

Abstract
The goal of the study was to improve the methodology of HIV genotyping for analysis of HIV drug resistance and HIV clustering. Using the protocol of Gall et al. (A. Gall, B. Ferns, C. Morris, S. Watson, M. Cotten, M. Robinson, N. Berry, D. Pillay, and P. Kellam, J Clin Microbiol 50:3838–3844, 2012, doi:10.1128/JCM.01516-12), we developed a robust methodology for amplification of two large fragments of viral genome covering about 80% of the unique HIV-1 genome sequence. Importantly, this method can be applied to both viral RNA and proviral DNA amplification templates, allowing genotyping in HIV-infected subjects with suppressed viral loads (e.g., subjects on antiretroviral therapy [ART]). The two amplicons cover critical regions across the HIV-1 genome (including pol and env), allowing analysis of mutations associated with resistance to protease inhibitors, reverse transcriptase inhibitors (nucleoside reverse transcriptase inhibitors [NRTIs] and nonnucleoside reverse transcriptase inhibitors [NNRTIs]), integrase strand transfer inhibitors, and virus entry inhibitors. The two amplicons generated span 7,124 bp, providing substantial sequence length and numbers of informative sites for comprehensive phylogenic analysis and greater refinement of viral linkage analyses in HIV prevention studies. The long-range HIV genotyping from proviral DNA was successful in about 90% of 212 targeted blood specimens collected in a cohort where the majority of patients had suppressed viral loads, including 65% of patients with undetectable levels of HIV-1 RNA loads. The generated amplicons could be sequenced by different methods, such as population Sanger sequencing, single-genome sequencing, or next-generation ultradeep sequencing. The developed method is cost-effective—the cost of the long-range HIV genotyping is under $140 per subject (by Sanger sequencing)—and has the potential to enable the scale up of public health HIV prevention interventions.
INTRODUCTION
HIV genotyping is a critical tool for antiviral drug resistance testing that has revolutionized HIV care and advanced HIV-related research. Routine antiretroviral (ARV) drug resistance testing is useful in choosing an optimal treatment regimen and monitoring its efficiency in clinical practice (1–12). HIV genotyping has been used successfully in research on HIV transmission clusters and HIV transmission dynamics (13–35).
Initial broadly used ARV regimens included combinations of nucleoside reverse transcriptase (RT) inhibitors (NRTIs) and nonnucleoside reverse transcriptase inhibitors (NNRTIs). To monitor the emergence of drug resistance mutations associated with NRTIs and NNRTIs, HIV genotyping targeted viral sequences spanning an approximately 1,000- to 1,300-bp region of the HIV-1 genome encoding viral protease and partial RT, using viral RNA as a template for amplification. While the RNA-based approach works well in antiretroviral therapy (ART)-naive individuals, it is less successful if levels of viral replication are low, such as in individuals on ART. The sequence length of traditional RNA-based HIV genotyping for drug resistance is relatively short and does not cover the HIV-1 region encoding viral integrase or the viral envelope, hindering analysis of drug resistance mutations associated with integrase strand transfer inhibitors or entry inhibitors. The global scale up of ARV treatment and successful introduction of integrase strand transfer inhibitors and entry inhibitors into clinical trials and clinical practice necessitate modification of traditional methods of HIV genotyping.
Two commercial genotyping assays, ViroSeq HIV-1 from Abbott Molecular and TruGene HIV-1 from Siemens Molecular Diagnostics, have been widely used for analysis of HIV-1-associated drug resistance. Both genotyping kits were extensively tested and validated (36–45). While the ViroSeq HIV-1 kit is still on the market, Siemens discontinued selling and supporting the TruGene HIV-1 kit in 2014. The ViroSeq HIV-1 kit covers the entire protease-coding region and the RT region encoding the first 320 amino acids. The TruGene HIV-1 sequences span the protease (amino acids 4 to 99)- and RT (amino acids 40 to 240)-coding regions. The CDC supplies WHO-designated and CDC-supported President's Emergency Plan for AIDS Relief (PEPFAR) Genotyping Laboratories with the ATCC HIV-1 Drug Resistance Genotyping kit (46) for drug resistance testing. Many experienced genotyping laboratories have developed their own in-house amplification and sequencing protocols (11, 47–56), including identification of minor viral variants that are normally missed by commercial genotyping kits (57–61). All of these approaches generally include smaller and more restricted regions for testing HIV-1 drug resistance.
Recently, the protocol developed by Gall et al. (62) has enabled high-throughput, nearly full-length HIV-1 genome genotyping in individuals infected with multiple HIV-1 subtypes. The method has become the cornerstone of the PANGEA (Phylogenetics and Networks for Generalized HIV Epidemics in Africa)-HIV Consortium (http://www.pangea-hiv.org/) aiming to establish worldwide scientific collaborations across phylogenetics, public health, and epidemiology. The Gall protocol (62) targets viral RNA as a template for cDNA synthesis and amplification and is very robust and reproducible when the HIV-1 RNA load is high (e.g., above 10,000 cps/ml). However, specimens with levels of HIV-1 RNA below 1,000 cps/ml, or lower thresholds, present a substantial challenge, and few of those samples could be genotyped. This is consistent with the commercially available assays for HIV drug resistance genotyping, ViroSeq and TrueGene, which are unable to genotype specimens with low or undetectable HIV-1 RNA loads.
In HIV infection, proviral DNA presents an alternative template for HIV genotyping. Drug resistance mutations detected in viral RNA from plasma and proviral DNA from peripheral blood mononuclear cells (PBMCs) or dried blood spots (DBS) show substantial correlation in treated patients, suggesting that either compartment is suitable for the detection of mutations as a virological guide for clinical care (63–65).
It is known that amplified HIV sequences and sequences from proviral DNA could have substantial numbers of guanine-to-adenine transitions. Such an inordinate number of identical G-to-A transitions is a retroviral signature known as hypermutation (66–69). G-to-A hypermutations produce multiple stop codons and reduce HIV replication, leading to an evolutionary dead end. It is an innate host intracellular defense mechanism. The host factors APOBEC3F and APOBEC3G induce G-to-A substitutions in reverse-transcribed nascent retroviral DNA (70). G-to-A hypermutations play an important role in the evolution of antiretroviral drug resistance (71, 72) and could be associated with ART failure (73). The extent of G-to-A hypermutations is not associated with levels of HIV-1 RNA (74), although hypermutations are frequent in viremic controllers (75). For sequence quality control, it is important that G-to-A hypermutations are not products of PCR amplification (76).
In this study, we present a technique for long-range HIV genotyping using proviral DNA, as well as viral RNA, as a template for amplification and sequencing. The outcome of the long-range HIV genotyping is two large fragments that span about 80% of the unique full-length HIV-1 genome sequence. The proposed technique is a modification of the method of Gall et al. (62). The key modifications include using (i) a proviral DNA template, (ii) an extra round of PCR, (iii) selection of robust primers, and (iv) modified running conditions. To illustrate the potential utility of long-range HIV genotyping, the technique was applied to a set of specimens collected in Botswana.
MATERIALS AND METHODS
Study subjects.
The technique of long-range HIV genotyping was applied to specimens collected within three Botswana-Harvard AIDS Institute Partnership (BHP) studies performing viral genotyping: the HIV Prevention Program for Mochudi, Botswana (Mochudi Prevention Project [MPP]; R01 AI083036; principal investigator, M. Essex) (34, 35); the GWAS on Determinants of HIV-1 Subtype C Infection study (RC4 AI092715; principal investigator, M. Essex); and the Botswana Combination Prevention Project (BCPP, or Ya Tsie; U01 GH000447; principal investigator, M. Essex) (77). All studies were approved by the Health Research and Development Committee (HRDC) of the Republic of Botswana and the Office of Human Research Administration (OHRA) of the Harvard T.H. Chan School of Public Health. All study subjects signed a consent form and donated a blood sample for viral genotyping. The first large fragment of the HIV-1 genome, amplicon 1, was amplified and sequenced in 649 HIV-infected subjects (a single sequence per subject) originating from eight geographic localities in Botswana: Digawana, Gaborone, Lobatse, Mochudi, Molapowabojang, Molepolole, Otse, and Ranaka. The second large fragment of the HIV-1 genome, amplicon 2, was amplified and sequenced in 90 subjects (the work is still in progress) originating from Mochudi, Molapowabojang, Otse, and Ranaka.
A total of 212 specimens from the BCPP study were used for analysis of genotyping efficiency. These samples were collected consecutively from subjects participating in the BCPP baseline household survey (20% of households) in the first four communities, Ranaka, Digawana, Molapowabojang, and Otse, from November 2013 to June 2014. Specimens from two other studies, MPP and GWAS, represented subsets successfully amplified in the past for a shorter region of HIV-1 env gp120, V1C5. Due to potential selection bias, the MPP and GWAS specimens were not used in analysis of genotyping efficiency.
Analyzed regions of the HIV-1 genome.
The extent of HIV clustering was analyzed by using the following subgenomic regions across the HIV-1 genome: (i) amplicon 1, spanning the 3′ end of gag and almost the entire pol and corresponding to amplicon 2 in the study of Gall et al. (62), nucleotide positions 1486 to 5058; (ii) amplicon 2, spanning vpu, env, nef, and the TATA box in the U3 region of the 3′ long terminal repeat (LTR) and corresponding to amplicon 4 in the study of Gall et al. (62), nucleotide positions 5967 to 9517; (iii) ViroSeq, a partial pol sequence spanning the region encoding HIV-1 protease and the first 335 amino acids of reverse transcriptase and corresponding to the sequence produced by ViroSeq (39, 44, 45, 78), nucleotide positions 2253 to 3554; and (iv) V1C5, a partial env sequence spanning the region encoding gp120 V1C5 (34, 79, 80), nucleotide positions 6570 to 7757. In addition, the following combinations of the subgenomic regions included concatenated amplicon 1 plus amplicon 2 and amplicon 1 plus V1C5. All multiple-sequence codon-based alignments were generated using MUSCLE (81) in MEGA6 (82).
To prevent sample contamination, basic laboratory rules were enforced, including controlled flow of specimens, use of dedicated areas and equipment, proper training, and routine implementation of a quality assurance/quality control (QA/QC) program.
Analysis of drug resistance.
The WHO 2009 list of mutations for surveillance of transmitted drug-resistant HIV strains was used for analysis of protease inhibitor (PI)-, NRTI-, and NNRTI-associated mutations (2). The list of PI-associated mutations included 40 mutations at 18 positions across protease. The list of NRTI mutations included 34 mutations at 15 positions in RT. The list of NNRTI mutations included 19 mutations at 10 positions across RT. The International AIDS Society (IAS)-USA list (2014 update) of drug resistance mutations in HIV-1 was used for analysis of integrase strand transfer inhibitors (20 mutations at 11 positions in integrase) and entry inhibitors (10 mutations at 7 positions in gp41) (3).
APOBEC-induced hypermutations.
The APOBEC-induced hypermutations were assessed by Hypermut (83) at the Los Alamos National Laboratory (LANL) HIV Database (http://www.hiv.lanl.gov/). The HIV-1 subtype C (HIV-1C) consensus sequence was used as a reference. Two parameters related to APOBEC-induced hypermutations were analyzed: adjusted hypermutations and the hypermutation ratio. The adjusted hypermutations were expressed as a number of identified hypermutations adjusted by sequence length. The hypermutation ratio was computed as the ratio between weighted mutations (matched mutations out of potential mutations) and weighted controls (control mutations out of potential controls) and was derived as a statistical outcome of the Hypermut package (83).
Definition of the HIV cluster.
An HIV cluster was defined as a viral lineage that gives rise to a monophyletic subtree of the overall phylogeny with strong statistical support. The bootstrapped maximum-likelihood (ML) method (84–86) was used to determine the statistical support of clusters. The four bootstrap thresholds for identification of HIV clusters were ≥0.7, ≥0.8, ≥0.9, and 1.0. A viral lineage (group or subtree) with at least two viral sequences and specified statistical support was considered to be an HIV cluster. Clusters were identified using a depth-first algorithm (87, 88), a method for traversing or searching tree or graph data structures starting from the root. This approach eliminated double counting of viral sequences in clusters when the clusters had internal structure with strong support.
Confidentiality.
The sharing of data, including generated HIV sequences, with the scientific community for the purpose of research is of key importance in ensuring continued progress in our understanding of how to contain the HIV epidemic. The confidentiality of study subjects was protected by recoding of HIV sequences deposited in GenBank at the country level (with no community or village data).
Phylogenetic inference.
The ML tree inference was implemented in RAxML (89, 90) under the GAMMA model of rate heterogeneity. The statistical support for each node was assessed by bootstrap analysis from 100 bootstrap replicates performed with the rapid bootstrap algorithm implemented in RAxML (89). The RAxML runs were performed using RAxML version 8.0.20 at the high-performance computing cluster Odyssey (https://rc.fas.harvard.edu/resources/odyssey-architecture/) at the Faculty of Arts and Sciences, Harvard University (https://rc.fas.harvard.edu/).
Proportion of HIV-1C sequences in clusters.
To test whether the extent of HIV clustering is associated with any subgenomic region, the proportion of clustered sequences was compared between long (amplicon 1, amplicon 2, concatenated amplicons 1 plus 2, and concatenated amplicon 1 plus V1C5) and short (ViroSeq and V1C5) HIV-1C sequences. The proportion of HIV sequences in clusters was estimated at the bootstrap thresholds for cluster definition from 0.7 to 1.0 under ML inference.
Statistical analysis.
The HIV sequences in clusters were enumerated with PhyloPart v.2 (88) using bootstrap thresholds of 0.7, 0.8, 0.9, and 1.0. All confidence intervals (CI) of estimated proportions are asymptotic 95% binomial confidence intervals (95% CI) computed with the prop.test() function in R version 3.1.2 (91). Comparisons of continuous outcomes between two groups were performed using the Wilcoxon rank sum test. P values of less than 0.05 were considered statistically significant. All reported P values are 2 sided. Proportions of viral sequences in clusters between targeted loci were compared by McNemar's test in R, and P values of less than 1.0E−04 were considered statistically significant. All plots were produced in R. All figures were finalized in Adobe Illustrator CS6.
Nucleotide sequence accession numbers.
The generated HIV sequences were deposited in GenBank. For sequences used in this study, the accession numbers are KR860607 to KR861255 for 649 amplicon 1 sequences and KR861256 to KR861345 for 90 amplicon 2 sequences.
RESULTS
Long-range HIV genotyping.
The original protocol for nearly full-length HIV-1 genome genotyping by amplification of four large overlapping amplicons in a single round of RT-PCR using viral RNA as a template was developed by Gall et al. (62). The protocol of Gall et al. (62) is robust and highly reproducible for samples with relatively high HIV-1 RNA levels. However, specimens with low or undetectable levels of HIV-1 RNA presented a substantial challenge for amplification from viral RNA. Attempts to apply the original protocol to proviral DNA produced large numbers of nonspecific products evident from smeared “ladders” on the electrophoretic gel (data not shown).
The modifications of the protocol of Gall et al. (62) included the following steps: (i) focus on 2 (amplicons 2 and 4 in the original protocols) instead of 4 amplicons, (ii) an extra round of PCR where the amplified ∼8.3-kb product was used as a template for the second round of PCR, (iii) highly specific primers for the first round of PCR and for cDNA synthesis (for viral RNA templates), and (iv) modified PCR running conditions.
The rationale for focusing on two instead of four amplicons was driven by a balance between sequencing data and cost. The two amplicons have lengths of 3,574 bp and 3,550 bp (HXB2 nucleotide length), which cumulatively covers about 80% of the unique full-length HIV-1 genome sequence (Fig. 1). The first amplicon (corresponding to amplicon 2 in the study by Gall et al. [62]) spans partial gag at the 3′ end and almost the entire pol (HXB2 nucleotide positions 1,486 to 5,058). The second amplicon (corresponding to amplicon 4 in the study by Gall et al. [62]) spans vpu, env, nef, and the 3′ LTR up to the TATA box in the U3 region (HXB2 nucleotide positions 5,967 to 9,517).
FIG 1.
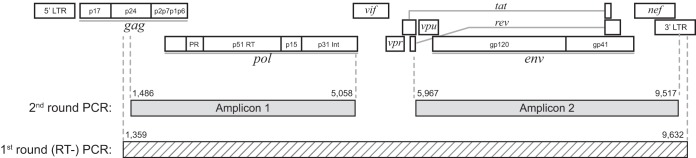
Overview of long-range HIV genotyping. The 1st- and 2nd-round products are mapped against the HIV-1 genome structure. The 1st-round (RT) PCR product is shown at the bottom as a hatched bar. The 2nd-round PCR products, amplicon 1 and amplicon 2, are shown as gray bars.
Amplification of a large fragment spanning almost the entire HIV-1 genome (Fig. 1, hatched bar) was introduced as the first round of PCR (RT-PCR for the RNA template). Primers OFM19 and SK145 (see Table S1 in the supplemental material) substantially increased the specificity of viral amplification. For the proviral DNA template, the 1st round of PCR was run with primers SK145 and OFM19. The PrimeStar GXL DNA polymerase (TaKaRa; catalog number R050A) was used in 30 amplification cycles with the annealing temperature at 62°C (98°C for 10 s, 62°C for 15 s, and 68°C for 9 min cycling). For the RNA template, cDNA synthesis with primer OFM19 was followed by PCR with primers SK145 and OFM19 in a single-tube RT-PCR. The SuperScript III One-Step RT-PCR High Fidelity enzyme (Invitrogen; catalog number 12574035) was used with a cDNA synthesis step of incubation at 50°C for 60 min and 94°C for 2 min, followed by 30 cycles of amplification in the 1st PCR round (94°C for 15 s, 62°C for 30 s, and 68°C for 9 min cycling). In cases of specimens from subjects with low viral loads, a lower annealing temperature between 58°C and 60°C was used in the 1st round.
The 1st-round product was used as the template in two separate 2nd-round PCRs with specific primers (see Table S1 in the supplemental material) to obtain amplicon 1 and amplicon 2 (Fig. 1, gray bars). The PrimeStar GXL DNA polymerase (TaKaRa; catalog number R050A) was used in 30 amplification cycles with the annealing temperature at 62°C (98°C for 10 s, 62°C for 15 s, and 68°C for 4 min cycling). No additional extension step was performed at the end of the run.
After standard purification with USB ExoSap-It (92) (Affymetrix; catalog number 782011ML), amplicon 1 was subjected to direct Sanger sequencing on both strands using a total of 12 sequencing primers (see Table S2 in the supplemental material). In about 30% of cases, direct sequencing of amplicon 1 failed, apparently due to the heterogeneity of the amplified products. These cases were cloned and Sanger sequenced on both strands. All amplicon 2 products were cloned before Sanger sequencing on both strands, with a total of 12 sequencing primers (see Table S2 in the supplemental material). Direct Sanger sequencing was performed on the ABI 3730 DNA analyzer using BigDye technology.
The high diversity of HIV presents a challenge for direct Sanger sequencing. Samples collected during the early stage of HIV infection are relatively homogeneous (in the case of transmission of a single HIV variant). In contrast, samples obtained from chronically infected individuals are likely to include a heterogeneous pool of viral quasispecies. High heterogeneity of viral quasispecies combined with numerous insertions and deletions (indels) could result in low quality of the directly sequenced specimens. In this case, cloning may be considered an alternative solution to direct sequencing. If the time of HIV infection is unknown, the diversity of the targeted region, or subregion, could guide the initial sequencing strategy. Amplicon 1 spans a relatively conserved region of the HIV-1 genome. In contrast, amplicon 2 includes the most variable regions of the HIV-1 genome, with multiple indels. Our preliminary results suggest that applying cloning to about 30% of amplicon 1 sequences and to 100% of amplicon 2 sequences is the most efficient sequencing strategy to overcome the complexity of HIV quasispecies. The goal of this study was to obtain a single HIV sequence per subject. Therefore, generation of a single amplicon 1 and a single amplicon 2 sequence was considered a success. If a study aimed to address the multiplicity of HIV infection or the diversity of viral quasispecies, multiple sequences (e.g., 20 per targeted region per subject) could be generated by appropriate amplification methods.
Cloning was performed with a PCR cloning kit (NEB; catalog number E1202S) using Fast-Media Amp XGal (Invivogen; catalog number fas-am-x). Ligation, transformation, and plating were performed according to the manufacturer's instructions. Colonies were checked for inserts with EmeraldAmp GT PCR master mix (TaKaRa; catalog number RR310A) and submitted to GENEWIZ for colony sequencing. A list of sequencing primers used with clones is presented in Table S3 in the supplemental material.
All sequence contigs were assembled with SeqScape v.2.7.
Troubleshooting.
Some amplification issues during long-range HIV genotyping, such as lack of or insufficient amplification, an overamplified product, or the presence of multiple bands, could be resolved by troubleshooting. The initial amplification results could guide troubleshooting. A lack of visible bands (or weak bands) on the gel after second-round PCR could be resolved by decreasing the annealing temperature in the first-round PCR to 58°C and/or increasing the number of cycles in the first-round PCR to 35 or increasing the amount of RNA template (e.g., up to 5 μl). The overamplified products could be overcome by reducing the number of cycles in the first-round PCR to 25 or in the second-round PCR to 20 to 25 or by decreasing the amount of input template. Multiple bands on the gel could be resolved by either extracting the band of the right size from the gel using the Wizard SV Gel and PCR Clean-Up System (Promega; catalog number A9281) or rerunning the first-round PCR in replicates and with serial dilutions.
HIV genotyping results.
Amplicon 1 was amplified and sequenced in 649 HIV-infected subjects (a single sequence per subject), while amplicon 2 was amplified and sequenced in 90 subjects.
The long-range HIV genotyping from proviral DNA was applied to 212 specimens collected from subjects participating in the BCPP baseline household survey in the first four communities, Ranaka, Digawana, Molapowabojang, and Otse, from November 2013 until June 2014. The distribution of amplified and sequenced samples from proviral DNA is presented in Table 1. Amplicon 1 was successfully amplified in 89.6% (95% CI, 84.5% to 93.2%) of the cases. Viral sequences were obtained for all amplified samples. The majority of amplified amplicon 1 sequences, 144 of 167 (86.2%; 95% CI, 79.8% to 90.9%), were obtained by direct Sanger sequencing. In 23 cases (12.1%; 95% CI, 8.0% to 17.8%), amplicon 1 sequences obtained by direct Sanger sequencing had gaps that did not exceed 10% of the amplicon 1 length. Cloning followed by Sanger sequencing helped to resolve the gaps in all 23 cases.
TABLE 1.
Summary of HIV genotyping from proviral DNA, amplicon 1 (BCPP)
| Parameter | Value for proviral DNA specimens |
|||
|---|---|---|---|---|
| Total |
Subset with available HIV-1 RNA load data |
|||
| n | Proportion (95% CI) | n | Proportion (95% CI) | |
| Attempted cases | 212 | 202 | ||
| Amplified cases | 190 | 0.896 (0.845–0.932) | 181 | 0.896 (0.843–0.933) |
| Cases in which amplification faileda | 22 | 0.104 (0.068–0.155) | 21 | 0.104 (0.067–0.157) |
| Cases sequenced by direct Sanger sequencingb | 167 | 0.879 (0.822–0.920) | 160 | 0.884 (0.826–0.925) |
| Cases with partial sequences by direct Sanger sequencingb,c | 23 | 0.121 (0.080–0.178) | 21 | 0.116 (0.075–0.174) |
The proportion of failed cases was calculated from the number of attempted cases.
The proportion of sequenced cases was calculated from the number of amplified cases.
Gaps at ≤10% of sequence length; resolved by cloning.
The levels and distribution of the HIV-1 RNA load in amplified and nonamplified specimens from proviral DNA were of particular interest. HIV-1 RNA load data were available for a subset of 202 HIV-positive subjects from the BCPP study. The proportion of successfully amplified cases was 89.6% (95% CI, 84.3% to 93.3%) (Table 1). Sequences were obtained for all amplified products. Partial sequences (less than 10% missing data) were obtained in 11.6% (95% CI, 7.5% to 17.4%) of the amplified cases.
The distribution of the HIV-1 RNA load among specimens amplified and sequenced from proviral DNA is presented in Fig. 2. The histogram shows the distribution of HIV-1 RNA among 202 specimens with available viral load data (both amplified and failed specimens). The distribution indicates that a high proportion of subjects participating in the baseline household survey in four BCPP communities had suppressed levels of HIV-1 RNA, primarily due to a high proportion of HIV-infected individuals receiving ART. In fact, 71.3% (95% CI, 64.4% to 77.3%) of HIV-infected subjects had HIV-1 RNA levels below 1,000 cps/ml, including 65.3% (95% CI, 58.3% to 71.8%) with undetectable HIV-1 RNA, below 40 cps/ml. Distributions of HIV-1 RNA were similar among specimens amplified from proviral DNA (n = 181) and failed specimens (n = 21) (Fig. 2, pie charts).
FIG 2.

Distribution of the HIV-1 RNA load in BCPP samples (n = 202) that were subjects of long-range HIV genotyping using proviral DNA as a template for amplification. The histogram depicts the distribution of HIV-1 RNA in all samples (n = 202). The x axis shows HIV-1 RNA on a log10 scale. The two pie charts illustrate the distributions of HIV-1 RNA among successfully amplified (n = 181) and failed (n = 21) samples. The legend on the right outlines the breakdown intervals of HIV-1 RNA presented in the pie charts.
Amplification and sequencing of amplicon 2 were completed for 90 subjects. Given that the first-round (RT) PCR product is used for amplification of both amplicons 1 and 2, obtaining amplicon 1 suggests a successful amplification of amplicon 2. Amplification of the overlapping product, designated “amplicon 3” in the study by Gall et al. (62), should be possible, as the first-round PCR product completely covers amplicon 3. This strategy has not been explored yet.
Overall, the long-range HIV genotyping from proviral DNA (for amplicon 1) was successful in about 90% of targeted blood specimens collected in a cohort where a majority of the patients had suppressed viral loads, including 65% of patients with undetectable HIV-1 RNA loads.
Amplification from viral RNA.
To assess the utility of long-range HIV genotyping for amplification and sequencing from a viral RNA template, we performed small-scale genotyping (n = 32) from viral RNA in plasma (Table 2). The HIV-1 RNA load was available for 31 of these samples and was above 1,000 cps/ml in 29 cases. A subset of 23 specimens were successfully amplified and sequenced. Interestingly, two of nine specimens that failed amplification from proviral DNA (HIV-1 RNA loads, 1,576 cps/ml and 5,620 cps/ml), were successfully amplified from viral RNA.
TABLE 2.
Summary of HIV genotyping from viral RNA, amplicon 1 (BCPP)
| Parameter | Value for viral RNA specimens |
|||
|---|---|---|---|---|
| Total |
Subset with available HIV-1 RNA load data |
|||
| n | Proportion (95% CI) | n | Proportion (95% CI) | |
| Attempted cases | 32 | 31 | ||
| Amplified cases | 23 | 0.719 (0.530–0.856) | 23 | 0.742 (0.551–0.875) |
| Cases in which amplification faileda | 9 | 0.281 (0.144–0.470) | 8 | 0.258 (0.125–0.449) |
| Sequenced casesb | 23 | 1.0 (0.822–1.0) | 23 | 1.0 (0.822–1.0) |
The proportion of failed cases was calculated from the number of attempted cases.
The proportion of sequenced cases was calculated from the number of amplified cases.
The nine failed cases included one sample with unknown and eight specimens with available viral loads. Among the latter group, two samples had viral loads below 1,000 cps/ml (181 and 497 cps/ml), 5 samples had viral loads between 1,191 and 8,528 cps/ml, and 1 sample had a viral load of 156,821 cps/ml. The last failed sample, with a high viral load, also failed amplification from proviral DNA, apparently suggesting an intrinsic problem with mismatch of amplification primers.
Analysis of mutations associated with antiretroviral drug resistance.
Amplicon 1 covers almost the entire HIV-1 pol gene and allows analysis of mutations associated with antiretroviral drug resistance to PIs, NRTIs, NNRTIs, and integrase strand transfer inhibitors. Amplicon 2 covers the entire HIV-1 env gene and allows analysis of mutations associated with drug resistance to virus entry inhibitors.
To illustrate the validity of long-range HIV genotyping for analysis of mutations associated with antiretroviral drug resistance, we estimated drug resistance profiles within two groups of specimens originating from the MPP and BCPP studies. Amplicon 1 sets included 192 MPP sequences and 186 BCPP sequences. Amplicon 2 sets included 35 MPP and 55 BCPP sequences.
Despite relatively rare use of protease inhibitors in Botswana, mutations associated with resistance to PIs were detected at five positions in protease: D30N (5% in MPP and 6% in BCPP), M46I (5% in MPP and 10% in BCPP), G73S (10% in MPP and 9% in BCPP), I85V (1% in MPP), and N88S (1% in BCPP). The encoding analysis revealed that all 22 D30N mutations were caused by GAT (Asp)-to-AAT (Asn) substitutions, 26 of 27 M46I mutations were due to ATG (Met)-to-ATA (Ile) substitutions, and 35 of 36 G73S mutations were found because of GGT (Gly)-to-AGT (Ser) substitutions. Thus, it is likely that the majority of identified mutations in the protease gene were caused by G-to-A hypermutations.
NRTIs and NNRTIs have been part of the national antiretroviral program in Botswana since 2002. Viral mutations associated with resistance to NRTIs were found at the following positions across RT: M41L (1% in BCPP), D67N (1% in MPP and 2% in BCPP), K70R (1% in BCPP), K70E (1% in BCPP), V75M (1% in BCPP), M184V (2% in BCPP), M184I (16% in BCPP), and T215Y (1% in BCPP). Almost all (60 out of 61) M184I mutations were caused by ATG (Met)-to-ATA (Ile) substitutions. Mutations to NNRTIs were observed at multiple RT positions and demonstrated low frequency: K101E (1% in MPP and 1% in BCPP), K103N (1% in MPP and 3% in BCPP), K103S (1% MPP in and 1% in BCPP), Y181C (1% in BCPP), Y188C (1% in BCPP), G190A (1% in BCPP), G190S (1% in BCPP), G190E (1% in MPP and 1% in BCPP), and P225H (1% in BCPP).
HIV mutations associated with integrase strand transfer inhibitors were detected at three positions in integrase: L74M (1% in MPP), T97A (3% in MPP and 1% in BCPP), and E138K (3% in MPP and 6% in BCPP). Mutations to entry inhibitors were found at the following positions in gp41: G36S (24% in BCPP) and V38M (2% in BCPP). All 13 out of 55 G36S mutations were caused by a switch from GGT (Gly) to AGT (Ser), which is a likely effect of G-to-A hypermutation.
G-to-A hypermutations.
The presence of G-to-A hypermutations in the products amplified from proviral DNA is not surprising, as massive APOBEC-induced G-to-A transitions in retroviruses are well recognized as a key innate defense by the host. The distribution of identified APOBEC-induced hypermutations in HIV-1C sequences amplified from proviral DNA is presented in Fig. 3.
FIG 3.

Distributions of APOBEC-induced hypermutations in sequences amplified from proviral DNA (histograms). The horizontal box plots outline the distributions of APOBEC-induced hypermutations in subsets of sequences with identified drug resistance mutations. The box plots are drawn to the x-axis scale. The left and right box boundaries indicate lower and upper quartiles, the line within the box is the median, and the left and right whiskers indicate minimum and maximum values without outliers. (A and B) Amplicon 1 (n = 649), distribution of hypermutations adjusted by sequence length (A) and distribution of hypermutation ratio data (B) (see Materials and Methods). (C and D) Amplicon 2 (n = 90), distribution of hypermutations adjusted by sequence length (C) and distribution of hypermutation ratio data (D) (see Materials and Methods).
The sequence lengths among the 649 cases analyzed differed, ranging from 3,190 bp to 3,625 bp. Therefore, the number of potentially G-to-A-hypermutated sites (compared to the HIV-1 subtype C consensus sequence) was adjusted for the sequence length and expressed as a proportion. Two cutoff values, 0.02 and 0.05, were used to demonstrate the proportion of viral sequences in the analyzed set with potential G-to-A hypermutations. Figures 3A and C demonstrate the distribution of G-to-A hypermutations adjusted by sequence length for amplicons 1 (n = 649) and 2 (n = 90), respectively. For example, 125 of 649 amplicon 1 sequences (19.3%; 95% CI, 16.3% to 22.6%) and 37 of 90 amplicon 2 sequences (41.1%; 95% CI, 31.0% to 52.0%) exceeded the 0.02 level of adjusted hypermutations. On the other hand, 23 of 649 amplicon 1 sequences (3.5%; 95% CI, 2.3% to 5.4%) and 11 of 90 amplicon 2 sequences (12.2%; 95% CI, 6.6% to 21.2%) were above the 0.05 level of adjusted hypermutations. Figures 3B and D show the distributions of the hypermutation ratios estimated with Hypermut (83). Both metrics indicate the presence of APOBEC-induced hypermutations among amplicon 1 and amplicon 2 sequences amplified from proviral DNA.
The majority of viral sequences with antiretroviral mutations had high rates of APOBEC-induced hypermutations, suggesting association between hypermutations and drug resistance mutations. The distribution of APOBEC-induced hypermutations among 36 MPP sequences with drug resistance mutations within protease, RT, and integrase is presented in Table S4 in the supplemental material, while hypermutations among 46 BCPP sequences with drug resistance mutations are presented in Table S5 in the supplemental material. It is evident that many hypermutated sequences have multiple drug resistance mutations due to G-to-A transitions.
HIV-1C sequences with identified drug resistance mutations demonstrated high rates of APOBEC-induced hypermutations. The horizontal box plots in Fig. 3 indicate the distributions of the adjusted numbers of hypermutations (Fig. 3A and C) and the hypermutation ratios (Fig. 3B and D) among viral sequences with drug resistance mutations in relation to the distribution of hypermutation parameters in the entire set of sequences. Comparison of these distributions indicates an association between APOBEC-induced hypermutations and drug resistance mutations.
To further address how G-to-A hypermutations can affect drug resistance mutations, we compared hypermutations between two groups with and without M184I mutations. Individuals with M184I mutations have higher adjusted numbers of hypermutations (Fig. 4A) and higher hypermutation ratios (Fig. 4B). Summary statistics are presented at the bottom of Fig. 4. The differences were highly significant for both comparisons (P < 0.0001; Wilcoxon rank sum test).
FIG 4.

G-to-A hypermutations in HIV-1C sequences with and without the M184I mutation. Bean plots (a combination of a box plot, a density plot, and a rug with ticks for each value in the middle) are shown (96). Comparison between groups was performed by a Wilcoxon signed-rank test. (A) Hypermutations adjusted by sequence length. (B) Hypermutation ratios. Summary statistics are presented at the bottom.
HIV cluster analysis.
To demonstrate the utility of the long-range HIV genotyping for analysis of HIV transmission dynamics and viral linkage, we compared the extents of clustering within viral sequences generated in this study. The concatenated amplicons 1 and 2 span over 80% of the unique HIV-1 genome sequence. In this study, the number of matched amplicon 1 plus 2 sequences was limited to 83. The extents of HIV clustering within this small set were compared for three long loci—amplicon 1 (3,574 bp), amplicon 2 (3,550 bp), and the concatenated amplicons 1 plus 2 (7,124 bp)—and two short loci, ViroSeq (1,263 bp) and V1C5 (1,188 bp).
The proportions of clustered HIV sequences were compatible within long loci (Table 3). For example, at a bootstrap support of ≥0.80, the proportions of clustered HIV sequences were 0.265, 0.289, and 0.337 for amplicon 1, amplicon 2, and concatenated amplicons 1 plus 2, respectively. For the short loci ViroSeq and V1C5 at the same bootstrap support of ≥0.80, the proportions of HIV sequences in clusters were 0.157 and 0.145, respectively. The proportion of clustered sequences seemed to be higher for long regions than for short loci, although the difference reached significance at the 0.05 level in selected comparisons only.
TABLE 3.
Observed proportions of HIV-1C sequences in clusters in a small set of sequences (n = 83)
| Locus | No. (proportion) of HIV-1C sequences in clusters at a bootstrap support of splits of: |
|||
|---|---|---|---|---|
| ≥0.70 | ≥0.8 | ≥0.9 | 1.0 | |
| Amplicon 1 | 32 (0.386)a,b | 22 (0.265) | 19 (0.229) | 11 (0.133) |
| Amplicon 2 | 32 (0.386)a,b | 24 (0.289)b | 24 (0.289)a,b | 12 (0.145) |
| Amplicons 1 + 2 | 33 (0.398)a,b | 28 (0.337)a,b | 24 (0.289)a,b | 11 (0.133) |
| ViroSeq | 16 (0.193) | 13 (0.157) | 11 (0.133) | 7 (0.084) |
| V1C5 | 16 (0.193) | 12 (0.145) | 9 (0.108) | 4 (0.048) |
P < 0.05 for comparison to ViroSeq (Fisher exact test).
P < 0.05 for comparison to V1C5 (Fisher exact test).
A larger set of available HIV-1C sequences (n = 547) included matched viral sequences for amplicon 1 and the V1C5 region of gp120 generated in our previous studies (34, 35, 80). Clustering patterns were compared for two long loci, amplicon 1 and concatenated amplicon 1 plus V1C5, and for two short regions across the HIV-1 genome, ViroSeq and V1C5. Similar to the small set of HIV sequences (n = 83), the proportion of clustered sequences in the large set (n = 547) was higher for long loci than for short regions (Table 4).
TABLE 4.
Observed proportions of HIV-1C sequences in clusters in a large set of sequences (n = 547)
| Locus | No. (proportion) of HIV-1C sequences in clusters at a bootstrap support of splits of: |
|||
|---|---|---|---|---|
| ≥0.70 | ≥0.8 | ≥0.9 | 1.0 | |
| Amplicon 1 | 251 (0.459)a,b | 215 (0.393)a,b | 159 (0.291)a,b | 88 (0.161)a,b |
| Amplicon 1 + V1C5 | 267 (0.488)a | 220 (0.402)a,b | 181 (0.331)a,b | 122 (0.223)a,b |
| ViroSeq | 120 (0.219) | 90 (0.165) | 73 (0.133) | 34 (0.062) |
| V1C5 | 135 (0.247) | 114 (0.208) | 88 (0.161) | 44 (0.080) |
P < 0.001 for comparison to ViroSeq (Fisher exact test).
P < 0.001 for comparison to V1C5 (Fisher exact test).
To address whether longer loci are associated with a greater extent of HIV clustering, we analyzed congruent (++ and −−) and discordant (+− and −+) clustering between different combinations of long and short HIV-1C sequences (Fig. 5). At all bootstrap thresholds from 0.70 to 1.0, amplicon 1 and concatenated amplicon 1 plus V1C5 demonstrated greater extents of HIV clustering than ViroSeq and V1C5 sequences (Fig. 5, gray background, indicating a significant difference).
FIG 5.

Clustering of HIV-1C sequences by locus (n = 547). The proportions of HIV-1C sequences in clusters were estimated by bootstrapped ML inference. The extent of HIV clustering was analyzed at bootstrap thresholds for cluster definition of ≥0.70, ≥0.80, ≥0.90, and 1.0. The numbers of viral sequences found in clusters for a specified locus and at a specified bootstrap support were compared between loci. Four loci were used: amplicon 1 concatenated with the V1C5 region of gp120 (Amp 1 + V1c5), amplicon 1 alone (Amp 1), the ViroSeq sequence (ViroSeq), and the V1C5 region of gp120 (V1C5). The pie charts show concordant (++ and −−) and discordant (+− and −+) clustering between specified sequence loci (the first sign corresponds to the first sequence locus listed). Cases of significantly different clustering between loci with P values of less than 1.0E−04 in McNemar's test are highlighted with gray backgrounds.
Estimated cost of long-range HIV genotyping.
The estimated cost for amplification and Sanger sequencing of both amplicons 1 and 2 in this study was $137.50 for proviral DNA and $139.75 for viral RNA. This includes the cost of reagents, materials, and disposables for nucleic acid isolation, amplification (RT-PCR and PCR), purification of amplicons, cloning up to 30% of amplicon 1 products and 100% of amplicon 2 products, and Sanger sequencing. The estimated cost does not include labor, training, supervision, or indirect costs.
DISCUSSION
A technique for long-range HIV genotyping from both viral RNA and proviral DNA has been presented. Using proviral DNA as a template, long-range HIV genotyping was successfully performed in one of the BCPP cohorts with a high proportion of virologically suppressed individuals, with a success rate of about 90%.
Both clinical trials and clinical care could benefit from routine use of long-range HIV genotyping. The proposed long-range HIV genotyping has the potential to improve the methodology of drug resistance testing, to broaden the spectrum of monitored ARVs, and to enable surveillance of transmitted drug resistance. Mapping of HIV transmission networks performed by long-range genotyping could help reveal transmitting viral variants in treatment-as-prevention studies. Implementation of long-range HIV genotyping could allow greater refinement of viral linkage analyses in HIV prevention studies and better coordination with evaluation of prevention strategies based on such interventions as behavior change, male circumcision, and treatment as prevention. The cost-effective long-range HIV genotyping technique has the potential to enable the scale up of public health HIV prevention interventions across communities.
In this study, we demonstrated that long-range HIV genotyping using proviral DNA could be successfully applied to a population with a high level of ART-experienced individuals, which normally presents a challenge for HIV genotyping from viral RNA. In fact, more than 70% of individuals in the BCPP cohort participating in the baseline household survey in the first four communities had HIV-1 RNA levels below 1,000 cps/ml, including 65% with undetectable levels of HIV-1 RNA, below 40 cps/ml. The ongoing scale up of national ARV programs in Africa has led to a growing number of individuals with suppressed HIV-1 RNA loads across communities. The presented technique of long-range HIV genotyping from proviral DNA should alleviate challenges and enable analysis of HIV drug resistance and HIV transmission dynamics using samples collected from individuals on ART.
A comprehensive strategy of HIV genotyping could include two steps. First, a viral RNA template for amplification could be targeted, if the HIV-1 RNA load is relatively high (e.g., above 1,000 cps/ml). If amplification is successful, there is no need for proviral DNA. However, if amplification from viral RNA does not work, or the HIV-1 RNA load is below 1,000 cps/ml or undetectable, using proviral DNA is a logical step toward successful HIV genotyping. Complementary use of both viral RNA and proviral DNA templates could be an efficient and cost-effective approach for HIV genotyping.
Long-range HIV genotyping enables analysis of drug resistance (both transmitted and acquired) for all major groups of ARVs, including protease inhibitors, NRTIs, NNRTIs, integrase strand transfer inhibitors, and virus entry inhibitors. A comprehensive analysis of HIV drug resistance is feasible due to the long sequence length of generated amplicons that span the HIV-1 pol and env genes. While long-range HIV genotyping is able to identify drug resistance mutations, it cannot distinguish transmitted and acquired drug resistance mutations without additional information on the sampling strategy and stage of HIV infection. For example, drug resistance mutations identified in individuals during early stages of HIV infection (e.g., in seroconverters) are likely to represent transmitted drug resistance mutations. In contrast, specimens collected in chronic HIV infection, or from individuals on ART, are more likely to be associated with acquired HIV drug resistance.
The G-to-A hypermutations observed in sequences amplified from proviral DNA and their relation to drug resistance mutations should be interpreted cautiously in the context of a specific study. Our data suggest that G-to-A hypermutations are likely to contribute to critical drug resistance mutations, such as M184I. We recommend controlling viral sequences generated from proviral DNA for the adjusted number of hypermutations and/or the hypermutation ratio using the online package Hypermut (83) at the LANL HIV Database (http://www.hiv.lanl.gov/) and the subtype consensus sequence as a reference. Based on interquartile range (IQR) boundaries in our data, the adjusted number of hypermutations above 2.8% (the 1st IQR in individuals with M184I) indicates a hypermutated sequence and that below 0.5% (the 3rd IQR in individuals without M184I) suggests a nonhypermutated sequence. Whether HIV-associated drug resistance mutations should be interpreted differently depending on the extent of G-to-A hypermutations still needs to be addressed in future studies.
The study showed the utility of long-range genotyping for analysis of HIV transmission dynamics and HIV clustering. A greater extent of clustering for longer HIV sequences in this study corroborates the results of our recent study (93), which used a set of nearly full-length HIV-1C sequences from the LANL HIV Database (http://www.hiv.lanl.gov/). Longer HIV sequences are more informative for HIV cluster analysis due to a larger number of informative sites (93). The technique of long-range HIV genotyping allows the use of amplicon 1 and amplicon 2 sequences either separately or in concatenation for a powerful cluster analysis. The concatenated amplicons 1 and 2 span about 80% of the unique HIV-1 genome sequence and could be considered a cheaper alternative to nearly full-length HIV-1 sequencing. A combination of conserved (amplicon 1) and variable (amplicon 2) regions could help to deal with different and/or unknown stages of HIV infection in an analyzed set of viral sequences. The choice of a particular bootstrap value and filtering by the threshold of pairwise distances and/or internode certainty (94, 95) could depend on the specific scientific question and take into account the specifics of an analyzed set of sequences, including sampling density (35).
In summary, the presented technique of long-range HIV genotyping using viral RNA and proviral DNA can help in analysis of HIV drug resistance and HIV clustering in cohorts and populations on ART when amplification from viral RNA is unsuccessful due to low HIV-1 RNA loads.
Supplementary Material
ACKNOWLEDGMENTS
We are very grateful to all participants in the BHP projects in Botswana. We thank the CDC and the Botswana Ministry of Health for their collaboration. We thank Lendsey Melton for excellent editorial assistance.
The Mochudi Prevention Project in Botswana was supported and funded by NIH grant R01 AI083036, an HIV Prevention Program for Mochudi, Botswana. The GWAS on Determinants of HIV-1 Subtype C Infection study was supported and funded by NIH grant RC4 AI092715. The Botswana Combination Prevention Project (BCPP) has been supported by the President's Emergency Plan for AIDS Relief (PEPFAR) through the U.S. Centers for Disease Control and Prevention under the terms of grant number U01 GH000447.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JCM.00756-15.
REFERENCES
- 1.Johnson VA, Calvez V, Gunthard HF, Paredes R, Pillay D, Shafer RW, Wensing AM, Richman DD. 2013. Update of the drug resistance mutations in HIV-1: March 2013. Top Antivir Med 21:6–14. [PMC free article] [PubMed] [Google Scholar]
- 2.Bennett DE, Camacho RJ, Otelea D, Kuritzkes DR, Fleury H, Kiuchi M, Heneine W, Kantor R, Jordan MR, Schapiro JM, Vandamme AM, Sandstrom P, Boucher CA, van de Vijver D, Rhee SY, Liu TF, Pillay D, Shafer RW. 2009. Drug resistance mutations for surveillance of transmitted HIV-1 drug-resistance: 2009 update. PLoS One 4:e4724. doi: 10.1371/journal.pone.0004724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wensing AM, Calvez V, Gunthard HF, Johnson VA, Paredes R, Pillay D, Shafer RW, Richman DD. 2014. 2014 update of the drug resistance mutations in HIV-1. Top Antivir Med 22:642–650. [PMC free article] [PubMed] [Google Scholar]
- 4.Rojas Sanchez P, Holguin A. 2014. Drug resistance in the HIV-1-infected paediatric population worldwide: a systematic review. J Antimicrob Chemother 69:2032–2042. doi: 10.1093/jac/dku104. [DOI] [PubMed] [Google Scholar]
- 5.Ssemwanga D, Lihana RW, Ugoji C, Abimiku A, Nkengasong J, Dakum P, Ndembi N. 2015. Update on HIV-1 acquired and transmitted drug resistance in Africa. AIDS Rev 17:3–20. [PubMed] [Google Scholar]
- 6.Smit E. 2014. Antiviral resistance testing. Curr Opin Infect Dis 27:566–572. doi: 10.1097/QCO.0000000000000108. [DOI] [PubMed] [Google Scholar]
- 7.Bhargava M, Cajas JM, Wainberg MA, Klein MB, Pant Pai N. 2014. Do HIV-1 non-B subtypes differentially impact resistance mutations and clinical disease progression in treated populations? Evidence from a systematic review. J Int AIDS Soc 17:18944. doi: 10.7448/IAS.17.1.18944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Snedecor SJ, Sudharshan L, Nedrow K, Bhanegaonkar A, Simpson KN, Haider S, Chambers R, Craig C, Stephens J. 2014. Burden of nonnucleoside reverse transcriptase inhibitor resistance in HIV-1-infected patients: a systematic review and meta-analysis. AIDS Res Hum Retroviruses 30:753–768. doi: 10.1089/aid.2013.0262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ambrosioni J, Nicolas D, Sued O, Aguero F, Manzardo C, Miro JM. 2014. Update on antiretroviral treatment during primary HIV infection. Expert Rev Anti Infect Ther 12:793–807. doi: 10.1586/14787210.2014.913981. [DOI] [PubMed] [Google Scholar]
- 10.Iwuji CC, Orne-Gliemann J, Tanser F, Boyer S, Lessells RJ, Lert F, Imrie J, Barnighausen T, Rekacewicz C, Bazin B, Newell ML, Dabis F, ANRS 12249 TasP Study Group . 2013. Evaluation of the impact of immediate versus WHO recommendations-guided antiretroviral therapy initiation on HIV incidence: the ANRS 12249 TasP (Treatment as Prevention) trial in Hlabisa sub-district, KwaZulu-Natal, South Africa: study protocol for a cluster randomised controlled trial. Trials 14:230. doi: 10.1186/1745-6215-14-230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Manasa J, Danaviah S, Pillay S, Padayachee P, Mthiyane H, Mkhize C, Lessells RJ, Seebregts C, de Wit TF, Viljoen J, Katzenstein D, De Oliveira T. 30 March 2014. An affordable HIV-1 drug resistance monitoring method for resource limited settings. J Vis Exp doi: 10.3791/51242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Siliciano JD, Siliciano RF. 2013. Recent trends in HIV-1 drug resistance. Curr Opin Virol 3:487–494. doi: 10.1016/j.coviro.2013.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brenner BG, Roger M, Moisi DD, Oliveira M, Hardy I, Turgel R, Charest H, Routy JP, Wainberg MA. 2008. Transmission networks of drug resistance acquired in primary/early stage HIV infection. AIDS 22:2509–2515. doi: 10.1097/QAD.0b013e3283121c90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brenner BG, Roger M, Routy JP, Moisi D, Ntemgwa M, Matte C, Baril JG, Thomas R, Rouleau D, Bruneau J, Leblanc R, Legault M, Tremblay C, Charest H, Wainberg MA. 2007. High rates of forward transmission events after acute/early HIV-1 infection. J Infect Dis 195:951–959. doi: 10.1086/512088. [DOI] [PubMed] [Google Scholar]
- 15.Brenner BG, Roger M, Stephens D, Moisi D, Hardy I, Weinberg J, Turgel R, Charest H, Koopman J, Wainberg MA. 2011. Transmission clustering drives the onward spread of the HIV epidemic among men who have sex with men in Quebec. J Infect Dis 204:1115–1119. doi: 10.1093/infdis/jir468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brenner BG, Wainberg MA. 2013. Future of phylogeny in HIV prevention. J Acquir Immune Defic Syndr 63(Suppl 2):S248–S254. doi: 10.1097/QAI.0b013e3182986f96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Leventhal GE, Gunthard HF, Bonhoeffer S, Stadler T. 2014. Using an epidemiological model for phylogenetic inference reveals density dependence in HIV transmission. Mol Biol Evol 31:6–17. doi: 10.1093/molbev/mst172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Leventhal GE, Kouyos R, Stadler T, Wyl V, Yerly S, Boni J, Cellerai C, Klimkait T, Gunthard HF, Bonhoeffer S. 2012. Inferring epidemic contact structure from phylogenetic trees. PLoS Comput Biol 8:e1002413. doi: 10.1371/journal.pcbi.1002413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stadler T, Bonhoeffer S. 2013. Uncovering epidemiological dynamics in heterogeneous host populations using phylogenetic methods. Philos Trans R Soc Lond B Biol Sci 368:20120198. doi: 10.1098/rstb.2012.0198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stadler T, Kouyos R, von Wyl V, Yerly S, Boni J, Burgisser P, Klimkait T, Joos B, Rieder P, Xie D, Gunthard HF, Drummond AJ, Bonhoeffer S. 2012. Estimating the basic reproductive number from viral sequence data. Mol Biol Evol 29:347–357. doi: 10.1093/molbev/msr217. [DOI] [PubMed] [Google Scholar]
- 21.Stadler T, Kuhnert D, Bonhoeffer S, Drummond AJ. 2013. Birth-death skyline plot reveals temporal changes of epidemic spread in HIV and hepatitis C virus (HCV). Proc Natl Acad Sci U S A 110:228–233. doi: 10.1073/pnas.1207965110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Frost SD, Volz EM. 2010. Viral phylodynamics and the search for an ‘effective number of infections.’ Philos Trans R Soc Lond B Biol Sci 365:1879–1890. doi: 10.1098/rstb.2010.0060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Volz EM, Ionides E, Romero-Severson EO, Brandt MG, Mokotoff E, Koopman JS. 2013. HIV-1 transmission during early infection in men who have sex with men: a phylodynamic analysis. PLoS Med 10:e1001568. doi: 10.1371/journal.pmed.1001568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Volz EM, Koelle K, Bedford T. 2013. Viral phylodynamics. PLoS Comput Biol 9:e1002947. doi: 10.1371/journal.pcbi.1002947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Volz EM, Koopman JS, Ward MJ, Brown AL, Frost SD. 2012. Simple epidemiological dynamics explain phylogenetic clustering of HIV from patients with recent infection. PLoS Comput Biol 8:e1002552. doi: 10.1371/journal.pcbi.1002552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Volz EM, Kosakovsky Pond SL, Ward MJ, Leigh Brown AJ, Frost SD. 2009. Phylodynamics of infectious disease epidemics. Genetics 183:1421–1430. doi: 10.1534/genetics.109.106021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Leigh Brown AJ, Lycett SJ, Weinert L, Hughes GJ, Fearnhill E, Dunn DT, UK HIV Drug Resistance Collaboration . 2011. Transmission network parameters estimated from HIV sequences for a nationwide epidemic. J Infect Dis 204:1463–1469. doi: 10.1093/infdis/jir550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wertheim JO, Leigh Brown AJ, Hepler NL, Mehta SR, Richman DD, Smith DM, Kosakovsky Pond SL. 2014. The global transmission network of HIV-1. J Infect Dis 209:304–313. doi: 10.1093/infdis/jit524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kuhnert D, Stadler T, Vaughan TG, Drummond AJ. 2014. Simultaneous reconstruction of evolutionary history and epidemiological dynamics from viral sequences with the birth-death SIR model. J R Soc Interface 11:20131106. doi: 10.1098/rsif.2013.1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bezemer D, Faria NR, Hassan AS, Hamers RL, Mutua G, Anzala O, Mandaliya KN, Cane PA, Berkley JA, Rinke de Wit TF, Wallis CL, Graham SM, Price MA, Coutinho R, Sanders EJ. 17 September 2013. HIV-1 transmission networks amongst men having sex with men and heterosexuals in Kenya. AIDS Res Hum Retroviruses doi: 10.1089/AID.2013.0171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bezemer D, Ratmann O, van Sighem A, Dutilh BE, Faria N, van den Hengel R, Gras L, Reiss P, de Wolf F, Fraser C, ATHENA Observational Cohort . 2014. Ongoing HIV-1 subtype B transmission networks in the Netherlands, abstr 205. CROI 2014, Boston, MA: http://www.croiconference.org/sessions/ongoing-hiv-1-subtype-b-transmission-networks-netherlands-0. [Google Scholar]
- 32.Bezemer D, van Sighem A, Lukashov VV, van der Hoek L, Back N, Schuurman R, Boucher CA, Claas EC, Boerlijst MC, Coutinho RA, de Wolf F, ATHENA Observational Cohort . 2010. Transmission networks of HIV-1 among men having sex with men in the Netherlands. AIDS 24:271–282. doi: 10.1097/QAD.0b013e328333ddee. [DOI] [PubMed] [Google Scholar]
- 33.Carnegie NB, Wang R, Novitsky V, De Gruttola V. 2014. Linkage of viral sequences among HIV-infected village residents in Botswana: estimation of linkage rates in the presence of missing data. PLoS Comput Biol 10:e1003430. doi: 10.1371/journal.pcbi.1003430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Novitsky V, Bussmann H, Logan A, Moyo S, van Widenfelt E, Okui L, Mmalane M, Baca J, Buck L, Phillips E, Tim D, McLane MF, Lei Q, Wang R, Makhema J, Lockman S, DeGruttola V, Essex M. 2013. Phylogenetic relatedness of circulating HIV-1C variants in Mochudi, Botswana. PLoS One 8:e80589. doi: 10.1371/journal.pone.0080589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Novitsky V, Moyo S, Lei Q, DeGruttola V, Essex M. 2014. Impact of sampling density on the extent of HIV clustering. AIDS Res Hum Retroviruses 30:1226–1235. doi: 10.1089/aid.2014.0173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Maes B, Schrooten Y, Snoeck J, Derdelinckx I, Van Ranst M, Vandamme AM, Van Laethem K. 2004. Performance of ViroSeq HIV-1 Genotyping System in routine practice at a Belgian clinical laboratory. J Virol Methods 119:45–49. doi: 10.1016/j.jviromet.2004.02.005. [DOI] [PubMed] [Google Scholar]
- 37.Ribas SG, Heyndrickx L, Ondoa P, Fransen K. 2006. Performance evaluation of the two protease sequencing primers of the Trugene HIV-1 genotyping kit. J Virol Methods 135:137–142. doi: 10.1016/j.jviromet.2006.05.010. [DOI] [PubMed] [Google Scholar]
- 38.Church JD, Mwatha A, Bagenda D, Omer SB, Donnell D, Musoke P, Nakabiito C, Eure C, Bakaki P, Matovu F, Thigpen MC, Guay LA, McConnell M, Fowler MG, Jackson JB, Eshleman SH. 2009. In utero HIV infection is associated with an increased risk of nevirapine resistance in Ugandan infants who were exposed to perinatal single dose nevirapine. AIDS Res Hum Retroviruses 25:673–677. doi: 10.1089/aid.2009.0003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cunningham S, Ank B, Lewis D, Lu W, Wantman M, Dileanis JA, Jackson JB, Palumbo P, Krogstad P, Eshleman SH. 2001. Performance of the applied biosystems ViroSeq human immunodeficiency virus type 1 (HIV-1) genotyping system for sequence-based analysis of HIV-1 in pediatric plasma samples. J Clin Microbiol 39:1254–1257. doi: 10.1128/JCM.39.4.1254-1257.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Eshleman SH, Crutcher G, Petrauskene O, Kunstman K, Cunningham SP, Trevino C, Davis C, Kennedy J, Fairman J, Foley B, Kop J. 2005. Sensitivity and specificity of the ViroSeq human immunodeficiency virus type 1 (HIV-1) genotyping system for detection of HIV-1 drug resistance mutations by use of an ABI PRISM 3100 genetic analyzer. J Clin Microbiol 43:813–817. doi: 10.1128/JCM.43.2.813-817.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Eshleman SH, Guay LA, Mwatha A, Brown ER, Cunningham SP, Musoke P, Mmiro F, Jackson JB. 2004. Characterization of nevirapine resistance mutations in women with subtype A vs. D HIV-1 6-8 weeks after single-dose nevirapine (HIVNET 012). J Acquir Immune Defic Syndr 35:126–130. [DOI] [PubMed] [Google Scholar]
- 42.Eshleman SH, Hackett J Jr, Swanson P, Cunningham SP, Drews B, Brennan C, Devare SG, Zekeng L, Kaptue L, Marlowe N. 2004. Performance of the Celera Diagnostics ViroSeq HIV-1 Genotyping System for sequence-based analysis of diverse human immunodeficiency virus type 1 strains. J Clin Microbiol 42:2711–2717. doi: 10.1128/JCM.42.6.2711-2717.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Eshleman SH, Hoover DR, Chen S, Hudelson SE, Guay LA, Mwatha A, Fiscus SA, Mmiro F, Musoke P, Jackson JB, Kumwenda N, Taha T. 2005. Nevirapine (NVP) resistance in women with HIV-1 subtype C, compared with subtypes A and D, after the administration of single-dose NVP. J Infect Dis 192:30–36. doi: 10.1086/430764. [DOI] [PubMed] [Google Scholar]
- 44.Mracna M, Becker-Pergola G, Dileanis J, Guay LA, Cunningham S, Jackson JB, Eshleman SH. 2001. Performance of Applied Biosystems ViroSeq HIV-1 Genotyping System for sequence-based analysis of non-subtype B human immunodeficiency virus type 1 from Uganda. J Clin Microbiol 39:4323–4327. doi: 10.1128/JCM.39.12.4323-4327.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sturmer M, Berger A, Doerr HW. 2003. Modifications and substitutions of the RNA extraction module in the ViroSeq HIV-1 genotyping system version 2: effects on sensitivity and complexity of the assay. J Med Virol 71:475–479. doi: 10.1002/jmv.10527. [DOI] [PubMed] [Google Scholar]
- 46.CDC. Accessed 6 January 2015. ATCC HIV-1 drug resistance genotyping kit. CDC, Atlanta, GA: http://www.atcc.org/products/cells_and_microorganisms/hiv-1_drug_resistance_genotyping_kit.aspx. [Google Scholar]
- 47.Wallis CL, Papathanasopoulos MA, Lakhi S, Karita E, Kamali A, Kaleebu P, Sanders E, Anzala O, Bekker LG, Stevens G, de Wit TF, Stevens W. 2010. Affordable in-house antiretroviral drug resistance assay with good performance in non-subtype B HIV-1. J Virol Methods 163:505–508. doi: 10.1016/j.jviromet.2009.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Youngpairoj AS, Masciotra S, Garrido C, Zahonero N, de Mendoza C, Garcia-Lerma JG. 2008. HIV-1 drug resistance genotyping from dried blood spots stored for 1 year at 4 degrees C. J Antimicrob Chemother 61:1217–1220. doi: 10.1093/jac/dkn100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Devereux HL, Youle M, Johnson MA, Loveday C. 1999. Rapid decline in detectability of HIV-1 drug resistance mutations after stopping therapy. AIDS 13:F123–F127. [PubMed] [Google Scholar]
- 50.Neogi U, Sahoo PN, De Costa A, Shet A. 2012. High viremia and low level of transmitted drug resistance in anti-retroviral therapy-naive perinatally-infected children and adolescents with HIV-1 subtype C infection. BMC Infect Dis 12:317. doi: 10.1186/1471-2334-12-317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhou Z, Wagar N, DeVos JR, Rottinghaus E, Diallo K, Nguyen DB, Bassey O, Ugbena R, Wadonda-Kabondo N, McConnell MS, Zulu I, Chilima B, Nkengasong J, Yang C. 2011. Optimization of a low cost and broadly sensitive genotyping assay for HIV-1 drug resistance surveillance and monitoring in resource-limited settings. PLoS One 6:e28184. doi: 10.1371/journal.pone.0028184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Inzaule S, Yang C, Kasembeli A, Nafisa L, Okonji J, Oyaro B, Lando R, Mills LA, Laserson K, Thomas T, Nkengasong J, Zeh C. 2013. Field evaluation of a broadly sensitive HIV-1 in-house genotyping assay for use with both plasma and dried blood spot specimens in a resource-limited country. J Clin Microbiol 51:529–539. doi: 10.1128/JCM.02347-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang G, Cai F, Zhou Z, DeVos J, Wagar N, Diallo K, Zulu I, Wadonda-Kabondo N, Stringer JS, Weidle PJ, Ndongmo CB, Sikazwe I, Sarr A, Kagoli M, Nkengasong J, Gao F, Yang C. 2013. Simultaneous detection of major drug resistance mutations in the protease and reverse transcriptase genes for HIV-1 subtype C by use of a multiplex allele-specific assay. J Clin Microbiol 51:3666–3674. doi: 10.1128/JCM.01669-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Acharya A, Vaniawala S, Shah P, Misra RN, Wani M, Mukhopadhyaya PN. 2014. Development, validation and clinical evaluation of a low cost in-house HIV-1 drug resistance genotyping assay for Indian patients. PLoS One 9:e105790. doi: 10.1371/journal.pone.0105790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chen JH, Wong KH, Chan K, Lam HY, Lee SS, Li P, Lee MP, Tsang DN, Zheng BJ, Yuen KY, Yam WC. 2007. Evaluation of an in-house genotyping resistance test for HIV-1 drug resistance interpretation and genotyping. J Clin Virol 39:125–131. doi: 10.1016/j.jcv.2007.03.008. [DOI] [PubMed] [Google Scholar]
- 56.Steegen K, Demecheleer E, De Cabooter N, Nges D, Temmerman M, Ndumbe P, Mandaliya K, Plum J, Verhofstede C. 2006. A sensitive in-house RT-PCR genotyping system for combined detection of plasma HIV-1 and assessment of drug resistance. J Virol Methods 133:137–145. doi: 10.1016/j.jviromet.2005.11.004. [DOI] [PubMed] [Google Scholar]
- 57.MacLeod IJ, Rowley CF, Thior I, Wester C, Makhema J, Essex M, Lockman S. 2010. Minor resistant variants in nevirapine-exposed infants may predict virologic failure on nevirapine-containing ART. J Clin Virol 48:162–167. doi: 10.1016/j.jcv.2010.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rowley CF, Boutwell CL, Lee EJ, MacLeod IJ, Ribaudo HJ, Essex M, Lockman S. 2010. Ultrasensitive detection of minor drug-resistant variants for HIV after nevirapine exposure using allele-specific PCR: clinical significance. AIDS Res Hum Retroviruses 26:293–300. doi: 10.1089/aid.2009.0082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rowley CF, Boutwell CL, Lockman S, Essex M. 2008. Improvement in allele-specific PCR assay with the use of polymorphism-specific primers for the analysis of minor variant drug resistance in HIV-1 subtype C. J Virol Methods 149:69–75. doi: 10.1016/j.jviromet.2008.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rozera G, Abbate I, Bruselles A, Vlassi C, D'Offizi G, Narciso P, Chillemi G, Prosperi M, Ippolito G, Capobianchi MR. 2009. Massively parallel pyrosequencing highlights minority variants in the HIV-1 env quasispecies deriving from lymphomonocyte sub-populations. Retrovirology 6:15. doi: 10.1186/1742-4690-6-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wang C, Mitsuya Y, Gharizadeh B, Ronaghi M, Shafer RW. 2007. Characterization of mutation spectra with ultra-deep pyrosequencing: application to HIV-1 drug resistance. Genome Res 17:1195–1201. doi: 10.1101/gr.6468307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gall A, Ferns B, Morris C, Watson S, Cotten M, Robinson M, Berry N, Pillay D, Kellam P. 2012. Universal amplification, next-generation sequencing, and assembly of HIV-1 genomes. J Clin Microbiol 50:3838–3844. doi: 10.1128/JCM.01516-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Devereux HL, Loveday C, Youle M, Sabin CA, Burke A, Johnson M. 2000. Substantial correlation between HIV type 1 drug-associated resistance mutations in plasma and peripheral blood mononuclear cells in treatment-experienced patients. AIDS Res Hum Retroviruses 16:1025–1030. doi: 10.1089/08892220050075273. [DOI] [PubMed] [Google Scholar]
- 64.Steegen K, Luchters S, Demecheleer E, Dauwe K, Mandaliya K, Jaoko W, Plum J, Temmerman M, Verhofstede C. 2007. Feasibility of detecting human immunodeficiency virus type 1 drug resistance in DNA extracted from whole blood or dried blood spots. J Clin Microbiol 45:3342–3351. doi: 10.1128/JCM.00814-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Diallo K, Murillo WE, de Rivera IL, Albert J, Zhou Z, Nkengasong J, Zhang G, Sabatier JF, Yang C. 2012. Comparison of HIV-1 resistance profiles in plasma RNA versus PBMC DNA in heavily treated patients in Honduras, a resource-limited country. Int J Mol Epidemiol Genet 3:56–65. [PMC free article] [PubMed] [Google Scholar]
- 66.Vartanian JP, Henry M, Wain-Hobson S. 2002. Sustained G→A hypermutation during reverse transcription of an entire human immunodeficiency virus type 1 strain Vau group O genome. J Gen Virol 83:801–805. [DOI] [PubMed] [Google Scholar]
- 67.Vartanian JP, Meyerhans A, Asjo B, Wain-Hobson S. 1991. Selection, recombination, and G-A hypermutation of human immunodeficiency virus type 1 genomes. J Virol 65:1779–1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Vartanian JP, Meyerhans A, Sala M, Wain-Hobson S. 1994. G→A hypermutation of the human immunodeficiency virus type 1 genome: evidence for dCTP pool imbalance during reverse transcription. Proc Natl Acad Sci U S A 91:3092–3096. doi: 10.1073/pnas.91.8.3092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wain-Hobson S, Sonigo P, Guyader M, Gazit A, Henry M. 1995. Erratic G→A hypermutation within a complete caprine arthritis-encephalitis virus (CAEV) provirus. Virology 209:297–303. doi: 10.1006/viro.1995.1261. [DOI] [PubMed] [Google Scholar]
- 70.Harris RS, Liddament MT. 2004. Retroviral restriction by APOBEC proteins. Nat Rev Immunol 4:868–877. doi: 10.1038/nri1489. [DOI] [PubMed] [Google Scholar]
- 71.McCallum M, Oliveira M, Ibanescu RI, Kramer VG, Moisi D, Asahchop EL, Brenner BG, Harrigan PR, Xu H, Wainberg MA. 2013. Basis for early and preferential selection of the E138K mutation in HIV-1 reverse transcriptase. Antimicrob Agents Chemother 57:4681–4688. doi: 10.1128/AAC.01029-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Fourati S, Malet I, Lambert S, Soulie C, Wirden M, Flandre P, Fofana DB, Sayon S, Simon A, Katlama C, Calvez V, Marcelin AG. 2012. E138K and M184I mutations in HIV-1 reverse transcriptase coemerge as a result of APOBEC3 editing in the absence of drug exposure. AIDS 26:1619–1624. doi: 10.1097/QAD.0b013e3283560703. [DOI] [PubMed] [Google Scholar]
- 73.Neogi U, Shet A, Sahoo PN, Bontell I, Ekstrand ML, Banerjea AC, Sonnerborg A. 2013. Human APOBEC3G-mediated hypermutation is associated with antiretroviral therapy failure in HIV-1 subtype C-infected individuals. J Int AIDS Soc 16:18472. doi: 10.7448/IAS.16.1.18472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ulenga NK, Sarr AD, Hamel D, Sankale JL, Mboup S, Kanki PJ. 2008. The level of APOBEC3G (hA3G)-related G-to-A mutations does not correlate with viral load in HIV type 1-infected individuals. AIDS Res Hum Retroviruses 24:1285–1290. doi: 10.1089/aid.2008.0072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Eyzaguirre LM, Charurat M, Redfield RR, Blattner WA, Carr JK, Sajadi MM. 2013. Elevated hypermutation levels in HIV-1 natural viral suppressors. Virology 443:306–312. doi: 10.1016/j.virol.2013.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Cheynier R, Gratton S, Vartanian JP, Meyerhans A, Wain-Hobson S. 1997. G→A hypermutation does not result from polymerase chain reaction. AIDS Res Hum Retroviruses 13:985–986. doi: 10.1089/aid.1997.13.985. [DOI] [PubMed] [Google Scholar]
- 77.Wang R, Goyal R, Lei Q, Essex M, De Gruttola V. 2014. Sample size considerations in the design of cluster randomized trials of combination HIV prevention. Clin Trials 11:309–318. doi: 10.1177/1740774514523351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Eshleman SH, Jones D, Flys T, Petrauskene O, Jackson JB. 2003. Analysis of HIV-1 variants by cloning DNA generated with the ViroSeq HIV-1 Genotyping System. Biotechniques 35:614–618, 620-622. [DOI] [PubMed] [Google Scholar]
- 79.Novitsky V, Lagakos S, Herzig M, Bonney C, Kebaabetswe L, Rossenkhan R, Nkwe D, Margolin L, Musonda R, Moyo S, Woldegabriel E, van Widenfelt E, Makhema J, Essex M. 2009. Evolution of proviral gp120 over the first year of HIV-1 subtype C infection. Virology 383:47–59. doi: 10.1016/j.virol.2008.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Novitsky V, Wang R, Rossenkhan R, Moyo S, Essex M. 2013. Intra-host evolutionary rates in HIV-1C env and gag during primary infection. Infect Genet Evol 19:361–368. doi: 10.1016/j.meegid.2013.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. 2013. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol Biol Evol 30:2725–2729. doi: 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Rose PP, Korber BT. 2000. Detecting hypermutations in viral sequences with an emphasis on G→A hypermutation. Bioinformatics 16:400–401. doi: 10.1093/bioinformatics/16.4.400. [DOI] [PubMed] [Google Scholar]
- 84.Felsenstein J. 1985. Confidence limits on phylogenies: an approach using a bootstrap. Evolution 39:783–791. doi: 10.2307/2408678. [DOI] [PubMed] [Google Scholar]
- 85.Felsenstein J. 2004. Inferring phylogenies. Sinauer Associates, Inc., Sunderland, MA. [Google Scholar]
- 86.Nei M, Kumar S. 2000. Molecular evolution and phylogenetics. Oxford University Press, New York, NY. [Google Scholar]
- 87.Even S. 2011. Graph algorithms, 2nd ed Cambridge University Press, New York, NY. [Google Scholar]
- 88.Prosperi MC, Ciccozzi M, Fanti I, Saladini F, Pecorari M, Borghi V, Di Giambenedetto S, Bruzzone B, Capetti A, Vivarelli A, Rusconi S, Re MC, Gismondo MR, Sighinolfi L, Gray RR, Salemi M, Zazzi M, De Luca A, ARCA Collaborative Group. 2011. A novel methodology for large-scale phylogeny partition. Nat Commun 2:321. doi: 10.1038/ncomms1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Stamatakis A. 2006. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22:2688–2690. doi: 10.1093/bioinformatics/btl446. [DOI] [PubMed] [Google Scholar]
- 90.Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30:1312–1313. doi: 10.1093/bioinformatics/btu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.R Core Team. 2014. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria: http://www.R-project.org/. [Google Scholar]
- 92.Dugan KA, Lawrence HS, Hares DR, Fisher CL, Budowle B. 2002. An improved method for post-PCR purification for mtDNA sequence analysis. J Forensic Sci 47:811–818. [PubMed] [Google Scholar]
- 93.Novitsky V, Moyo S, Lei Q, DeGruttola V, Essex M. 6 February 2015. Importance of viral sequence length and number of variable and informative sites in analysis of HIV clustering. AIDS Res Hum Retroviruses doi: 10.1089/AID.2014.0211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Salichos L, Rokas A. 2013. Inferring ancient divergences requires genes with strong phylogenetic signals. Nature 497:327–331. doi: 10.1038/nature12130. [DOI] [PubMed] [Google Scholar]
- 95.Salichos L, Stamatakis A, Rokas A. 2014. Novel information theory-based measures for quantifying incongruence among phylogenetic trees. Mol Biol Evol 31:1261–1271. doi: 10.1093/molbev/msu061. [DOI] [PubMed] [Google Scholar]
- 96.Kampstra P. 2008. Beanplot: a boxplot alternative for visual comparison of distributions. J Stat Software 28:1–9. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.