglion cell bodies and beta-III tubulin (red) to stain ganglion axonal processes. In each panel, the image on the left is from a wild-type mouse retina and the image on the right is from an RBP4-Tg mouse retina. Confocal images were acquired under a 63× (A to G) or 40× (H) magnification objective, but the secretagogin (C) and calbindin (F) images are shown with an additional digital zoom of ×2. All images shown represent the maximum stack of z-plane acquisition. All images were acquired from central retina sections and are representative of those from 3 to 7 independent samples from mice of each genotype. Bars, 10 μm.
To further evaluate retinal integrity, we investigated retinal synaptic connectivity by immunohistochemical labeling of retinal cryosections. Presynaptic photoreceptor ribbon synapses were significantly reduced in RBP4-Tg mice, as shown by reduced labeling for ribeye and bassoon and the loss of a horseshoe-like morphology (Fig. 8A and B). Bipolar (PKC-α) and horizontal (calbindin) cell postsynaptic processes were normal (Fig. 8C and D). Vesicular glutamate transporter 1 (VGLUT1) labeling in photoreceptors was abnormal, showing trails of VGLUT1 that localized within the ONL layer instead of at the synaptic outer plexiform layer (Fig. 8E). ZNP-1 labeling revealed a significant loss of cone bipolar cell terminals at the synaptic inner plexiform layer by 6 months of age (Fig. 8E). A similar loss of rod bipolar cell terminals was also observed at 6 months of age (Fig. 7A and B). Additional analyses showed that rod and cone bipolar cell numbers and the terminal morphology were initially normal at 1 month of age (data not shown). The degradation of rod and cone bipolar cell synapses chronologically coincided with reduced rod and cone bipolar cell density at 6 months of age (Fig. 7A to C), suggesting that cell loss accounts for the loss of bipolar cell terminals. In contrast, there was already a noticeable deficiency of the presynaptic photoreceptor ribbon synapses at 1 month of age (Fig. 8A), an age at which there is no measurable photoreceptor degeneration or a loss of intrinsic photoreceptor function, according to ERG a-wave recordings (Fig. 3A and 4A). Taken together, these data show that the earliest observable retinal abnormality is deficient photoreceptor ribbon synapse formation at 1 month of age, which likely contributes to subsequent rod and cone bipolar cell loss.
FIG 8.

RBP4-Tg mice have deficiencies in photoreceptor ribbon synapses and cone bipolar terminals. (A to E) Representative confocal images of retinal cryosections from 1-month-old (A) or 6-month-old (B to E) mice (WT, wild type) immunolabeled for presynaptic photoreceptor ribbon synapses (red, bassoon; green, ribeye) (A and B), photoreceptor ribbon synapse connection to postsynaptic rod bipolar cell dendrites (red, bassoon; green, PKC-α) (C), photoreceptor ribbon synapse connection to postsynaptic horizontal cell dendrites (red, bassoon; green, calbindin) (D), and photoreceptor presynaptic terminals (red, VLGUT1) and cone bipolar axonal and dendritic terminals (green, ZNP-1) (E). Confocal images were acquired under a 63× magnification objective, but panels A to C are shown with an additional digital zoom of ×4, and panel D is shown with an additional zoom of ×6. All images shown represent the maximum stack of z-plane acquisition. DAPI counterstaining is shown in blue. Arrowheads, displaced VGLUT1; arrows, thinning or loss of ZNP-1 labeling. All images were acquired from central retina sections and are representative of those from 4 to 8 independent samples from mice of each genotype. Bars, 10 μm.
RBP4-Tg mice have normal retinal vasculature.
Since RBP4-Tg mice have a primary inner retinal degeneration and we have previously shown that RBP4 has proinflammatory activity in human retinal capillary endothelial cells (27), we investigated whether RBP4-Tg mice have an abnormal retinal vasculature, including retinal leukostasis, that may contribute to inner retinal dysfunction. Fluorescein angiography showed that RBP4-Tg mice have a normal retinal vascular tree with no signs of vascular leakage (Fig. 9A). Retinal leukostasis assays were performed to assess retinal vascular inflammation characteristic of retinopathy. There was no significant difference in retinal leukostasis between genotypes, and both wild-type and RBP4-Tg mice had higher levels of retinal leukostasis at 1 month of age than at 6 months of age (Fig. 9B and C).
FIG 9.

RBP4-Tg mice have normal retinal vasculature. (A) Fluorescein angiogram images from wild-type (WT) and RBP4-Tg mice aged 6 months. Each image shown is from a different mouse. (B) Epifluorescent images from whole-mount retinas following a retinal leukostasis procedure in which mice were perfused with FITC-concanavalin A to label adherent leukocytes. Arrows, adherent leukocytes. Magnification, ×20. (C) Quantification of retinal leukostasis. Values are means ± SEMs for ≥3 mice (6 eyes) per genotype-age combination. There was no statistically significant difference between genotypes by two-way analysis of variance.
RBP4-Tg mice have early-onset retinal gliosis and microglia activation.
To investigate the mechanisms underlying retinal degeneration, we performed immunohistochemical labeling of retinas for the gliosis stress marker glial fibrillary acidic protein (GFAP) and the microglia markers CD11b and Iba-1. GFAP expression was noticeably increased in both astrocytes and Müller cells as early as 1 month of age (Fig. 10A), and gliosis was widespread throughout Müller cell bodies by 6 months of age (Fig. 10B). CD11b staining of retinal whole mounts showed that RBP4-Tg mice had a thickening of microglia cell bodies, which is characteristic of an activated state, by 1 month of age (Fig. 10C and D), and the activated microglia morphology was maintained at 6 months of age (Fig. 11A and B), at which time retinal microglia cell numbers were increased in RBP4-Tg mouse retinas (Fig. 10E and F). 3D confocal processing also showed that microglia processes often extend into the ONL in RBP4-Tg mice, a phenomenon that was never observed in wild-type mouse retinas (Fig. 11C and D).
FIG 10.

RBP4-Tg mice have early-onset retinal gliosis and microglia activation. (A and B) Representative confocal images of retinal cryosections from 1-month-old mice (A) and 6-month-old (B) mice immunofluorescently labeled for GFAP, a marker of gliosis (WT, wild type). DAPI counterstaining is shown in blue. Confocal images were acquired under a 40× magnification objective and represent the maximum stack of z-plane acquisition. All images were acquired from central retina sections and are representative of those from 3 to 5 independent samples from mice of each genotype. Bars, 10 μm. (C and D) Representative confocal images of retinal whole mounts from 1-month-old mice immunofluorescently labeled for microglia marker CD11b. Confocal images were acquired under a 40× magnification objective, and each image represents a single layer of z-plane acquisition. Arrows, microglia cells with enlarged cell bodies characteristic of activated microglia. All images were acquired from the midperiphery and are representative of those from 3 to 6 independent samples from mice of each genotype. Bars, 40 μm. (E and F) Microglia cell bodies were counted in retinal whole mounts under a 40× magnification objective. Each field of view corresponds to a surface area of 0.14 mm2. Microglia were counted in 3 to 5 retinas per genotype. Values are means ± SEMs. **, P < 0.01 by Student's t test.
FIG 11.

Microglia projections extend into the outer nuclear layer in RBP4-Tg mice. (A and B) Representative confocal images of retinal whole mounts from 6-month-old mice immunofluorescently labeled for the microglia marker Iba-1. Confocal images were acquired under a 63× magnification objective, and each image represents the z-series of the inner plexus layer (GCL, ganglion cell layer). Numbered boxes denote cells that are shown at higher magnification to the right side of the original image. All images were acquired from the midperiphery and are representative of those from 3 to 5 independent samples from mice of each genotype. Note the enlarged cell bodies in the microglia of RBP4-Tg mouse retinas. Bars, 40 μm. (C and D) Representative 3D confocal images of retinal whole mounts from 6-month-old mice immunofluorescently labeled for CD31 (red) and either Iba-1 (green) (C) or CD11b (green) (D). All images were acquired from the midperiphery and are representative of those from 3 to 5 independent samples from mice of each genotype. Note the microglia extensions into the ONL in the retinas of RBP4-Tg mice only.
RBP4-Tg mice have retinal neuroinflammation.
To investigate whether neuroinflammation could be driving retinal degeneration, we performed quantitative PCR analyses to look for changes in retinal proinflammatory cytokine expression. We found a significant increase in IL-18 mRNA levels in the retinas of RBP4-Tg mice compared to those of wild-type mice (Fig. 12A). Interestingly, although IL-18 and IL-1β are often coactivated during neuroinflammation (61), we did not find a similar increase in IL-1β mRNA levels but instead found a significant reduction (Fig. 12B). Western blotting for protein levels also showed an increase in activated (cleaved) IL-18 protein, but no activation of IL-1β protein (Fig. 12C to E). The IL-18 protein level was also significantly increased in the serum of RBP4-Tg mice (Fig. 12F), indicating ongoing systemic inflammation, in addition to retinal inflammation. Most typically, IL-18 and IL-1β are activated by inflammasome-mediated caspase-1 cleavage of their pro-forms to generate the cleaved active forms (61). However, the mRNA level of the key inflammasome component NLRP3 was significantly decreased in RBP4-Tg mouse retinas (Fig. 12H); moreover, there was no increase in caspase-1 activity in the retinas of RBP4-Tg mice compared to those of wild-type mice (Fig. 12G). This suggests that IL-18 activation is inflammasome and caspase-1 independent, a novel phenomenon recently reported by others (62, 63). It is interesting that both IL-1β and NLRP3 mRNA levels were significantly decreased in RBP4-Tg mouse retinas, and it is unclear if this was a direct response to RBP4 or occurred as a compensatory response following RBP4-mediated induction of alternative inflammatory signaling pathways.
FIG 12.

RBP4-Tg mice have retinal neuroinflammation. qRT-PCR analyses showed increased levels of pro-IL-18 (A) and reduced levels of pro-IL-1β (B) mRNA expression in retinas from 6-month-old RBP4-Tg mice. (C) Western blots show no retinal expression of mature IL-1β in RBP4-Tg or wild-type mice at age 3 months. Lane +C, 1 μg recombinant mouse IL-1β as a positive control. The upper band near 45 kDa is β-actin, while the lower band near 15 kDa is mature IL-1β. (D and E) Western blot and densitometry (densitomet) analyses show significantly increased expression of mature IL-18 in retinas from RBP4-Tg mice at age 6 months. Densitometry is from 3 independent experiments consisting of 3 mice per genotype. (F) Serum IL-18 levels measured by ELISA. (G) The enzymatic activity of caspase-1 in lysates of retinas from 6-month-old mice was determined by incubation with the colorimetric substrate YVAD-pNA. (H) qRT-PCR analyses show decreased levels of NLRP3 mRNA expression in retinas from 6-month-old RBP4-Tg mice. Values are means ± SEMs for 4 to 6 mice per genotype. n.s., not significant by Student's t test; *, P < 0.05 by Student's t test; **, P < 0.01 by Student's t test; ***, P < 0.001 by Student's t test.
DISCUSSION
RBP4 is a proinflammatory adipokine associated with obesity (8, 9), insulin resistance (8–13), type 2 diabetes (9, 13), and cardiovascular disease (14–18). Moreover, a genetic or pharmacologic increase in the serum levels of RBP4 induces insulin resistance in mice (13). Obesity, diabetes, and cardiovascular disease can each lead to dysfunction in multiple organ systems, including the retina (38, 40–45), which can cause permanent vision loss, yet it is unknown whether increased serum levels of RBP4 contribute to the development of such comorbidities. Our study provides the first evidence that elevated levels of serum RBP4 can contribute to retinal neuroinflammation and neurodegeneration, even in the absence of obesity, dyslipidemia, or hyperglycemia.
The absolute level of RBP4 measured in patient serum varies between different clinical studies according to the methodology used for quantification, which includes Western blotting, various commercial ELISA kits, and mass spectrometry (reviewed in reference 64). Therefore, there is a rather broad definition of normal/healthy serum RBP4 levels, which range from 10 to 50 μg/ml, depending on the methodology used in a specific study. Likewise, the relative increase in the serum RBP4 level associated with various disease states, such as obesity, insulin resistance, cardiovascular disease, and type 2 diabetes, can range from 17 to 150 μg/ml (8, 9, 11–17, 64–67). In one particular study (11), about half of the patient samples in which RBP4 levels were measured had serum RBP4 levels above 100 μg/ml, which is similar to the serum RBP4 level in young RBP4-Tg mice (Fig. 1A). However, the same study reported normal patient serum RBP4 levels of 40 μg/ml, indicating that there was a maximum ∼4-fold increase in the serum RBP4 level. Therefore, since the RBP4-Tg mouse model had a 6- to 10-fold increase in serum RBP4 levels compared to those in wild-type mice, this model may be physiologically relevant only to extreme cases of long-duration obesity, insulin resistance, or type 2 diabetes. Nonetheless, it is important to remember that in chronic diseases, such as insulin resistance, cardiovascular disease, and type 2 diabetes, a multitude of factors contributes to disease progression. Some factors have independent activity that directly contributes to disease pathogenesis, whereas others must act in concert to produce significant effects. In order to reveal the potential effects of one specific factor, such as RBP4, a supraphysiological dose may be necessary. The present study is striking in that it shows that increased levels of RBP4 alone, without any additional insult, can result in very significant retinal degeneration and vision loss. This warrants further investigations to understand how a more physiologically relevant 2- to 4-fold increase in serum RBP4 levels may impact the progression of retinal diseases, including diabetic retinopathy, when patients have a combination of other pathological factors contributing to disease progression.
In contrast to two previous reports (13, 20), our results show no significant reduction in insulin sensitivity in RBP4-Tg mice compared to that in wild-type mice. In one of the previous reports, RBP4-Tg mice had reduced insulin sensitivity when 12-week-old male mice (n = 7 per group) were dosed with 0.9 U/kg insulin by i.p. injection 4 h after food removal (13). In the second report, RBP4-Tg mice had reduced insulin sensitivity when 12- to 19-week-old male mice (n = 13 to 15 per group) were dosed with 0.8 U/kg insulin by i.p. injection 5 h after food removal (20). However, in the second study the reduction in insulin sensitivity in RBP4-Tg mice was less pronounced and significant (P < 0.05 in the second study [20] compared to P < 0.01 in the first study [13]). Moreover, similar to our results, no other research group has reported fasting hyperglycemia in RBP4-Tg mice. In our study, 17- to 21-week-old male mice (n = 7 to 8 mice per group) were dosed with 0.75 U/kg insulin by i.p. injection 6 h after food removal. Since we used an established procedure (47) and evaluated mice of the same sex and age as the mice used in the previous studies, it is difficult to explain why we found no significant difference in insulin sensitivity, except that there seems to be variability in the severity of systemic insulin resistance in RBP4-Tg mice, according to the results of the previous studies (13, 20). In addition, previous studies have shown that the proinflammatory activity of RBP4 indirectly inhibits insulin signaling in adipocytes (19, 20), so that RBP4 primarily acts to promote inflammation, which secondarily inhibits insulin signaling. Therefore, variability in the degree of the inflammatory response in a particular animal may account for the variability in the systemic insulin response, whereas RBP4-induced inflammation in localized tissues, such as the retina, may have a less variable and more potent effect to perturb tissue homeostasis and function.
We show that retinoid and bisretinoid A2E levels are normal in the eyes of RBP4-Tg mice, suggesting that a retinoid-independent mechanism underlies retinal dysfunction and degeneration. Although there was a slight trend toward higher A2E levels in RBP4-Tg mice, this was below the level of significance, and the levels of A2E were 4- to 12-fold below the level of A2E measured in the eyecups of abcr−/− and mutant Elovl4-Tg mice, which have retinal degeneration driven by A2E accumulation (53–55). Additionally, mice with A2E-associated retinal degeneration have very significant A2E accumulation beginning as early as 2 months of age (53–55), whereas the relatively low level of A2E in the eyecups of RBP4-Tg mice at the older age of 9 months further suggests that A2E accumulation is not a factor contributing to retinal dysfunction and degeneration that occurs between 1 and 6 months of age. Furthermore, A2E from the visual cycle accumulates within lipid-rich deposits in the retinal pigment epithelium layer and consequently contributes to outer retinal (RPE and photoreceptor) degeneration (53, 68, 69). In contrast, RBP4-Tg mice have a predominant inner retinal degeneration.
Previous studies have shown that RBP4-Tg mice have a measurable level of insulin resistance, on the basis of the results of insulin and glucose tolerance tests (13, 20). Here we show that RBP4-Tg mice do not develop hyperglycemia and do not always have detectable systemic insulin resistance, yet they consistently develop progressive retinal degeneration. Thus, elevation of RBP4 levels can cause deranged function and damage in specific peripheral tissues well before the onset of chronic and severe systemic metabolic disease. Similar observations have been made in human studies, which show that subclinical symptoms and insulin resistance are often present for several years prior to the onset of clinical diabetes. In fact, although diabetic retinopathy is clinically monitored, characterized, and treated according to the presence and severity of retinal vascular pathology, multiple studies in animals and humans have documented significant retinal neuronal dysfunction and degeneration in diabetes, which often precedes any detectable changes in retinal vasculature (70–73). Interestingly, retinal neuronal dysfunction and degeneration in RBP4-Tg mice are similar to those in diabetic patients with retinopathy, in that both show predominantly inner rather than outer retinal dysfunction and degeneration (70, 71, 73, 74). On the basis of our findings, it would be interesting to evaluate whether serum RBP4 levels in diabetic patients correlate with subclinical retinal neuronal dysfunction.
Insulin receptor signaling in the retina is neuroprotective (75–78), and blocking retinal insulin receptor signaling causes retinal dysfunction and degeneration in mice (79, 80). Retinal insulin resistance contributes to diabetic retinopathy in rodents with streptozotocin (STZ)-induced diabetes, in which insulin receptor signaling is downregulated (81) and systemic or ocular injection of insulin significantly reduced proinflammatory gene expression and neurodegeneration (70, 82, 83). Intriguingly, disruption of insulin receptor signaling in cone photoreceptors causes disorganized and reduced photoreceptor ribbon synapses (79), similar to the types observed in RBP4-Tg mice. Likewise, insulin-deficient diabetic mice also develop a progressive loss of photoreceptor ribbon synapses (84) and decreased expression of presynaptic proteins (85). Since an elevation of RBP4 levels can inhibit insulin receptor signaling in adipose tissue, it is plausible that an elevation of RBP4 levels could similarly disrupt insulin receptor signaling in the retina as an underlying mechanism contributing to neuronal dysfunction and degeneration in RBP4-Tg mice, and this should be a subject for future investigation.
Neuroinflammation is a major contributing factor in several neurodegenerative diseases, including Alzheimer's disease (86) and Parkinson's disease (87–89), as well as retinal degeneration (90, 91). Neuroinflammation is characterized by elevation of proinflammatory cytokine levels and activation of microglia (92, 93). There are multiple reports implicating inflammasomes in mediating neuroinflammation (reviewed in references 94 and 95). Inflammasomes activate proinflammatory caspase-1, which then cleaves precursor forms of the IL-1 family of proinflammatory cytokines into their active forms (61). We show that IL-18 but not IL-1β mRNA expression was increased in the retinas of RBP4-Tg mice. Likewise, the level of active (cleaved) IL-18 protein was increased, while IL-1β was nearly undetectable in RBP4-Tg mouse retinas. Although IL-18 and IL-1β are typically coregulated, there are recent reports of inflammasome- and caspase-1-independent activation of IL-18 without a coincident activation of IL-1β (62, 63). We also show that retinas from RBP4-Tg mice had no increase in the mRNA or protein level of the major inflammasome subtype NLRP3 and no increase in caspase-1 activity. Thus, the mechanism of IL-18 protein activation in RBP4-Tg mice is likely inflammasome and caspase-1 independent.
The cellular source of IL-18 in the retina of RBP4-Tg mice is unknown; however, microglia cells, which are the cellular source of IL-18 and other cytokines in brain neurodegenerative diseases (94), possess an activated morphology in the retinas of RBP4-Tg mice. Furthermore, microglia express Toll-like receptor 4 (TLR4) (96, 97), which is involved in the pathogenesis of different types of retinal degeneration (91, 96–99). TLR4 activation has been shown to induce IL-18 mRNA and protein activation in renal tubular epithelial cells during renal obstruction (100). Since RBP4 acts through a TLR4-dependent mechanism to induce proinflammatory cytokine expression in adipose tissue macrophages (19), RBP4 may similarly activate TLR4 signaling in retinal microglia to release IL-18, but this is yet to be determined.
It was somewhat surprising to find retinal neuronal dysfunction in the absence of retinal vascular pathology in RBP4-Tg mice, on the basis of our previous finding that high levels of RBP4 can induce the expression of proinflammatory cytokines and cell adhesion molecules to promote leukostasis in primary human retinal capillary endothelial cells (27). In addition, two relatively small clinical studies reported a modest increase in serum RBP4 levels in human patients with proliferative diabetic retinopathy, the most advanced vascular disease stage, compared to those in diabetic patients with mild or no retinopathy (66, 67). Since cultured cells can have altered properties compared to their properties in their in vivo state, it may be that culturing conditions primed endothelial cells to respond pathologically to increasing RBP4 levels in our previous study. Interestingly, hyperglycemia increases TLR4 expression in endothelial cells (101) and the retinas of rats with STZ-induced diabetes (99), which could prime the endothelium to respond to proinflammatory levels of RBP4 during diabetes. Further studies are necessary to determine whether the elevation of RBP4 levels exacerbates retinal vascular inflammation in a mouse model of diabetic retinopathy, such as mice with db/db- or STZ-induced diabetes.
In conclusion, this study demonstrates that RBP4-Tg mice develop progressive retinal degeneration through a retinoid-independent mechanism. Retinal microglia activation and increased levels of the active proinflammatory cytokine IL-18 protein indicate that neuroinflammation promotes retinal degeneration in RBP4-Tg mice. Neither chronic systemic metabolic disease nor other retinal insults are required for elevated RBP4 levels to promote retinal neurodegeneration, since RBP4-Tg mice do not have coincident retinal vascular pathology, obesity, dyslipidemia, or hyperglycemia. These findings suggest that elevated serum RBP4 levels could be a risk factor for retinal damage and vision loss in nondiabetic as well as diabetic patients.
ACKNOWLEDGMENTS
We thank Loredana Quadro for providing RBP4-Tg mice and David Sherry and Michael Elliott for recommendations for immunohistochemistry. We thank the following core facilities for their assistance with tissue processing: the University of Oklahoma Health Sciences Center (OUHSC) CoBRE Diabetes Histology Core and the Oklahoma Medical Research Foundation Imaging Core. We thank the OUHSC CoBRE Diabetic Animal Core for providing extra wild-type mice and tissues as needed for method development. We thank the OUHSC Flow Cytometry and Imaging Core for providing access and support in using the confocal microscope. We thank the OUHSC Vision Core facility for providing access and support in using the core ERG equipment.
The OUHSC Vision Core facility is supported by National Eye Institute core grant EY021725. The research reported in this study was supported by an institutional development award (IDeA) from the National Institute of General Medical Sciences of the National Institutes of Health under grant number P20GM104934 (K.M.F. received funding as a promising junior investigator on this grant), a macular degeneration research grant from the BrightFocus Foundation (M2012057; principal investigator, K.M.F.), and an American Heart Association beginning grant-in-aid (13BGIA16920097; principal investigator, K.M.F.).
REFERENCES
- 1.Kanai M, Raz A, Goodman DS. 1968. Retinol-binding protein: the transport protein for vitamin A in human plasma. J Clin Invest 47:2025–2044. doi: 10.1172/JCI105889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Raz A, Shiratori T, Goodman DS. 1970. Studies on the protein-protein and protein-ligand interactions involved in retinol transport in plasma. J Biol Chem 245:1903–1912. [PubMed] [Google Scholar]
- 3.Vahlquist A, Nilsson SF, Peterson PA. 1971. Isolation of the human retinol binding protein by affinity chromatography. Eur J Biochem 20:160–168. doi: 10.1111/j.1432-1033.1971.tb01374.x. [DOI] [PubMed] [Google Scholar]
- 4.Blomhoff R, Blomhoff HK. 2006. Overview of retinoid metabolism and function. J Neurobiol 66:606–630. doi: 10.1002/neu.20242. [DOI] [PubMed] [Google Scholar]
- 5.Quadro L, Blaner WS, Salchow DJ, Vogel S, Piantedosi R, Gouras P, Freeman S, Cosma MP, Colantuoni V, Gottesman ME. 1999. Impaired retinal function and vitamin A availability in mice lacking retinol-binding protein. EMBO J 18:4633–4644. doi: 10.1093/emboj/18.17.4633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Biesalski HK, Frank J, Beck SC, Heinrich F, Illek B, Reifen R, Gollnick H, Seeliger MW, Wissinger B, Zrenner E. 1999. Biochemical but not clinical vitamin A deficiency results from mutations in the gene for retinol binding protein. Am J Clin Nutr 69:931–936. [DOI] [PubMed] [Google Scholar]
- 7.Seeliger MW, Biesalski HK, Wissinger B, Gollnick H, Gielen S, Frank J, Beck S, Zrenner E. 1999. Phenotype in retinol deficiency due to a hereditary defect in retinol binding protein synthesis. Invest Ophthalmol Vis Sci 40:3–11. [PubMed] [Google Scholar]
- 8.Aeberli I, Biebinger R, Lehmann R, L'Allemand D, Spinas GA, Zimmermann MB. 2007. Serum retinol-binding protein 4 concentration and its ratio to serum retinol are associated with obesity and metabolic syndrome components in children. J Clin Endocrinol Metab 92:4359–4365. doi: 10.1210/jc.2007-0468. [DOI] [PubMed] [Google Scholar]
- 9.Graham TE, Yang Q, Bluher M, Hammarstedt A, Ciaraldi TP, Henry RR, Wason CJ, Oberbach A, Jansson PA, Smith U, Kahn BB. 2006. Retinol-binding protein 4 and insulin resistance in lean, obese, and diabetic subjects. N Engl J Med 354:2552–2563. doi: 10.1056/NEJMoa054862. [DOI] [PubMed] [Google Scholar]
- 10.Bremer AA, Devaraj S, Afify A, Jialal I. 2011. Adipose tissue dysregulation in patients with metabolic syndrome. J Clin Endocrinol Metab 96:E1782–E1788. doi: 10.1210/jc.2011-1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kowalska I, Straczkowski M, Adamska A, Nikolajuk A, Karczewska-Kupczewska M, Otziomek E, Gorska M. 2008. Serum retinol binding protein 4 is related to insulin resistance and nonoxidative glucose metabolism in lean and obese women with normal glucose tolerance. J Clin Endocrinol Metab 93:2786–2789. doi: 10.1210/jc.2008-0077. [DOI] [PubMed] [Google Scholar]
- 12.Mostafaie N, Sebesta C, Zehetmayer S, Jungwirth S, Huber KR, Hinterberger M, Leitha T, Hofman J, Hejtman M, Schrattbauer K, Krugluger W, Tragl KH, Fischer P. 2011. Circulating retinol-binding protein 4 and metabolic syndrome in the elderly. Wien Med Wochenschr 161:505–510. doi: 10.1007/s10354-011-0885-7. [DOI] [PubMed] [Google Scholar]
- 13.Yang Q, Graham TE, Mody N, Preitner F, Peroni OD, Zabolotny JM, Kotani K, Quadro L, Kahn BB. 2005. Serum retinol binding protein 4 contributes to insulin resistance in obesity and type 2 diabetes. Nature 436:356–362. doi: 10.1038/nature03711. [DOI] [PubMed] [Google Scholar]
- 14.Bobbert T, Raila J, Schwarz F, Mai K, Henze A, Pfeiffer AF, Schweigert FJ, Spranger J. 2010. Relation between retinol, retinol-binding protein 4, transthyretin and carotid intima media thickness. Atherosclerosis 213:549–551. doi: 10.1016/j.atherosclerosis.2010.07.063. [DOI] [PubMed] [Google Scholar]
- 15.Ingelsson E, Sundstrom J, Melhus H, Michaelsson K, Berne C, Vasan RS, Riserus U, Blomhoff R, Lind L, Arnlov J. 2009. Circulating retinol-binding protein 4, cardiovascular risk factors and prevalent cardiovascular disease in elderly. Atherosclerosis 206:239–244. doi: 10.1016/j.atherosclerosis.2009.02.029. [DOI] [PubMed] [Google Scholar]
- 16.Sasaki M, Otani T, Kawakami M, Ishikawa SE. 2010. Elevation of plasma retinol-binding protein 4 and reduction of plasma adiponectin in subjects with cerebral infarction. Metabolism 59:527–532. doi: 10.1016/j.metabol.2009.08.015. [DOI] [PubMed] [Google Scholar]
- 17.Solini A, Santini E, Madec S, Rossi C, Muscelli E. 2009. Retinol-binding protein-4 in women with untreated essential hypertension. Am J Hypertens 22:1001–1006. doi: 10.1038/ajh.2009.116. [DOI] [PubMed] [Google Scholar]
- 18.Bobbert P, Weithauser A, Andres J, Bobbert T, Kuhl U, Schultheiss HP, Rauch U, Skurk C. 2009. Increased plasma retinol binding protein 4 levels in patients with inflammatory cardiomyopathy. Eur J Heart Fail 11:1163–1168. doi: 10.1093/eurjhf/hfp153. [DOI] [PubMed] [Google Scholar]
- 19.Norseen J, Hosooka T, Hammarstedt A, Yore MM, Kant S, Aryal P, Kiernan UA, Phillips DA, Maruyama H, Kraus BJ, Usheva A, Davis RJ, Smith U, Kahn BB. 2012. Retinol-binding protein 4 inhibits insulin signaling in adipocytes by inducing proinflammatory cytokines in macrophages through a c-Jun N-terminal kinase- and Toll-like receptor 4-dependent and retinol-independent mechanism. Mol Cell Biol 32:2010–2019. doi: 10.1128/MCB.06193-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Moraes-Vieira PM, Yore MM, Dwyer PM, Syed I, Aryal P, Kahn BB. 2014. RBP4 activates antigen-presenting cells, leading to adipose tissue inflammation and systemic insulin resistance. Cell Metab 19:512–526. doi: 10.1016/j.cmet.2014.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Deng ZB, Poliakov A, Hardy RW, Clements R, Liu C, Liu Y, Wang J, Xiang X, Zhang S, Zhuang X, Shah SV, Sun D, Michalek S, Grizzle WE, Garvey T, Mobley J, Zhang HG. 2009. Adipose tissue exosome-like vesicles mediate activation of macrophage-induced insulin resistance. Diabetes 58:2498–2505. doi: 10.2337/db09-0216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.van Hoek M, Dehghan A, Zillikens MC, Hofman A, Witteman JC, Sijbrands EJ. 2008. An RBP4 promoter polymorphism increases risk of type 2 diabetes. Diabetologia 51:1423–1428. doi: 10.1007/s00125-008-1042-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Munkhtulga L, Nagashima S, Nakayama K, Utsumi N, Yanagisawa Y, Gotoh T, Omi T, Kumada M, Zolzaya K, Lkhagvasuren T, Kagawa Y, Fujiwara H, Hosoya Y, Hyodo M, Horie H, Kojima M, Ishibashi S, Iwamoto S. 2010. Regulatory SNP in the RBP4 gene modified the expression in adipocytes and associated with BMI. Obesity (Silver Spring) 18:1006–1014. doi: 10.1038/oby.2009.358. [DOI] [PubMed] [Google Scholar]
- 24.Zemany L, Bhanot S, Peroni OD, Murray SF, Moraes-Vieira PM, Castoldi A, Manchem P, Guo S, Monia BP, Kahn BB. 2015. Transthyretin antisense oligonucleotides lower circulating RBP4 levels and improve insulin sensitivity in obese mice. Diabetes 64:1603–1604. doi: 10.2337/db14-0970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Berry DC, Jin H, Majumdar A, Noy N. 2011. Signaling by vitamin A and retinol-binding protein regulates gene expression to inhibit insulin responses. Proc Natl Acad Sci U S A 108:4340–4345. doi: 10.1073/pnas.1011115108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Berry DC, Noy N. 2012. Signaling by vitamin A and retinol-binding protein in regulation of insulin responses and lipid homeostasis. Biochim Biophys Acta 1821:168–176. doi: 10.1016/j.bbalip.2011.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Farjo KM, Farjo RA, Halsey S, Moiseyev G, Ma JX. 2012. Retinol-binding protein 4 induces inflammation in human endothelial cells by an NADPH oxidase- and nuclear factor kappa B-dependent and retinol-independent mechanism. Mol Cell Biol 32:5103–5115. doi: 10.1128/MCB.00820-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mathew B, Francis L, Kayalar A, Cone J. 2008. Obesity: effects on cardiovascular disease and its diagnosis. J Am Board Fam Med 21:562–568. doi: 10.3122/jabfm.2008.06.080080. [DOI] [PubMed] [Google Scholar]
- 29.Rexrode KM, Manson JE, Hennekens CH. 1996. Obesity and cardiovascular disease. Curr Opin Cardiol 11:490–495. doi: 10.1097/00001573-199609000-00007. [DOI] [PubMed] [Google Scholar]
- 30.Sowers JR. 1998. Obesity and cardiovascular disease. Clin Chem 44:1821–1825. [PubMed] [Google Scholar]
- 31.Sowers JR. 2003. Obesity as a cardiovascular risk factor. Am J Med 115(Suppl 8A):37S–41S. [DOI] [PubMed] [Google Scholar]
- 32.Lastra G, Sowers JR. 2013. Obesity and cardiovascular disease: role of adipose tissue, inflammation, and the renin-angiotensin-aldosterone system. Horm Mol Biol Clin Investig 15:49–57. doi: 10.1515/hmbci-2013-0025. [DOI] [PubMed] [Google Scholar]
- 33.Beckman JA, Creager MA, Libby P. 2002. Diabetes and atherosclerosis: epidemiology, pathophysiology, and management. JAMA 287:2570–2581. doi: 10.1001/jama.287.19.2570. [DOI] [PubMed] [Google Scholar]
- 34.Kannel WB, McGee DL. 1979. Diabetes and cardiovascular disease. The Framingham Study. JAMA 241:2035–2038. [DOI] [PubMed] [Google Scholar]
- 35.Kannel WB, McGee DL. 1979. Diabetes and glucose tolerance as risk factors for cardiovascular disease: the Framingham Study. Diabetes Care 2:120–126. doi: 10.2337/diacare.2.2.120. [DOI] [PubMed] [Google Scholar]
- 36.Fong DS, Aiello L, Gardner TW, King GL, Blankenship G, Cavallerano JD, Ferris FL III, Klein R. 2004. Retinopathy in diabetes. Diabetes Care 27(Suppl 1):S84–S87. doi: 10.2337/diacare.27.2007.S84. [DOI] [PubMed] [Google Scholar]
- 37.Stratton IM, Adler AI, Neil HA, Matthews DR, Manley SE, Cull CA, Hadden D, Turner RC, Holman RR. 2000. Association of glycaemia with macrovascular and microvascular complications of type 2 diabetes (UKPDS 35): prospective observational study. BMJ 321:405–412. doi: 10.1136/bmj.321.7258.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fong DS, Aiello LP, Ferris FL III, Klein R. 2004. Diabetic retinopathy. Diabetes Care 27:2540–2553. doi: 10.2337/diacare.27.10.2540. [DOI] [PubMed] [Google Scholar]
- 39.Fong DS, Aiello L, Gardner TW, King GL, Blankenship G, Cavallerano JD, Ferris FL III, Klein R. 2003. Diabetic retinopathy. Diabetes Care 26(Suppl 1):S99–S102. doi: 10.2337/diacare.26.2007.S99. [DOI] [PubMed] [Google Scholar]
- 40.Wong TY, Liew G, Tapp RJ, Schmidt MI, Wang JJ, Mitchell P, Klein R, Klein BE, Zimmet P, Shaw J. 2008. Relation between fasting glucose and retinopathy for diagnosis of diabetes: three population-based cross-sectional studies. Lancet 371:736–743. doi: 10.1016/S0140-6736(08)60343-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang S, Xu L, Jonas JB, Wang YS, Wang YX, You QS, Yang H, Zhou JQ. 2012. Five-year incidence of retinal microvascular abnormalities and associations with arterial hypertension: the Beijing Eye Study 2001/2006. Ophthalmology 119:2592–2599. doi: 10.1016/j.ophtha.2012.06.031. [DOI] [PubMed] [Google Scholar]
- 42.Scheie HG. 1952. Retinal changes associated with hypertension and arteriosclerosis. Ill Med J 101:126–129. [PubMed] [Google Scholar]
- 43.Scheie HG. 1953. Evaluation of ophthalmoscopic changes of hypertension and arteriolar sclerosis. AMA Arch Ophthalmol 49:117–138. doi: 10.1001/archopht.1953.00920020122001. [DOI] [PubMed] [Google Scholar]
- 44.Munch IC, Linneberg A, Larsen M. 2013. Precursors of age-related macular degeneration: associations with physical activity, obesity, and serum lipids in the inter99 eye study. Invest Ophthalmol Vis Sci 54:3932–3940. doi: 10.1167/iovs.12-10785. [DOI] [PubMed] [Google Scholar]
- 45.Seddon JM, Cote J, Davis N, Rosner B. 2003. Progression of age-related macular degeneration: association with body mass index, waist circumference, and waist-hip ratio. Arch Ophthalmol 121:785–792. doi: 10.1001/archopht.121.6.785. [DOI] [PubMed] [Google Scholar]
- 46.Quadro L, Blaner WS, Hamberger L, Van Gelder RN, Vogel S, Piantedosi R, Gouras P, Colantuoni V, Gottesman ME. 2002. Muscle expression of human retinol-binding protein (RBP). Suppression of the visual defect of RBP knockout mice. J Biol Chem 277:30191–30197. [DOI] [PubMed] [Google Scholar]
- 47.Wang Q, Xie Z, Zhang W, Zhou J, Wu Y, Zhang M, Zhu H, Zou MH. 2014. Myeloperoxidase deletion prevents high-fat diet-induced obesity and insulin resistance. Diabetes 63:4172–4185. doi: 10.2337/db14-0026. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 48.Prusky GT, Alam NM, Beekman S, Douglas RM. 2004. Rapid quantification of adult and developing mouse spatial vision using a virtual optomotor system. Invest Ophthalmol Vis Sci 45:4611–4616. doi: 10.1167/iovs.04-0541. [DOI] [PubMed] [Google Scholar]
- 49.Du M, Wu M, Fu D, Yang S, Chen J, Wilson K, Lyons TJ. 2013. Effects of modified LDL and HDL on retinal pigment epithelial cells: a role in diabetic retinopathy? Diabetologia 56:2318–2328. doi: 10.1007/s00125-013-2986-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ishida S, Usui T, Yamashiro K, Kaji Y, Ahmed E, Carrasquillo KG, Amano S, Hida T, Oguchi Y, Adamis AP. 2003. VEGF164 is proinflammatory in the diabetic retina. Invest Ophthalmol Vis Sci 44:2155–2162. doi: 10.1167/iovs.02-0807. [DOI] [PubMed] [Google Scholar]
- 51.Liu J, Itagaki Y, Ben-Shabat S, Nakanishi K, Sparrow JR. 2000. The biosynthesis of A2E, a fluorophore of aging retina, involves the formation of the precursor, A2-PE, in the photoreceptor outer segment membrane. J Biol Chem 275:29354–29360. doi: 10.1074/jbc.M910191199. [DOI] [PubMed] [Google Scholar]
- 52.Sparrow JR, Fishkin N, Zhou J, Cai B, Jang YP, Krane S, Itagaki Y, Nakanishi K. 2003. A2E, a byproduct of the visual cycle. Vision Res 43:2983–2990. doi: 10.1016/S0042-6989(03)00475-9. [DOI] [PubMed] [Google Scholar]
- 53.Karan G, Lillo C, Yang Z, Cameron DJ, Locke KG, Zhao Y, Thirumalaichary S, Li C, Birch DG, Vollmer-Snarr HR, Williams DS, Zhang K. 2005. Lipofuscin accumulation, abnormal electrophysiology, and photoreceptor degeneration in mutant ELOVL4 transgenic mice: a model for macular degeneration. Proc Natl Acad Sci U S A 102:4164–4169. doi: 10.1073/pnas.0407698102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Radu RA, Mata NL, Bagla A, Travis GH. 2004. Light exposure stimulates formation of A2E oxiranes in a mouse model of Stargardt's macular degeneration. Proc Natl Acad Sci U S A 101:5928–5933. doi: 10.1073/pnas.0308302101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Weng J, Mata NL, Azarian SM, Tzekov RT, Birch DG, Travis GH. 1999. Insights into the function of Rim protein in photoreceptors and etiology of Stargardt's disease from the phenotype in abcr knockout mice. Cell 98:13–23. doi: 10.1016/S0092-8674(00)80602-9. [DOI] [PubMed] [Google Scholar]
- 56.Balmer JE, Blomhoff R. 2002. Gene expression regulation by retinoic acid. J Lipid Res 43:1773–1808. doi: 10.1194/jlr.R100015-JLR200. [DOI] [PubMed] [Google Scholar]
- 57.de The H, Marchio A, Tiollais P, Dejean A. 1989. Differential expression and ligand regulation of the retinoic acid receptor alpha and beta genes. EMBO J 8:429–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.de The H, Vivanco-Ruiz MM, Tiollais P, Stunnenberg H, Dejean A. 1990. Identification of a retinoic acid responsive element in the retinoic acid receptor beta gene. Nature 343:177–180. doi: 10.1038/343177a0. [DOI] [PubMed] [Google Scholar]
- 59.Hoffmann B, Lehmann JM, Zhang XK, Hermann T, Husmann M, Graupner G, Pfahl M. 1990. A retinoic acid receptor-specific element controls the retinoic acid receptor-beta promoter. Mol Endocrinol 4:1727–1736. doi: 10.1210/mend-4-11-1727. [DOI] [PubMed] [Google Scholar]
- 60.Sucov HM, Murakami KK, Evans RM. 1990. Characterization of an autoregulated response element in the mouse retinoic acid receptor type beta gene. Proc Natl Acad Sci U S A 87:5392–5396. doi: 10.1073/pnas.87.14.5392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lamkanfi M. 2011. Emerging inflammasome effector mechanisms. Nat Rev Immunol 11:213–220. doi: 10.1038/nri2936. [DOI] [PubMed] [Google Scholar]
- 62.Hanamsagar R, Torres V, Kielian T. 2011. Inflammasome activation and IL-1beta/IL-18 processing are influenced by distinct pathways in microglia. J Neurochem 119:736–748. doi: 10.1111/j.1471-4159.2011.07481.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Eltom S, Belvisi MG, Yew-Booth L, Dekkak B, Maher SA, Dubuis ED, Jones V, Fitzgerald KA, Birrell MA. 2014. TLR4 activation induces IL-1ss release via an IPAF dependent but caspase 1/11/8 independent pathway in the lung. Respir Res 15:87. doi: 10.1186/s12931-014-0087-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Graham TE, Wason CJ, Bluher M, Kahn BB. 2007. Shortcomings in methodology complicate measurements of serum retinol binding protein (RBP4) in insulin-resistant human subjects. Diabetologia 50:814–823. doi: 10.1007/s00125-006-0557-0. [DOI] [PubMed] [Google Scholar]
- 65.Erikstrup C, Mortensen OH, Nielsen AR, Fischer CP, Plomgaard P, Petersen AM, Krogh-Madsen R, Lindegaard B, Erhardt JG, Ullum H, Benn CS, Pedersen BK. 2009. RBP-to-retinol ratio, but not total RBP, is elevated in patients with type 2 diabetes. Diabetes Obes Metab 11:204–212. doi: 10.1111/j.1463-1326.2008.00901.x. [DOI] [PubMed] [Google Scholar]
- 66.Li ZZ, Lu XZ, Liu JB, Chen L. 2010. Serum retinol-binding protein 4 levels in patients with diabetic retinopathy. J Int Med Res 38:95–99. doi: 10.1177/147323001003800111. [DOI] [PubMed] [Google Scholar]
- 67.Takebayashi K, Suetsugu M, Wakabayashi S, Aso Y, Inukai T. 2007. Retinol binding protein-4 levels and clinical features of type 2 diabetes patients. J Clin Endocrinol Metab 92:2712–2719. doi: 10.1210/jc.2006-1249. [DOI] [PubMed] [Google Scholar]
- 68.Wu L, Nagasaki T, Sparrow JR. 2010. Photoreceptor cell degeneration in Abcr(−/−) mice. Adv Exp Med Biol 664:533–539. doi: 10.1007/978-1-4419-1399-9_61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ramkumar HL, Zhang J, Chan CC. 2010. Retinal ultrastructure of murine models of dry age-related macular degeneration (AMD). Prog Retin Eye Res 29:169–190. doi: 10.1016/j.preteyeres.2010.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Barber AJ, Lieth E, Khin SA, Antonetti DA, Buchanan AG, Gardner TW. 1998. Neural apoptosis in the retina during experimental and human diabetes. Early onset and effect of insulin. J Clin Invest 102:783–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Parisi V, Uccioli L. 2001. Visual electrophysiological responses in persons with type 1 diabetes. Diabetes Metab Res Rev 17:12–18. doi: 10.1002/dmrr.177. [DOI] [PubMed] [Google Scholar]
- 72.Bresnick GH. 1986. Diabetic retinopathy viewed as a neurosensory disorder. Arch Ophthalmol 104:989–990. doi: 10.1001/archopht.1986.01050190047037. [DOI] [PubMed] [Google Scholar]
- 73.Mizutani M, Gerhardinger C, Lorenzi M. 1998. Muller cell changes in human diabetic retinopathy. Diabetes 47:445–449. doi: 10.2337/diabetes.47.3.445. [DOI] [PubMed] [Google Scholar]
- 74.Zeng XX, Ng YK, Ling EA. 2000. Neuronal and microglial response in the retina of streptozotocin-induced diabetic rats. Vis Neurosci 17:463–471. doi: 10.1017/S0952523800173122. [DOI] [PubMed] [Google Scholar]
- 75.Rajala A, Tanito M, Le YZ, Kahn CR, Rajala RV. 2008. Loss of neuroprotective survival signal in mice lacking insulin receptor gene in rod photoreceptor cells. J Biol Chem 283:19781–19792. doi: 10.1074/jbc.M802374200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Barber AJ, Nakamura M, Wolpert EB, Reiter CE, Seigel GM, Antonetti DA, Gardner TW. 2001. Insulin rescues retinal neurons from apoptosis by a phosphatidylinositol 3-kinase/Akt-mediated mechanism that reduces the activation of caspase-3. J Biol Chem 276:32814–32821. doi: 10.1074/jbc.M104738200. [DOI] [PubMed] [Google Scholar]
- 77.Reiter CE, Sandirasegarane L, Wolpert EB, Klinger M, Simpson IA, Barber AJ, Antonetti DA, Kester M, Gardner TW. 2003. Characterization of insulin signaling in rat retina in vivo and ex vivo. Am J Physiol Endocrinol Metab 285:E763–E774. doi: 10.1152/ajpendo.00507.2002. [DOI] [PubMed] [Google Scholar]
- 78.Reiter CE, Gardner TW. 2003. Functions of insulin and insulin receptor signaling in retina: possible implications for diabetic retinopathy. Prog Retin Eye Res 22:545–562. doi: 10.1016/S1350-9462(03)00035-1. [DOI] [PubMed] [Google Scholar]
- 79.Ivanovic I, Anderson RE, Le YZ, Fliesler SJ, Sherry DM, Rajala RV. 2011. Deletion of the p85alpha regulatory subunit of phosphoinositide 3-kinase in cone photoreceptor cells results in cone photoreceptor degeneration. Invest Ophthalmol Vis Sci 52:3775–3783. doi: 10.1167/iovs.10-7139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Rajala A, Dighe R, Agbaga MP, Anderson RE, Rajala RV. 2013. Insulin receptor signaling in cones. J Biol Chem 288:19503–19515. doi: 10.1074/jbc.M113.469064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Rajala RV, Wiskur B, Tanito M, Callegan M, Rajala A. 2009. Diabetes reduces autophosphorylation of retinal insulin receptor and increases protein-tyrosine phosphatase-1B activity. Invest Ophthalmol Vis Sci 50:1033–1040. doi: 10.1167/iovs.08-2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Fort PE, Losiewicz MK, Reiter CE, Singh RS, Nakamura M, Abcouwer SF, Barber AJ, Gardner TW. 2011. Differential roles of hyperglycemia and hypoinsulinemia in diabetes induced retinal cell death: evidence for retinal insulin resistance. PLoS One 6:e26498. doi: 10.1371/journal.pone.0026498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Reiter CE, Wu X, Sandirasegarane L, Nakamura M, Gilbert KA, Singh RS, Fort PE, Antonetti DA, Gardner TW. 2006. Diabetes reduces basal retinal insulin receptor signaling: reversal with systemic and local insulin. Diabetes 55:1148–1156. doi: 10.2337/diabetes.55.04.06.db05-0744. [DOI] [PubMed] [Google Scholar]
- 84.Hombrebueno JR, Chen M, Penalva RG, Xu H. 2014. Loss of synaptic connectivity, particularly in second order neurons is a key feature of diabetic retinal neuropathy in the Ins2Akita mouse. PLoS One 9:e97970. doi: 10.1371/journal.pone.0097970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.VanGuilder HD, Brucklacher RM, Patel K, Ellis RW, Freeman WM, Barber AJ. 2008. Diabetes downregulates presynaptic proteins and reduces basal synapsin I phosphorylation in rat retina. Eur J Neurosci 28:1–11. doi: 10.1111/j.1460-9568.2008.06322.x. [DOI] [PubMed] [Google Scholar]
- 86.Pimplikar SW. 2014. Neuroinflammation in Alzheimer's disease: from pathogenesis to a therapeutic target. J Clin Immunol 34(Suppl 1):S64–S69. doi: 10.1007/s10875-014-0032-5. [DOI] [PubMed] [Google Scholar]
- 87.Hirsch EC, Vyas S, Hunot S. 2012. Neuroinflammation in Parkinson's disease. Parkinsonism Relat Disord 18(Suppl 1):S210–S212. doi: 10.1016/S1353-8020(11)70065-7. [DOI] [PubMed] [Google Scholar]
- 88.Hirsch EC, Hunot S. 2010. Pathophysiological involvement of neuroinflammation in various neurological disorders. J Neural Transm 117:897–898. doi: 10.1007/s00702-010-0439-y. [DOI] [PubMed] [Google Scholar]
- 89.Hirsch EC, Hunot S. 2009. Neuroinflammation in Parkinson's disease: a target for neuroprotection? Lancet Neurol 8:382–397. doi: 10.1016/S1474-4422(09)70062-6. [DOI] [PubMed] [Google Scholar]
- 90.Tarallo V, Hirano Y, Gelfand BD, Dridi S, Kerur N, Kim Y, Cho WG, Kaneko H, Fowler BJ, Bogdanovich S, Albuquerque RJ, Hauswirth WW, Chiodo VA, Kugel JF, Goodrich JA, Ponicsan SL, Chaudhuri G, Murphy MP, Dunaief JL, Ambati BK, Ogura Y, Yoo JW, Lee DK, Provost P, Hinton DR, Nunez G, Baffi JZ, Kleinman ME, Ambati J. 2012. DICER1 loss and Alu RNA induce age-related macular degeneration via the NLRP3 inflammasome and MyD88. Cell 149:847–859. doi: 10.1016/j.cell.2012.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Qi Y, Zhao M, Bai Y, Huang L, Yu W, Bian Z, Zhao M, Li X. 2014. Retinal ischemia/reperfusion injury is mediated by Toll-like receptor 4 activation of NLRP3 inflammasomes. Invest Ophthalmol Vis Sci 55:5466–5475. doi: 10.1167/iovs.14-14380. [DOI] [PubMed] [Google Scholar]
- 92.Frank-Cannon TC, Alto LT, McAlpine FE, Tansey MG. 2009. Does neuroinflammation fan the flame in neurodegenerative diseases? Mol Neurodegener 4:47. doi: 10.1186/1750-1326-4-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Streit WJ, Mrak RE, Griffin WS. 2004. Microglia and neuroinflammation: a pathological perspective. J Neuroinflammation 1:14. doi: 10.1186/1742-2094-1-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Singhal G, Jaehne EJ, Corrigan F, Toben C, Baune BT. 2014. Inflammasomes in neuroinflammation and changes in brain function: a focused review. Front Neurosci 8:315. doi: 10.3389/fnins.2014.00315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Walsh JG, Muruve DA, Power C. 2014. Inflammasomes in the CNS. Nat Rev Neurosci 15:84–97. doi: 10.1038/nrn3638. [DOI] [PubMed] [Google Scholar]
- 96.Halder SK, Matsunaga H, Ishii KJ, Akira S, Miyake K, Ueda H. 2013. Retinal cell type-specific prevention of ischemia-induced damages by LPS-TLR4 signaling through microglia. J Neurochem 126:243–260. doi: 10.1111/jnc.12262. [DOI] [PubMed] [Google Scholar]
- 97.Kohno H, Chen Y, Kevany BM, Pearlman E, Miyagi M, Maeda T, Palczewski K, Maeda A. 2013. Photoreceptor proteins initiate microglial activation via Toll-like receptor 4 in retinal degeneration mediated by all-trans-retinal. J Biol Chem 288:15326–15341. doi: 10.1074/jbc.M112.448712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Wang H, Shi H, Zhang J, Wang G, Zhang J, Jiang F, Xiao Q. 2014. Toll-like receptor 4 in bone marrow-derived cells contributes to the progression of diabetic retinopathy. Mediators Inflamm 2014:858763. doi: 10.1155/2014/858763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Wang YL, Wang K, Yu SJ, Li Q, Li N, Lin PY, Li MM, Guo JY. 2015. Association of the TLR4 signaling pathway in the retina of streptozotocin-induced diabetic rats. Graefes Arch Clin Exp Ophthalmol 253:389–398. doi: 10.1007/s00417-014-2832-y. [DOI] [PubMed] [Google Scholar]
- 100.Rhee AC, Cain AL, Hile KL, Zhang H, Matsui F, Meldrum KK. 2013. IL-18 activation is dependent on Toll-like receptor 4 during renal obstruction. J Surg Res 183:278–284. doi: 10.1016/j.jss.2012.11.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Rajamani U, Jialal I. 2014. Hyperglycemia induces Toll-like receptor-2 and -4 expression and activity in human microvascular retinal endothelial cells: implications for diabetic retinopathy. J Diabetes Res 2014:790902. doi: 10.1155/2014/790902. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]