

Summary
Lipid metabolism and receptor-mediated signaling are highly intertwined processes that cooperate to fulfill cellular functions and safeguard cellular homeostasis. Activation of Toll-like receptors (TLRs) leads to a complex cellular response, orchestrating a diverse range of inflammatory events that need to be tightly controlled. Here, we identified the GPI-anchored Sphingomyelin Phosphodiesterase, Acid-Like 3B (SMPDL3B) in a mass spectrometry screening campaign for membrane proteins co-purifying with TLRs. Deficiency of Smpdl3b in macrophages enhanced responsiveness to TLR stimulation and profoundly changed the cellular lipid composition and membrane fluidity. Increased cellular responses could be reverted by re-introducing affected ceramides, functionally linking membrane lipid composition and innate immune signaling. Finally, Smpdl3b-deficient mice displayed an intensified inflammatory response in TLR-dependent peritonitis models, establishing its negative regulatory role in vivo. Taken together, our results identify the membrane-modulating enzyme SMPDL3B as a negative regulator of TLR signaling that functions at the interface of membrane biology and innate immunity.
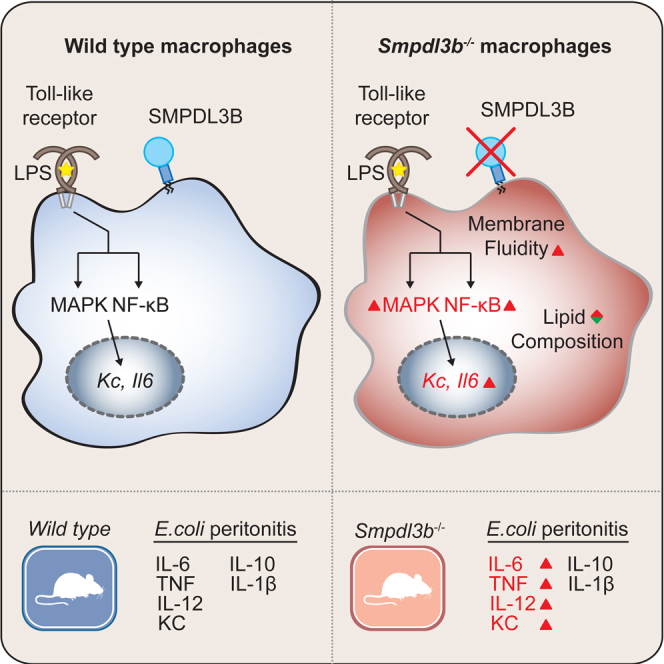
Graphical Abstract
Highlights
-
•
Identification of SMPDL3B as lipid-modulating phosphodiesterase on macrophages
-
•
Negative regulatory role for SMPDL3B in Toll-like receptor function
-
•
Strong influence of SMPDL3B on membrane lipid composition and fluidity
-
•
Smpdl3b-deficient mice show enhanced responsiveness in TLR-dependent peritonitis
Heinz et al. identify the lipid-modulating phosphodiesterase SMPDL3B as negative regulator of Toll-like receptor function. Smpdl3b-deficiency strongly affected macrophage lipid composition and fluidity and led to higher responsiveness to TLR stimulation. Peritonitis models in Smpdl3b-deficient mice confirmed the negative regulatory role in vivo.
Introduction
Toll-like receptors (TLRs) are important sensors of pathogens as well as cellular and environmental stress (Kawai and Akira, 2010; Moresco et al., 2011; O’Neill, 2008). Stimulation of these receptors leads to a complex inflammatory response, orchestrating a diverse range of cellular functions such as cytokine secretion, cell migration, and antigen presentation (Kawai and Akira, 2007). To avoid unnecessary tissue damage or manifestation of chronic inflammation, these events are tightly controlled (Kawai and Akira, 2010; Liew et al., 2005; Medzhitov, 2008). Many proteins have been identified previously as regulators of TLRs acting via different mechanisms (Kondo et al., 2012; Lee et al., 2012; Liew et al., 2005). These include accessory proteins involved in folding and vesicular transport, facilitators of receptor-ligand interactions, transcriptional regulators as well as intracellular proteins involved in activation of receptor-proximal signaling.
Lipids are involved in most cellular processes by serving at the same time as structural components of membranes, energy storage molecules, and second messengers in signaling events (Hannun and Obeid, 2008; van Meer et al., 2008). While the function of membrane-bound or associated proteins is strongly influenced by the lipid composition and state of the accommodating membrane, the molecular connections responsible for these relationships are only beginning to be elucidated (Ernst et al., 2010). Stimulation of macrophages with TLR ligands strongly impacts on cellular gene expression and morphology. This also involves regulation of the cellular lipid repertoire, further highlighting the high degree of interdependence of these processes (Andreyev et al., 2010; Dennis et al., 2010; Maurya et al., 2013). In addition to the TLRs themselves, several components of the receptor sorting and signaling machinery are associated with different cellular membranes (Gay et al., 2014). This extensive interconnection with membrane biology suggests that the receptor function is sensitive to changes in cellular lipid composition. Therefore, it is not surprising that perturbations influencing cellular lipid flux or membrane microdomain composition have been shown to affect TLR-dependent signaling events (Fessler and Parks, 2011; Triantafilou et al., 2002, 2004; Zhu et al., 2010).
Here, we report on the identification of SMPDL3B as a lipid-modifying enzyme and demonstrate its involvement in the regulation of TLR-induced signaling processes. We could show that this GPI-anchored glycoprotein is prominently expressed on macrophages and dendritic cells (DCs) and further strongly upregulated by TLR stimuli and interferon gamma (IFN-γ). Functionally, Smpdl3b-deficient macrophages and DCs showed hyper-responsiveness to TLR stimulation, suggesting a negative regulatory role for SMPDL3B in TLR-induced signaling. Enzymatic measurements revealed that SMPDL3B is a potent phosphodiesterase active on the surface of these cells. Identifying a role in lipid metabolism, Smpdl3b knockdown or knockout macrophages showed a strong reduction in membrane order. This was further highlighted by changes associated with SMPDL3B depletion on the global cellular lipid composition as assessed by lipidomics analysis. In particular, specific ceramide species appeared to be depleted. Supplementation of these lipids in Smpdl3b knockdown cells reverted their hyper-inflammatory phenotype, thus confirming an implication of these molecules in the regulation of TLR signaling. Finally, Smpdl3b-deficient mice manifested higher inflammatory responses in models of TLR-dependent peritonitis, establishing the importance of this enzyme in vivo. Taken together, our results identify SMPDL3B as lipid-modifying enzyme that acts as a negative regulator of TLR signaling at the interface of lipid metabolism and inflammatory signaling.
Results
Identification of SMPDL3B as GPI-Anchored TLR Interactor
The discovery of proteins modulating the activity of TLRs is crucial for the understanding of TLR-dependent immune responses. The characterization of TLR-associated protein complexes by affinity purification followed by mass spectrometry has previously led to the identification of CD14 as co-receptor for nucleic acid recognition by the endosomal TLRs 7 and 9 (Baumann et al., 2010). In the same screening campaign, SMPDL3B (UniProt: P58242, entry name: ASM3B_MOUSE) was consistently identified as membrane protein co-purifying with endosomal TLRs 3, 7, 8, and 9 with robust sequence coverage from RAW264.7 macrophages suggesting association with the same membrane compartments harboring TLRs (Figure 1A).
Figure 1.

Identification of SMPDL3B as GPI-Anchored TLR Interactor
(A) Average spectral counts (av spc) and average % sequence coverage (av%sc) of SMPDL3B detected by mass spectrometry in endosomal TLR tandem affinity purifications.
(B) Domain organization of SMPDL3B, SMPDL3A, and SMPD1/ASM. Gray triangles indicate predicted or validated N-linked glycosylation sites; black triangles indicate conserved motifs in metal coordination/substrate binding.
(C) HA-specific FACS analysis of HEK293T cells stably expressing murine SMPDL3B or a deletion mutant lacking the C-terminal GPI signal.
(D) Cells were lysed using 1% NP-40 or subjected to TX-114 phase separation. Proteins were analyzed by western blot for SMPDL3B, CD14, and IκBα. I, detergent-insoluble proteins; D, detergent phase, amphiphilic integral membrane proteins; A, aqueous phase, hydrophilic proteins.
(E) Cells were lysed with TX-114; lysates were divided in two and treated or not with PI-PLC. Proteins were subjected to phase separation, and fractions were analyzed by western blot for SMPDL3B, CD14, and IκBα.
(F) HEK293T cells were transfected with SMPDL3B and V5-tagged TLRs as indicated. Immunoprecipitates and extracts were analyzed by western blot using SMPDL3B- and V5-specific antibodies.
(C–F) Data are representative of at least two independent experiments. See also Figure S1.
SMPDL3B contains an N-terminal signal peptide (amino acid [aa] 1–18), a central metallo-phosphodiesterase domain (MPP_ASMase, aa 23–323) as well as a C-terminal GPI-membrane anchor signal and belongs to a small family of three evolutionarily related enzymes (Figure 1B) (Masuishi et al., 2013). Of these, Sphingomyelin Phosphodiesterase 1/Acid Sphingomyelinase (SMPD1/ASM) is a well-characterized lysosomal protein involved in the degradation of sphingomyelin to ceramide and phosphorylcholine (Hannun and Obeid, 2011; Milhas et al., 2010; Seto et al., 2004). In contrast, less is known about the precise functions of the related proteins SMPDL3A and SMPDL3B. SMPDL3A was shown to be regulated by the oxysterol-inducible transcription factor LXRα, linking it to lipid metabolism, and recently confirmed to harbor phosphodiesterase activity (Noto et al., 2012; Pehkonen et al., 2012; Traini et al., 2014). SMPDL3B was shown to be a membrane-associated protein proposed to play a role in podocytes in human kidney diseases including focal segmental glomerulosclerosis as well as diabetic kidney disease (Fornoni et al., 2011; Yoo et al., 2015).
To study the topology of SMPDL3B and related proteins in overexpression experiments, HEK293T cells were transiently transfected or stably transduced with the corresponding expression constructs (Figure S1A). SMPDL3B could be detected on the surface of HEK293T cells stably expressing the protein while mutants lacking the C-terminal GPI-membrane anchor signal (SMPDL3BΔGPI) accumulated in cell supernatants (Figures 1C, S1A, and S1B). While overexpressed SMPDL3A was also detected in cell supernatants, confirming a previous report that it is a secreted protein (Traini et al., 2014), SMPD1/ASM, a well-established lysosomal protein, was not found at this location (Figure S1B). Confocal microscopy of overexpressed SMPDL3B in HEK293T cells confirmed the predominantly surface-associated localization (Figure S1C). As the co-purification of SMPDL3B with several TLRs was initially observed in RAW264.7 macrophages, we used these cells to study the properties of the endogenous protein. In line with the human variant (Masuishi et al., 2013), endogenous murine SMPDL3B was enriched in the membrane fraction of these cells and showed sensitivity to phosphatidylinositol-specific phospholipase C (PI-PLC) treatment, an enzyme known to cleave GPI anchors (Figures 1D, 1E, and S1D). Additionally, we assessed the presence of N-linked glycosylation sites on murine or human SMPDL3B or murine SMPDL3A and found that all proteins were highly glycosylated as evidenced by sensitivity of proteins to PNGase F (Figure S1E).
GPI-anchored proteins such as the TLR co-receptor CD14 are known to localize to sphingomyelin- and cholesterol-enriched membrane subdomains that can be purified based on their insolubility in certain detergents (Mayor and Riezman, 2004). We found SMPDL3B and CD14 both enriched in these detergent resistant membranes isolated from RAW264.7 macrophages (Figure S1F). In this light, we also evaluated whether the association of SMPDL3B with TLRs could be confirmed by co-immunoprecipitation (Figure 1F). SMPDL3B co-purified with the TLRs 4, 7, 8, and 9 and only weakly with TLR3 (Figure 1F), thus indeed resembling the behavior of CD14 (Baumann et al., 2010; Lee et al., 2012). Although these data are compatible with physical proximity of SMPDL3B and TLRs, it is also possible that the observations made reflect the presence of these proteins in membrane and/or detergent complexes obtained under the experimental solubilization conditions used, without the involvement of direct protein-protein interactions.
Taken together, we found that SMPDL3B is a GPI-anchored glycoprotein with putative enzymatic function that efficiently co-purified with several TLRs from macrophages and was therefore chosen for further functional evaluation as potential modulator of TLR activity.
SMPDL3B Is a Negative Regulator of TLR Signaling
SMPDL3B expression was prominently observed in macrophages and DCs (Figure S1G). Consistent with a possible role for this enzyme in the course of inflammatory processes, Smpdl3b transcription in bone marrow-derived macrophages (BMDMs) and DCs (BMDCs) was robustly induced upon TLR stimulation (Figure 2A). To further study the role of this protein in primary cells, we decided to generate Smpdl3b-deficient mice. The knockout mice were viable and did not have any overt developmental phenotype. Interestingly, BMDMs and BMDCs from Smpdl3b-deficient mice showed higher expression and release of the chemokine KC/CXCL1 upon stimulation with TLR agonists in comparison to wild-type cells (Figures 2B–2F and S2A). In line with this, knockdown of Smpdl3b in RAW264.7 macrophages increased the release of interleukin 6 (IL-6) upon treatment with lipopolysaccharide (LPS), CpG-DNA (CpG), and imiquimod (IMQ) (Figures S2B and S2C). To evaluate whether other important membrane-dependent events that occur early upon TLR activation were affected by SMPDL3B depletion, we measured the internalization rates of TLR4 and the phagocytic uptake of CpG (Figures S2D–S2F). Endocytosis of TLR4 upon LPS stimulation was unchanged in RAW264.7 macrophages or primary BMDMs depleted or deficient in Smpdl3b, suggesting that the enhanced release of cytokines in SMPDL3B-depleted cells was not caused by retaining TLR4 at the plasma membrane or malfunctioning of receptor endocytosis (Figures S2D and S2E). Also, uptake of fluorescently labeled Cy3-CpG was unaltered in knockdown cells further indicating that endocytic functions were intact (Figure S2F). Taken together, these data show that Smpdl3b expression is induced upon TLR stimulation and reveal that SMPDL3B negatively affects TLR-dependent responses.
Figure 2.

SMPDL3B Is a Negative Regulator of TLR Signaling
(A) BMDMs or BMDCs were stimulated with 100 ng/ml LPS or 1 μM CpG-DNA for the indicated time and relative expression of Smpdl3b was measured by RT-PCR.
(B) Expression of SMPDL3B and tubulin in wild-type and Smpdl3b-deficient BMDMs was analyzed by western blot.
(C and E) BMDMs or BMDCs from wild-type (WT) or Smpdl3b-deficient (KO) mice were stimulated with 100 ng/ml LPS for the indicated time, and relative expression of KC was measured by RT-PCR.
(D and F) BMDMs or BMDCs from wild-type (WT) or Smpdl3b-deficient (KO) mice were stimulated with LPS for 8 hr, and supernatants were analyzed for KC by ELISA.
(G) The phosphorylation status of p38, JNK, and ERK and protein levels of IκBα in the lysates of control (shCTRL) and SMPDL3B-depleted RAW264.7 cells (shS3B) upon stimulation with LPS were analyzed by western blot.
(A and C–F) Data show mean ± SD of technical triplicates and are representative of at least two independent experiments. (G) Data are representative of two independent experiments. See also Figure S2.
SMPDL3B Affects TLR-Dependent Signaling Processes
TLR signaling proceeds through the activation of mitogen-activated protein kinases (MAPKs) and the transcription factor NF-κB (Arthur and Ley, 2013; Kawai and Akira, 2007). To determine the effect of Smpdl3b knockdown on these signaling events, we monitored phosphorylation of the MAPKs p38α, c-Jun N-terminal kinase (JNK) or extracellular signal-regulated kinase (ERK) (Figure 2G). SMPDL3B-depleted cells showed higher levels of phosphorylated p38α and JNK, whereas phosphorylation of ERK appeared rather reduced. In addition to the differences in these important phosphorylation events, SMPDL3B-depleted macrophages showed sustained degradation of IκBα, which is indicative of enhanced NF-κB activity (Figure 2G). Together, these results revealed that signaling processes occurring relatively early after TLR engagement are affected by the absence of SMPDL3B.
Identification of SMPDL3B as TLR-Inducible Neutral Phosphodiesterase
The similarity to other phosphodiesterases prompted us to assess SMPDL3B activity by monitoring phosphodiesterase-dependent hydrolysis of chromogenic bis(4-nitrophenyl)phosphate (bis-pNPP) (Figure S3A). Recombinant SMPDL3B lacking the C-terminal GPI signal, purified from supernatants of Sf9 insect cells or HEK293T cells, efficiently hydrolyzed the substrate, exerting highest activity at neutral pH (Figures 3A, 3B, and S3B). This indicated that the enzyme is fully active at the pH found at the plasma membrane location. Confirming specificity, a mutant (H135A) replacing a conserved histidine residue of SMPDL3B predicted to be involved in substrate hydrolysis (Seto et al., 2004), showed reduced enzymatic activity (Figure 3C). In line with the abundant expression on the plasma membrane, substrate hydrolysis could also be detected on HEK293T cells stably expressing murine or human SMPDL3B, indicating that this assay was well suited for measuring activity on the surface of intact cells (Figures 3D, S3C, and S3D). Indeed, RAW264.7 cells showed robustly detectable enzymatic activity, which was strongly reduced in the Smpdl3b knockdown cells, highlighting SMPDL3B as an important phosphodiesterase enzyme significantly contributing to substrate hydrolysis on macrophages (Figures 3E and S3E).
Figure 3.

Enzymatic Activity and Inducibility of SMPDL3B
(A) Substrate velocity curve and Lineweaver-Burk diagram for murine SMPDL3B produced in Sf9 insect cells.
(B) Influence of pH on the enzymatic activity of murine SMPDL3B produced in Sf9 insect cells.
(C) Coomassie staining of SDS-PAGE gel containing equal amounts of human or mouse SMPDL3B and H135A point mutants. Bar graphs show phosphodiesterase activity of the indicated proteins.
(D) Measurement of phosphodiesterase activity on HEK293T cells stably expressing an empty vector (mock) or murine SMPDL3B.
(E) Phosphodiesterase activity on control (shCTRL) and Smpdl3b-depleted (shSMPDL3B) RAW264.7 cells.
(F) Wild-type (wt) or Smpdl3b-deficient (ko) BMDMs were stimulated or not with 100 ng/ml LPS, 1 μM CpG, 2.5 μM IMQ, 200 ng/ml Pam3Csk4 (P3C4), and 1,000 U/ml IFN-β or 1,000 U/ml IFN-γ for 16 hr, and SMPDL3B or Actin protein levels were analyzed by western blotting. Bar graphs represent phosphodiesterase activity measured after stimulation.
(A and C) Data are representative of at least two independent experiments. (B and D–F) Data show mean ± SD of technical triplicates and are representative of at least two independent experiments. See also Figure S3.
Given that Smpdl3b transcription was upregulated upon TLR activation in macrophages and DCs (Figure 2A), we next determined the effect of inflammatory stimuli on protein levels and enzymatic activity. In line with enhanced transcription, SMPDL3B protein levels were upregulated in BMDMs upon stimulation with different TLR ligands, including LPS, CpG, IMQ, and Pam3CSK4, and also the pro-inflammatory cytokine interferon γ (Figure 3F). Consistent results were also obtained for RAW264.7 macrophages (Figure S3F). The cellular enzymatic activity correlated well with the protein levels and was strongly reduced in BMDMs from knockout mice, further highlighting SMPDL3B as the dominant phosphodiesterase on the surface of these immune cells (Figure 3F).
Deficiency in SMPDL3B Leads to a Global Increase in Membrane Fluidity
To evaluate the impact of SMPDL3B, a lipid-associated enzyme, on the cellular membrane environment, we measured membrane fluidity of macrophages. The lipid phase of membranes in intact cells can be studied using fluorescent probes that alter their spectral emission properties dependent on lipid packing and can thus reflect membrane order (Owen et al., 2012). The generalized polarization (GP) function can be used to calculate a normalized ratio between the measured intensities of ordered and unordered fractions in fluorescence microscopy images and therefore allows the quantification of changes in membrane order. In line with a role for SMPDL3B in membrane biology, microscopy-based measurements of membrane fluidity using the fluorescent probe di-4-ANEPPDHQ revealed a strong decrease of membrane order (i.e., increase of membrane fluidity) in SMPDL3B-depleted RAW264.7 macrophages (Figures 4A–4C). Treatment with the cholesterol-extracting agent methyl-β-cyclodextrin (MβCD) also led to reduced measureable membrane order and served as a positive control for this assay system. As an increase in membrane fluidity associated with SMPDL3B knockdown might affect the integrity of lipid rafts found on the cell surface, we measured the concentration of the raft marker GM1 ganglioside by flow cytometry. Interestingly, the concentration of GM1 was not affected by knockdown of Smpdl3b, suggesting no general modulatory effect on lipid raft abundance (Figure S4A). Highly consistent with the data obtained in RAW264.7 cells, also Smpdl3b-deficient BMDMs showed a decrease in membrane order, indeed assigning a role to SMPDL3B in membrane biology (Figure 4D).
Figure 4.

SMPDL3B Alters the Biophysical Properties and Composition of Cellular Membranes
(A–D) Membrane fluidity measurements. (A) Confocal images pseudocolored based on GP values (see color bar). Left: shCTRL; right: shSMPDL3B. Scale bar, 20 μm. (B) GP value distribution from representative experiment. Mean ± SEM, n = 7. (C) Fraction of ordered membrane of RAW264.7 relative to shCTRL. Mean ± SD of at least three biological replicates is shown. (D) Fraction of ordered membrane of BMDMs relative to WT. Mean ± SD, n = 5. Figure is representative of two biological replicates.
(E) Lipids were extracted from control (shCTLR) or SMPDL3B-depleted (shSMPDL3B) RAW264.7 macrophages and analyzed by MS for glycerophospho- and sphingolipids. Bubble plots represent mean log2-transformed fold-change differences between cell lines. SM, sphingomyelin; GluCer, glucosylceramide; Cer, ceramide; PC, (lyso-) phosphatidylcholine; PE, (lyso-) phosphatidylethanolamine; PA, phosphatidic acid; PI, (lyso-) phosphatidylinositol; PS, (lyso-) phosphatidylserine; PG, phosphatidylglycerol. Data are representative of two biological replicates each consisting of five technical replicates.
(F) Relative lipid levels for ceramide in control (shCTRL) and SMPDL3B-depleted RAW264.7 macrophages (shSMPDL3B). Cer d18:1/16:0 is abbreviated as C16:0, Cer d18:1/22:0 as C22:0, Cer d18:1/24:0 as C24:0, and Cer d18:1/24:1 as C24:1. Data represent mean ± SEM of two biological replicates each consisting of five technical replicates. ∗p ≤ 0.05.
(G) Control (shCTRL) and SMPDL3B-depleted RAW264.7 macrophages (shSMPDL3B) were pretreated for 30 min with vehicle (VEH) or the indicated synthetic ceramide species (15 μM) and then stimulated with 100 ng/ml LPS for 8 hr. IL-6 release was measured by ELISA.
Data show mean ± SD of technical triplicates and is representative of at least two independent experiments. See Figure S4.
Lipidomics Analysis
The clear changes of membrane fluidity in SMPDL3B knockdown or knockout cells strongly suggested that the TLR-modulating activity of SMPDL3B is associated with a change in macrophage lipid composition. To test this, we assessed the global impact of SMPDL3B on the cellular lipid repertoire by mass spectrometry-based lipidomics. RAW264.7 cells were incubated for 2 hr in serum-free medium and lipids from control (shCTRL) and SMPDL3B-depleted cells (shSMPDL3B) were extracted and analyzed for sphingo- and glycerophospholipid as well as cholesterol levels (Table S1). In line with the strong reduction in membrane order, knockdown of SMPDL3B led to a profound global change in the cellular lipid composition, with significant alterations observed for components of most analyzed lipid classes (Figures 4E, S4B, and S4C). Among sphingolipid species, significant decreases were observed for ceramide species upon knockdown of SMPDL3B, and two sphingomyelin species were significantly increased while five were decreased (Figures 4E and S4B). Also, glycerophospholipids were strongly affected by SMPDL3B depletion. Phosphatidylcholine species were rather increased, whereas most phosphatidylethanolamine, -inositol and -serine, were decreased (Figures 4E and S4B). Finally, cellular cholesterol levels were also significantly decreased in SMPDL3B-depleted cells (Figure S4D). Taken together, our membrane fluidity and lipidomics measurements identified SMPDL3B as regulator of macrophage membrane composition and order.
Silencing of Smpdl3b Leads to a Pro-inflammatory Lipid State
Given that the depletion of SMPDL3B in RAW264.7 cells did not only affect a subset of measured lipid species, but led to a global change in the cellular lipid repertoire, the interpretation of these data required further analysis. Based on a systematic perturbation screen targeting genes involved in sphingolipid metabolism combined with lipidomics analysis and measurement of TLR-related processes the function of 245 diverse lipid species could be inferred (see Köberlin et al., 2015). Utilizing this resource, we compared the changes in lipid abundance upon SMPDL3B knockdown with their predicted function in IL-6 release for those lipid species that were detected in both lipidomics data sets. Consistent with the increased inflammatory response observed in SMPDL3B-depleted cells, we found lipids associated with increased IL-6 release upon LPS or CpG stimulation to be increased in abundance while lipids associated with reduced IL-6 release were decreased in abundance (Figure S4E). Interestingly, several ceramide species predicted to dampen TLR-dependent IL-6 release (Cer d18:1/16:0, d18:1/22:0, and d18:1/24:1) were significantly reduced upon SMPDL3B knockdown (Figures 4F and S4E). To validate their potential involvement in regulating LPS-induced IL-6 release, we performed lipid supplementation experiments. Indeed, when control and SMPDL3B-depleted macrophages were pre-treated with the synthetic ceramide species Cer d18:1/16:0, Cer d18:1/22:0, and Cer d18:1/C24:0, the SMPDL3B-dependent increase in IL-6 release upon TLR stimulation could be reverted (Figures 4G and S4F). Similarly, the IL-6 release induced upon stimulation with CpG and IMQ was reduced by supplementation of ceramide Cer d18:1/24:0 further experimentally confirming the predicted roles on other TLRs (Figure S4G).
Taken together, changes in cellular lipid species abundance associated with SMPDL3B depletion were highly indicative of pro-inflammatory responses. Furthermore, our lipidomics analysis combined with lipid supplementation experiments allowed us to highlight several long-chained ceramide species as putative modulators of the enhanced inflammatory response upon SMPDL3B depletion.
SMPDL3B Negatively Regulates TLR Activity In Vivo
If, as all evidence so far would show, SMPDL3B is involved in homeostasis of membrane lipid composition and TLR signaling, then it should have a role in TLR-mediated inflammatory processes in vivo. To assess this, we used an LPS-dependent mouse inflammation model. Wild-type or Smpdl3b-deficient mice were injected intraperitoneally with LPS and peritoneal cell influx as well as serum cytokine levels were determined 6 hr later (Figures 5A and 5B). Smpdl3b-deficient mice contained higher numbers of immune cells in the peritoneal lavage fluid (PLF), due to a significantly enhanced influx of neutrophils, in line with an aberrantly strong inflammatory response (Figure 5A). Concomitantly, Smpdl3b-deficient mice showed increased IL-6 levels in the serum as compared to control mice (Figure 5B). Serum levels of TNF were not significantly affected under these conditions (Figure 5B). To test whether SMPDL3B also affects the host-response against viable pathogens, we used an E. coli-induced peritonitis model. Hence, wild-type and Smpdl3b-deficient mice were injected with bacteria in the peritoneum and serum cytokine levels were quantified 6 hr later. Serum levels of IL-6, TNF, IL-12p40, and KC were significantly elevated in Smpdl3b-deficient mice as compared to wild-type (Figure 5C). Interestingly, we did not detect significant alterations in the serum levels of the cytokines IL-10 and IL-1β, indicating that not all inflammatory mediators were equally affected by Smpdl3b deficiency (Figure 5C). Taken together, our experiments indeed establish SMPDL3B as negative regulator of TLR-dependent inflammatory responses in vivo.
Figure 5.

SMPDL3B Is a Negative Regulator of Inflammation In Vivo
(A and B) Wild-type (WT) and Smpdl3b-deficient (KO) mice were injected with 50 μg LPS i.p. After 6 hr, mice were sacrificed, and (A) peritoneal lavage was taken and total cells and neutrophils were counted, and (B) serum IL-6 and TNF were measured by ELISA.
(C) Wild-type (WT) and Smpdl3b-deficient (KO) mice were injected i.p. with 1 × 104 cfu of E. coli. After 6 hr, mice were sacrificed, and the serum cytokine levels of IL-6, TNF, IL-12p40, KC, IL-10, and IL-1β were analyzed by ELISA. Bars are means of each group ± SEM (∗p ≤ 0.05) and are representative of two independent experiments, each performed with eight mice/group.
Discussion
In the present study, we have identified the GPI-anchored lipid-modifying enzyme SMPDL3B as negative regulator of TLR signaling. We found that this sphingomyelinase-related protein is abundantly expressed on macrophages and DCs and can be further upregulated by inflammatory stimuli. Furthermore, enzymatic assays on intact cells revealed that SMPDL3B is responsible for a large fraction of measurable phosphodiesterase activity on these important immune cells. Loss-of-function of this enzyme led to increased cellular responses upon TLR stimulation while Smpdl3b-deficient mice showed enhanced inflammation in models of TLR-dependent peritonitis. Confirming the involvement of this protein in lipid metabolism, we found that lack of the cellular activity of this enzyme was associated with a significant change in membrane fluidity and the global cellular lipid composition. Supplementation of specific ceramide species that were reduced in SMPDL3B-depleted cells was able to revert the SMPDL3B-dependent hyper-inflammatory phenotype.
Bioactive lipids such as ceramides, sphingosine-1-phosphate and phosphatidylinositol derivatives as well as changes in structural components of cellular membranes have all been implicated in modulation of TLR signaling (Fessler and Parks, 2011; Liew et al., 2005). The inducibility upon TLR stimulation and impact of SMPDL3B on cellular lipid composition and consequently membrane fluidity is suggestive of a not yet described regulatory mode of macrophage function acting at the interface of membrane biology and inflammatory signaling. The lipid environment of TLRs, including membrane microdomains rich in sphingolipids and cholesterol, has been shown to play a role in receptor function (Fessler and Parks, 2011; Lingwood and Simons, 2010; Triantafilou et al., 2002, 2004; Zhu et al., 2010). Perturbations affecting the receptor-accommodating membranes, as observed in macrophages defective for the cholesterol efflux transporters ABCA1 and ABCG1 that display an increase in free cholesterol, lead to enhanced responsiveness of TLRs, presumably as a consequence of altered plasma membrane composition (Draper et al., 2010; Yvan-Charvet et al., 2008; Zhu et al., 2008, 2010). In cells deficient for SMPDL3B, we could not detect changes in the amount of GM1, a marker for sphingolipid- and cholesterol-enriched microdomains, indicating that these structures are intact. Instead, we observed a global decrease in membrane order and change in the cellular lipid repertoire that was associated with upregulation of TLR-triggered signaling, suggesting that SMPDL3B activity may affect innate immunity signaling by an alternative, possibly composite mechanism.
Interestingly, whereas TLR receptor-proximal signaling and cytokine production were clearly changed by SMPDL3B depletion, other processes, including TLR4 surface expression and endocytosis or CpG uptake, appeared normal. This indicates that membrane-dependent functions are differently sensitive to SMPDL3B depletion, which might also affect other TLR-associated processes including receptor-lipid interactions, adaptor recruitment dynamics, receptor diffusion and trafficking, or ligand dissociation (Ernst et al., 2010; Gay et al., 2014). While in the context of TLR signaling SMPDL3B-depleted or deficient macrophages manifested a stronger inflammatory response, the global changes in the cellular lipid repertoire are likely to have more pleiotropic functional effects and affect other cellular signaling pathways and processes that were not covered in our study. Therefore, future experiments will aim at the identification and elucidation of additional roles of SMPDL3B in macrophages and other cellular systems in which SMPDL3B is expressed.
Generation of ceramide has been observed in response to various stimuli, leading to the formation of ceramide-rich membrane platforms, thought to be involved in the regulation of transmembrane signaling processes (Hannun and Obeid, 2008; Milhas et al., 2010; Stancevic and Kolesnick, 2010). Based on observations in experimental lipid bilayers, an increase in ceramide species causes higher membrane order, highlighting a regulatory effect of these lipids on membrane biophysical properties (Stancevic and Kolesnick, 2010). Interestingly, several ceramide species (Cer d18:1/16:0, Cer d18:1/22:0, and Cer d18:1/24:1), which were found to be negatively correlated with TLR-induced IL-6 release, were also significantly reduced upon SMPDL3B depletion, potentially explaining the effect on membrane order observed in these cells.
Despite the complexity of the mechanism at play, the specific and high expression of SMPDL3B in immune cells, the inducibility of its expression and activity by immune stimuli, combined with the unequivocal phenotype observed in Smpdl3b-deficient mice, demonstrate an intimate link between a major cell surface lipid-modulating enzyme and the regulation of pro-inflammatory processes. Undoubtedly, more connections between metabolism, tissue homeostasis, and the resolution of inflammation are likely to emerge in the future and further elucidate these interdependent regulatory networks.
Experimental Procedures
Information on reagents, plasmids, cell culture, and protein purification can be found in the Supplemental Experimental Procedures.
FACS
All samples were analyzed on a BD Biosciences FACSCalibur or LSR Fortessa flow cytometer. See the Supplemental Experimental Procedures for further information.
RT-PCR
RNA was isolated with the QIAGEN RNeasy Mini Kit. 100 ng RNA/sample was reversely transcribed using RevertAid reverse transcriptase (Fermentas). cDNA was diluted 1:20, and relative transcript levels were analyzed using SYBR green (GeneXPress) using the following primers: Cyclophilin B 5′-CAG CAA GTT CCA TCG TGT CAT CAA GG-3′ and 5′-GGA AGC GCT CAC CAT AGA TGC TC-3′; Smpdl3b 5′-AAG TCT ATG CTG CTC TGG GAA-3′and 5′-TGC CAC CTG GTT ATA GAT GC-3′; mKC 5′CAATGAGCTGCGCTGTCAGTG-3′ and 5′-CTTGGGGACACCTTTTAGCATC-3′. Real-time PCR analysis was performed on a Rotor Gene 6000 (QIAGEN) in technical duplicates or triplicates. Expression of target genes was normalized to that of the housekeeping gene Cyclophilin B.
ELISA
All ELISA experiments were performed according to the manufacturers’ instructions. The kit for detection of KC was from R&D Systems, TNF was from BioLegend, and all others were from BD Biosciences.
Triton X-114 Phase Separation and PI-PLC Sensitivity Assays
Triton X-114 (TX114) phase separation experiments were performed according to Bordier (1981) and as described previously (Heinz et al., 2012). PI-PLC experiments were carried out by incubation of samples with 0.5 U/ml PI-PLC (Invitrogen) for 30 min on ice. See the Supplemental Experimental Procedures for further information.
Phosphodiesterase Enzymatic Activity
Generation of p-nitrophenol from bis-p-nitrophenolphosphate by phosphodiesterase activity was measured as absorbance at 405 nm in 96-well plates with 100 μl reaction volume. Kinetic measurements were carried out using 325 ng/ml enzyme purified from Sf9 insect cells in HEPES buffer (20 mM [pH 7.8]) with a substrate concentration as indicated. pH optima were determined by incubation of enzymes purified from Sf9 insect cells or HEK293T cells in the presence of different buffers adjusted to the indicated pH in the presence of 1 mM substrate. For determination of the impact of point mutations, enzymes were incubated with HEPES buffer (20 mM [pH 7.8]) in the presence of 1 mM substrate. For measurement of cell-associated enzymatic activity, cells were incubated in 96-well plates with isotonic Tris-buffered saline (TBS) in the presence of 1 mM substrate at 37°C and 5% CO2.
Membrane Fluidity Measurements
Membrane fluidity measurements were carried out using the fluorescent probe di-4-ANEPPDHQ following a modified protocol from Owen et al. (2012). See the Supplemental Experimental Procedures for additional information.
Lipidomics
1.5 × 107 RAW264.7 cells stably expressing SMPDL3B-specific or control small hairpin RNA (shRNAs) were seeded in 10-cm dishes in serum-free DMEM medium as five technical replicates and incubated for 2 hr at 37°C, 5% CO2. Cells were harvested into ice-cold PBS and centrifuged for 5 min at 300 × g, 4°C, and pellets were resuspended in 50 μl PBS, transferred to Eppendorf tubes, and frozen in liquid nitrogen for further processing. Samples were hydrated by adding 200 μl H2O and extracted by addition of 600 μl methanol/chloroform (1:2 v/v) by vigorously vortexing the samples three times for 1 min with 5-min intervals. After addition of 300 μl chloroform and 2 μl of 100 mM KCl samples were vortexed three times for 30 s with 1-min intervals. Samples were centrifuged at 9,000 rpm for 2 min at 4°C; the lower, organic phase was transferred to new tubes and dried in a SpeedVac centrifuge. Lipids were quantified by HPLC/MS as described previously with corresponding internal standards including C17-Cer, C12-SM, C8-GluCer, C20-LPC, and PC-14:0/14:0 (Avanti Polar Lipids) (Shui et al., 2011). Individual lipids were normalized to phosphatidylcholine and sphingomyelin levels.
In Vivo Experimental Procedures
Heterozygous Smpdl3b+/− mice (B6N;B6N-Smpdl3b < tm1a(EUCOMM)Wtsi > /H) were purchased from the EMMA consortium (EUCOMM) and shipped from the MRC. Smpdl3b homozygous knockout was confirmed by genotyping using the following primers: SMPDL3B WT 5′ -GTG TAA GCC TTC TCC CCC AG-3′; SMPDL3B WT 5′-CAG AAA AAG TTC TAC GGA CCA GC-3′; SMPDL3B MUT 5′-TTG GTG ATA TCG TGG TAT CGT T-5′. Wild-type C57BL/6J mice were obtained from Charles River Laboratories and bred at the same location as Smpdl3-deficient mice. Pathogen-free 9- to 11-week-old female C57BL/6J and homozygous B6N;B6N-Smpdl3btm1a(EUCOMM)Wtsi/H Smpdl3b−/− mice were used in all in vivo experiments, which were approved by the Animal Care and Use Committee of the Medical University of Vienna. Mice were injected intraperitoneally with 50 μg LPS in 200 μl of saline per mouse. After 6 hr, mice were sacrificed, peritoneal lavage was performed, and blood was taken. Cell counts were determined on each peritoneal lavage sample stained with Tuerks solution, and differential cell counts were performed on cytospin samples stained with Giemsa. E. coli-dependent peritonitis was induced as described previously (Knapp et al., 2007). In brief, E. coli O18:K1 was cultured in Luria-Bertani medium (Difco) at 37°C, harvested at mid-log phase, and washed twice before inoculation. Mice were injected i.p. with 1–2 × 104 cfu E. coli in 200 μl saline per mouse. The inoculum was plated on blood agar plates to determine viable counts. Mice were sacrificed, and blood was isolated after 6 hr.
Bioinformatic Analysis and General Statistics
Protein sequences and information were retrieved from UniProtKB database (2012). Additional information on domain organization was obtained through the NCBI conserved domain database (CDD) (Marchler-Bauer et al., 2011). Protein homology searches were carried out using NCBI Blast (Altschul et al., 1990). Expression data for murine SMPDL3B were extracted from the BioGPS gene annotation portal (Lattin et al., 2008; Wu et al., 2009). p values were calculated with two-tailed t tests, unless otherwise indicated.
Author Contributions
L.X.H., C.L.B., and G.S-F. conceived the study. C.L.B., R.G., O.S., and S.K. designed and performed in vivo experiments. A.C.M., K.L.B., F.P.B., J.C., R.K.K., C.L.B., L.X.H., and G.S.-F. designed and performed proteomics experiments and data analysis. G.S. and M.R.W. performed lipidomics analysis. B.S. performed image and lipidomics data analysis. L.X.H, C.L.B., M.S.K., and M.B. performed other experiments. L.X.H., C.L.B., and G.S.-F. designed other experiments. I.A., A.F., T.K., S.B., M.R., and A.P. generated reagents and contributed scientific insights. L.X.H., C.L.B., and G.-S.F. wrote the manuscript. All authors contributed to the discussion of results and participated in manuscript preparation.
Acknowledgments
We thank Gregory Vladimer for critically reading of the manuscript, Ruth Fuchs and Denise Barlow for help with mouse genotyping, and Sabine Jungwirth and Sarah Niggemeyer for breeding of mice. The work was funded by the Austrian Academy of Sciences (G.S.-F., M.K., I.A., A.C.M., S.B., T.K., and A.F.), the Medical University of Vienna (S.K. and O.S.), the European Research Council under the European Union’s Seventh Framework Programme (FP7/2007-2013)/ERC Grant agreement no. 250179 (G.S.-F., L.X.H., R.K.K., and A.P.), and Marie Curie Fellowship agreement no. 220596 (C.L.B.) and no. 301663 (M.R.), the EMBO Fellowships ATLF 2008-463 (C.L.B.), ALTF 1346-2011 (M.R.) and ATLF 314-2012 (R.K.K.), the Austrian Science Fund FWFI291B09 (M.B.) and FWF1205B09 (R.G.), and the Austrian Federal Ministry for Science and Research GEN-AU/BIN (J.C.) and GEN-AU/APP (K.L.B.). B.S. is supported by a fellowship from the Swiss National Science Foundation (Project no. P300P3_147897). M.R.W. is supported by grants from the National University of Singapore via the Life Sciences Institute (LSI), the Singapore National Research Foundation under CRP Award No. 2007-04, and the SystemsX.ch RTD project LipidX.
Published: June 18, 2015
Footnotes
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/).
Supplemental Information includes Supplemental Experimental Procedures, four figures, and one table and can be found with this article online at http://dx.doi.org/10.1016/j.celrep.2015.05.006.
Supplemental Information
References
- Altschul S.F., Gish W., Miller W., Myers E.W., Lipman D.J. Basic local alignment search tool. J. Mol. Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Andreyev A.Y., Fahy E., Guan Z., Kelly S., Li X., McDonald J.G., Milne S., Myers D., Park H., Ryan A. Subcellular organelle lipidomics in TLR-4-activated macrophages. J. Lipid Res. 2010;51:2785–2797. doi: 10.1194/jlr.M008748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arthur J.S., Ley S.C. Mitogen-activated protein kinases in innate immunity. Nat. Rev. Immunol. 2013;13:679–692. doi: 10.1038/nri3495. [DOI] [PubMed] [Google Scholar]
- Baumann C.L., Aspalter I.M., Sharif O., Pichlmair A., Blüml S., Grebien F., Bruckner M., Pasierbek P., Aumayr K., Planyavsky M. CD14 is a coreceptor of Toll-like receptors 7 and 9. J. Exp. Med. 2010;207:2689–2701. doi: 10.1084/jem.20101111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bordier C. Phase separation of integral membrane proteins in Triton X-114 solution. J. Biol. Chem. 1981;256:1604–1607. [PubMed] [Google Scholar]
- Dennis E.A., Deems R.A., Harkewicz R., Quehenberger O., Brown H.A., Milne S.B., Myers D.S., Glass C.K., Hardiman G., Reichart D. A mouse macrophage lipidome. J. Biol. Chem. 2010;285:39976–39985. doi: 10.1074/jbc.M110.182915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Draper D.W., Madenspacher J.H., Dixon D., King D.H., Remaley A.T., Fessler M.B. ATP-binding cassette transporter G1 deficiency dysregulates host defense in the lung. Am. J. Respir. Crit. Care Med. 2010;182:404–412. doi: 10.1164/rccm.200910-1580OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst A.M., Contreras F.X., Brügger B., Wieland F. Determinants of specificity at the protein-lipid interface in membranes. FEBS Lett. 2010;584:1713–1720. doi: 10.1016/j.febslet.2009.12.060. [DOI] [PubMed] [Google Scholar]
- Fessler M.B., Parks J.S. Intracellular lipid flux and membrane microdomains as organizing principles in inflammatory cell signaling. J. Immunol. 2011;187:1529–1535. doi: 10.4049/jimmunol.1100253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fornoni A., Sageshima J., Wei C., Merscher-Gomez S., Aguillon-Prada R., Jauregui A.N., Li J., Mattiazzi A., Ciancio G., Chen L. Rituximab targets podocytes in recurrent focal segmental glomerulosclerosis. Sci. Transl. Med. 2011;3:85ra46. doi: 10.1126/scitranslmed.3002231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gay N.J., Symmons M.F., Gangloff M., Bryant C.E. Assembly and localization of Toll-like receptor signalling complexes. Nat. Rev. Immunol. 2014;14:546–558. doi: 10.1038/nri3713. [DOI] [PubMed] [Google Scholar]
- Hannun Y.A., Obeid L.M. Principles of bioactive lipid signalling: lessons from sphingolipids. Nat. Rev. Mol. Cell Biol. 2008;9:139–150. doi: 10.1038/nrm2329. [DOI] [PubMed] [Google Scholar]
- Hannun Y.A., Obeid L.M. Many ceramides. J. Biol. Chem. 2011;286:27855–27862. doi: 10.1074/jbc.R111.254359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz L.X., Rebsamen M., Rossi D.C., Staehli F., Schroder K., Quadroni M., Gross O., Schneider P., Tschopp J. The death domain-containing protein Unc5CL is a novel MyD88-independent activator of the pro-inflammatory IRAK signaling cascade. Cell Death Differ. 2012;19:722–731. doi: 10.1038/cdd.2011.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawai T., Akira S. TLR signaling. Semin. Immunol. 2007;19:24–32. doi: 10.1016/j.smim.2006.12.004. [DOI] [PubMed] [Google Scholar]
- Kawai T., Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat. Immunol. 2010;11:373–384. doi: 10.1038/ni.1863. [DOI] [PubMed] [Google Scholar]
- Knapp S., Matt U., Leitinger N., van der Poll T. Oxidized phospholipids inhibit phagocytosis and impair outcome in gram-negative sepsis in vivo. J. Immunol. 2007;178:993–1001. doi: 10.4049/jimmunol.178.2.993. [DOI] [PubMed] [Google Scholar]
- Köberlin M.S., Snijder B., Heinz L.X., Baumann C.L., Fauster A., Vladimer G.I., Gavin A.-C., Superti-Furga G. A conserved circular network of coregulated lipids modulates innate immune responses. Cell. 2015 doi: 10.1016/j.cell.2015.05.051. Published online June 18, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo T., Kawai T., Akira S. Dissecting negative regulation of Toll-like receptor signaling. Trends Immunol. 2012;33:449–458. doi: 10.1016/j.it.2012.05.002. [DOI] [PubMed] [Google Scholar]
- Lattin J.E., Schroder K., Su A.I., Walker J.R., Zhang J., Wiltshire T., Saijo K., Glass C.K., Hume D.A., Kellie S., Sweet M.J. Expression analysis of G Protein-Coupled Receptors in mouse macrophages. Immunome Res. 2008;4:5. doi: 10.1186/1745-7580-4-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C.C., Avalos A.M., Ploegh H.L. Accessory molecules for Toll-like receptors and their function. Nat. Rev. Immunol. 2012;12:168–179. doi: 10.1038/nri3151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liew F.Y., Xu D., Brint E.K., O’Neill L.A. Negative regulation of toll-like receptor-mediated immune responses. Nat. Rev. Immunol. 2005;5:446–458. doi: 10.1038/nri1630. [DOI] [PubMed] [Google Scholar]
- Lingwood D., Simons K. Lipid rafts as a membrane-organizing principle. Science. 2010;327:46–50. doi: 10.1126/science.1174621. [DOI] [PubMed] [Google Scholar]
- Marchler-Bauer A., Lu S., Anderson J.B., Chitsaz F., Derbyshire M.K., DeWeese-Scott C., Fong J.H., Geer L.Y., Geer R.C., Gonzales N.R. CDD: a Conserved Domain Database for the functional annotation of proteins. Nucleic Acids Res. 2011;39:D225–D229. doi: 10.1093/nar/gkq1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuishi Y., Nomura A., Okayama A., Kimura Y., Arakawa N., Hirano H. Mass spectrometric identification of glycosylphosphatidylinositol-anchored peptides. J. Proteome Res. 2013;12:4617–4626. doi: 10.1021/pr4004807. [DOI] [PubMed] [Google Scholar]
- Maurya M.R., Gupta S., Li X., Fahy E., Dinasarapu A.R., Sud M., Brown H.A., Glass C.K., Murphy R.C., Russell D.W. Analysis of inflammatory and lipid metabolic networks across RAW264.7 and thioglycolate-elicited macrophages. J. Lipid Res. 2013;54:2525–2542. doi: 10.1194/jlr.M040212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayor S., Riezman H. Sorting GPI-anchored proteins. Nat. Rev. Mol. Cell Biol. 2004;5:110–120. doi: 10.1038/nrm1309. [DOI] [PubMed] [Google Scholar]
- Medzhitov R. Origin and physiological roles of inflammation. Nature. 2008;454:428–435. doi: 10.1038/nature07201. [DOI] [PubMed] [Google Scholar]
- Milhas D., Clarke C.J., Hannun Y.A. Sphingomyelin metabolism at the plasma membrane: implications for bioactive sphingolipids. FEBS Lett. 2010;584:1887–1894. doi: 10.1016/j.febslet.2009.10.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moresco E.M., LaVine D., Beutler B. Toll-like receptors. Curr. Biol. 2011;21:R488–R493. doi: 10.1016/j.cub.2011.05.039. [DOI] [PubMed] [Google Scholar]
- Noto P.B., Bukhtiyarov Y., Shi M., McKeever B.M., McGeehan G.M., Lala D.S. Regulation of sphingomyelin phosphodiesterase acid-like 3A gene (SMPDL3A) by liver X receptors. Mol. Pharmacol. 2012;82:719–727. doi: 10.1124/mol.112.078865. [DOI] [PubMed] [Google Scholar]
- O’Neill L.A. The interleukin-1 receptor/Toll-like receptor superfamily: 10 years of progress. Immunol. Rev. 2008;226:10–18. doi: 10.1111/j.1600-065X.2008.00701.x. [DOI] [PubMed] [Google Scholar]
- Owen D.M., Rentero C., Magenau A., Abu-Siniyeh A., Gaus K. Quantitative imaging of membrane lipid order in cells and organisms. Nat. Protoc. 2012;7:24–35. doi: 10.1038/nprot.2011.419. [DOI] [PubMed] [Google Scholar]
- Pehkonen P., Welter-Stahl L., Diwo J., Ryynänen J., Wienecke-Baldacchino A., Heikkinen S., Treuter E., Steffensen K.R., Carlberg C. Genome-wide landscape of liver X receptor chromatin binding and gene regulation in human macrophages. BMC Genomics. 2012;13:50. doi: 10.1186/1471-2164-13-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seto M., Whitlow M., McCarrick M.A., Srinivasan S., Zhu Y., Pagila R., Mintzer R., Light D., Johns A., Meurer-Ogden J.A. A model of the acid sphingomyelinase phosphoesterase domain based on its remote structural homolog purple acid phosphatase. Protein Sci. 2004;13:3172–3186. doi: 10.1110/ps.04966204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shui G., Stebbins J.W., Lam B.D., Cheong W.F., Lam S.M., Gregoire F., Kusonoki J., Wenk M.R. Comparative plasma lipidome between human and cynomolgus monkey: are plasma polar lipids good biomarkers for diabetic monkeys? PLoS ONE. 2011;6:e19731. doi: 10.1371/journal.pone.0019731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stancevic B., Kolesnick R. Ceramide-rich platforms in transmembrane signaling. FEBS Lett. 2010;584:1728–1740. doi: 10.1016/j.febslet.2010.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traini M., Quinn C.M., Sandoval C., Johansson E., Schroder K., Kockx M., Meikle P.J., Jessup W., Kritharides L. Sphingomyelin phosphodiesterase-like 3A (SMPDL3A) is a novel nucleotide phosphodiesterase regulated by cholesterol in human macrophages. J. Biol. Chem. 2014;289:32895–32913. doi: 10.1074/jbc.M114.612341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Triantafilou M., Miyake K., Golenbock D.T., Triantafilou K. Mediators of innate immune recognition of bacteria concentrate in lipid rafts and facilitate lipopolysaccharide-induced cell activation. J. Cell Sci. 2002;115:2603–2611. doi: 10.1242/jcs.115.12.2603. [DOI] [PubMed] [Google Scholar]
- Triantafilou M., Morath S., Mackie A., Hartung T., Triantafilou K. Lateral diffusion of Toll-like receptors reveals that they are transiently confined within lipid rafts on the plasma membrane. J. Cell Sci. 2004;117:4007–4014. doi: 10.1242/jcs.01270. [DOI] [PubMed] [Google Scholar]
- van Meer G., Voelker D.R., Feigenson G.W. Membrane lipids: where they are and how they behave. Nat. Rev. Mol. Cell Biol. 2008;9:112–124. doi: 10.1038/nrm2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C., Orozco C., Boyer J., Leglise M., Goodale J., Batalov S., Hodge C.L., Haase J., Janes J., Huss J.W., 3rd, Su A.I. BioGPS: an extensible and customizable portal for querying and organizing gene annotation resources. Genome Biol. 2009;10:R130. doi: 10.1186/gb-2009-10-11-r130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo T.H., Pedigo C.E., Guzman J., Correa-Medina M., Wei C., Villarreal R., Mitrofanova A., Leclercq F., Faul C., Li J. Sphingomyelinase-like phosphodiesterase 3b expression levels determine podocyte injury phenotypes in glomerular disease. J. Am. Soc. Nephrol. 2015;26:133–147. doi: 10.1681/ASN.2013111213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yvan-Charvet L., Welch C., Pagler T.A., Ranalletta M., Lamkanfi M., Han S., Ishibashi M., Li R., Wang N., Tall A.R. Increased inflammatory gene expression in ABC transporter-deficient macrophages: free cholesterol accumulation, increased signaling via toll-like receptors, and neutrophil infiltration of atherosclerotic lesions. Circulation. 2008;118:1837–1847. doi: 10.1161/CIRCULATIONAHA.108.793869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X., Lee J.Y., Timmins J.M., Brown J.M., Boudyguina E., Mulya A., Gebre A.K., Willingham M.C., Hiltbold E.M., Mishra N. Increased cellular free cholesterol in macrophage-specific Abca1 knock-out mice enhances pro-inflammatory response of macrophages. J. Biol. Chem. 2008;283:22930–22941. doi: 10.1074/jbc.M801408200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X., Owen J.S., Wilson M.D., Li H., Griffiths G.L., Thomas M.J., Hiltbold E.M., Fessler M.B., Parks J.S. Macrophage ABCA1 reduces MyD88-dependent Toll-like receptor trafficking to lipid rafts by reduction of lipid raft cholesterol. J. Lipid Res. 2010;51:3196–3206. doi: 10.1194/jlr.M006486. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.