

Abstract
Adult-onset hemophagocytic lymphohistiocytosis (HLH) has features that are distinct from that of HLH in pediatric patients. The clinical records at the Japanese Red Cross Kumamoto Hospital were reviewed. We retrospectively analyzed 34 patients who fulfilled the diagnostic criteria of HLH-2004. The median age of patients was 60.0 (range 15-86). Underlying diseases were diagnosed in 17 patients. They consisted of malignant lymphoma (n=3), other neoplastic disease (n=3), viral infection (n=4), collagen vascular disease (n=3), Kikuchi’s disease (n=3) and drug (n=1). Underlying diseases were not diagnosed in 17 patients despite examination. The treatments were steroids (n=18), dexamethasone + cyclosporine A (CSA) + etoposide (n=4), multidrug chemotherapy (n=2), steroids and CSA (n=3). Eleven patients died during observation. In a multivariate analysis, the significant predictor for death was age at onset (hazard ratio, 1.22; 95%CI, 1.02-1.44; P=0.027). Autopsy was performed in 4 cases, but the underlying disease remained unknown in 3 of those cases. Adult-onset HLH has high diversity and various outcomes. The mechanism of adult-onset HLH is not fully understood and further research is required.
Key words: Hemophagocytic lymphohistiocytosis, adult, prognosis, autopsy
Introduction
Hemophagocytic lymphohistiocytosis (HLH) is typically characterized by cytopenia and systemic symptoms including fever and splenomegaly.1 Other findings include hypertryglyceridemia, coagulopathy and neurological symptoms.2-4 Histopathological findings include accumulation of lymphocytes or red blood cells in the cytoplasm of mature macrophages in the spleen, lymph nodes, bone marrow or liver.
Hemophagocytic lymphohistiocytosis is classified into two categories, primary HLH and secondary HLH. Primary HLH, which is also called familial HLH, is caused by gene mutation associated with perforin-dependent cytotoxicity.5 Familial HLH usually occurs in infants and young children. The major causes of Japanese pediatric HLH are infection, autoimmune disease, and familial disease.6 On the other hand, secondary HLH typically occurs in adults. Secondary HLH in adults is associated with a variety of underlying conditions such as infection, malignant lymphoma and autoimmune disease. In contrast to pediatric HLH, there have been few reports published on the clinical features of adult-onset HLH. This retrospective study was carried out to clarify the clinical features and prognosis of adult-onset HLH.
Materials and Methods
The clinical records between November 2001 and September 2014 at the Japanese Red Cross Kumamoto Hospital were reviewed for the terms hemophagocytic syndrome and hemophagocytic lymphohistiocytosis. All adult patients aged 15 and over, whose medical record contained those words, were extracted. The patients were considered to have HLH when they fulfilled the diagnostic criteria in the guidelines of HLH-2004.7 The HLH-2004 guideline describes eight criteria for the diagnosis of HLH: i) fever, ii) splenomegaly, iii) cytopenia affecting at least two of three lineages in the peripheral blood, iv) hypertriglyceridemia (>265 mg/dL) and/or hypofibrinogenemia (<150 mg/dL), v) hemophagocytosis in the bone marrow, spleen, or lymph nodes, vi) low or absent NK cell activity, vii) hyperferritinemia (>500 g/L) and viii) high levels of sIL-2R (>2400 U/mL).7 Five of the eight criteria have to be fulfilled to diagnose HLH.
Overall survival (OS) was estimated by the Kaplan-Meier method. OS was defined as time from diagnosis to the date of death or date of last contact for those censored. All results were updated on October 21, 2014. The Cox regression model was used to assess predictors of death. The cutoff value of laboratory data used in the survival analysis was based on the diagnostic criteria in the HLH-2004. All medical statistical analysis was performed using SPSS ver.20. A P-value of <0.05 was considered significant.
This study was approved by the ethics committee of the Japanese Red Cross Kumamoto Hospital.
Results
Patient characteristics
A total of 34 patients were analyzed. Patient characteristics are shown in Table 1. Median age at diagnosis was 60.0 years old (range 15-86). Fifteen patients (44.1%) were male and 19 patients (55.9%) were female. The underlying conditions of HLH were diagnosed in 17 patients (50.0%) (Table 2). The underlying diseases were neoplastic disease (n=6; 17.6%), viral infection (n=4; 11.8%), Kikuchi’s disease (n=3; 8.8%), collagen vascular disease (n=3; 8.8%) and drugs (n=1; 2.9%). The subtype of neoplastic disease was diffuse large B cell lymphoma (DLBCL) (n=2), Hodgkin lymphoma (n=1), hepatocelluar carcinoma (n=2), cholangiocarcinoman (n=1). Among the 4 viral infection-associated HLH cases, the influenza virus was detected in 1 patient and cytomegalovirus (CMV) was detected in 1 patient. The influenza virus infection was diagnosed by a rapid antigen test and the CMV infection was diagnosed by an elevated CMV IgM titer. The etiology of the infection in the other 2 cases was not identified. The subtypes of the 3 cases of collagen disease associated HLH were systemic lupus erythematosus (n=1), adult-onset Still disease (n=1), granulomatosis with polyangitis (n=1). The offending drug in the drug associated HLH was carbamazepine. No patients underwent genetic testing for familial HLH.
Table 1.
Patient characteristics (n=34).
| Characteristic | No. of patients (%) |
|---|---|
| Age (years) | |
| Median | 60.0 |
| Range | 15-86 |
| Sex | |
| Male | 15 (44.1) |
| Female | 19 (55.9) |
| White cell count (×109/L)6* | 4.33±4.77 |
| Neutrophils (×109/L)6* | 2.69±3.52 |
| Hemoglobin (g/dL)6* | 10.3±2.5 |
| Platelet (×109/L)* | 79.0±48.0 |
| Lactate dehydrogenase (IU/L)6* | 776±590 |
| Triglyceride (mg/dL)6* | 190±99 |
| Soluble IL-2 receptor (U/mL)6* | 5450±6650 |
| Ferritin (ng/mL)6* | 12220±42490 |
| Treatment | |
| Corticosteroid | 18 (52.9) |
| Corticosteroid+CSA | 3 (8.8) |
| Corticosteroid+CSA+VP-16 (HLH-2004) | 4 (11.8) |
| Multidrug chemotherapy | 2 (5.9) |
| None | 7 (20.6) |
*Values are mean±SD. CSA, cyclosporine A.
Table 2.
Underlying disease of hemophagocytic lymphohistiocytosis (n=34).
| Characteristic | No. of patients (%) |
|---|---|
| Neoplastic disease (n=6; 17.6%) | |
| Diffuse large B cell lymphoma | 2 (33.3) |
| Hodgkin lymphoma | 1 (16.7) |
| Hepatocelluar carcinoma | 2 (33.3) |
| Cholangiocarcinoma | 1 (16.7) |
| Viral infection (n=4; 11.8%) | |
| Influenza | 1 (16.7) |
| Cytomegalovirus | 1 (16.7) |
| Not identified | 2 (33.3) |
| Kikuchi’s disease (n=3; 8.8%) | 3 (100) |
| Collagen vascular disease (n=3; 8.8%) | |
| Systemic lupus erythematosus | 1 (33.3) |
| Adult-onset Still disease | 1 (33.3) |
| Granulomatosis with polyangitis | 1 (33.3) |
| Drug (n=1; 2.9%) | 1 (100) |
| Unknown (n=17; 50.0%) | 17 (100) |
Underlying diseases were not diagnosed in 17 patients (50.0%). Among these 17 patients, six underwent biopsy with suspicion of intravascular lymphoma (IVL). The sites of the biopsy were skin and bone marrow (n=5) and liver (n=1). However, the results of these biopsies were non-contributory. Moreover, all patients underwent serologic examination for Epstein-Barr virus (EBV), CMV and two patients underwent serologic examination for herpes simplex virus type 1 (HSV-1). However, the results of these examinations were negative.
White blood cells count was 4.33±4.77×109/L (neutrophil count 2.69±3.52×109/L), hemoglobin was 10.3±2.5 g/dL, platelet count was 79.0±48.0×109/L, LDH value was 776±590 IU/L, triglyceride value was 190±99 mg/dL, sIL2R value was 5450±6650 U/mL and ferritin value was 12.220±42.490 ng/mL (mean±SD).
Treatment
Clinicians chose treatment according to the type of underlying diseases and the severity of HLH (Table 1). The treatments which the patients received were steroids (n=18; 52.9%), HLH-2004 protocol (dexamethasone + CSA + etoposide) (n=4; 11.8%), multidrug chemotherapy for malignant lymphoma (n=2; 5.9%), steroids and CSA (n=3; 8.8%). One patient with DLBCL was treated with chemotherapy that consisted of rituximab, cyclophosphamide, doxorubicin, vincristine and prednisone. One patient with Hodgkin lymphoma received chemotherapy that consisted of doxorubicin, bleomycin, vinblastine and dacarbazine. One out of the three patients with lymphoma did not receive chemotherapy because of a rapid clinical deterioration. Seven patients (20.6%) achieved spontaneous remission and received no treatment.
Survival
After a median follow up of 1.9 months (range 0.1-132.5), 11 of the 34 patients (32.4%) had died. The causes of death were multi-organ failure (n=3), respiratory failure (n=3), septic shock (n=2), malignant tumor (n=1), liver failure (n=1) and cerebral hemorrhage (n=1). The survival curve is shown in Figure 1. Median OS of the entire population was not reached and median OS of the fatal cases was 1.2 month (range 0.1-25.5). We conducted survival analysis using patient baseline characteristics. In the univariable analysis, age at onset was a significant predictor of death (hazard ratio, 1.11; 95%CI, 1.03-1.19; P=0.007) (Table 3). Other baseline characteristics were not associated with prognosis.
Figure 1.
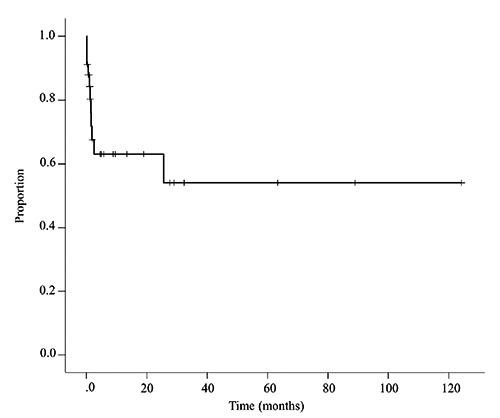
Kaplan Meier estimates of Odds Ratio (OS). Tick marks show censored data. Median OS was not reached.
Table 3.
Univariable and multivariable Cox regression model for predictors of death.
| Univariable analysis Hazard ratio (95%CI) | P | Multivariable analysis Hazard ratio (95%CI) | P | |
|---|---|---|---|---|
| Age (1-year increase) | 1.11 (1.03-1.19) | 0.007 | 1.22 (1.02-1.44) | 0.027 |
| Sex (women vs men) | 0.30 (0.08-1.13) | 0.075 | ||
| Neutrophil count (<109/L vs ≥109/L) | 0.54 (0.14-2.03) | 0.359 | ||
| Hemoglobin count (<9 g/dL vs ≥9 g/dL) | 1.88 (0.57-6.15) | 0.299 | ||
| Platelet count (<100×109/L vs≥100×109/L) | 4.81 (0.61-37.97) | 0.136 | ||
| Ferritin value (≥500 ng/mL vs <500 ng/mL) | 1.42 (0.30-6.59) | 0.657 | ||
| Splenomegaly6* | 0.723 (0.19-2.81) | 0.639 | ||
| Hemophagocytosis in bone marrow6* | 2.173 (0.274-17.24) | 0.463 | ||
| Underlying disease (known vs unknown) | 1.39 (0.42-4.57) | 0.592 | ||
| Underlying disease (neoplastic vs others) | 1.33 (0.17-10.40) | 0.828 |
*Presence versus absence.
We conducted multivariable analysis with variables used in the univariate analysis. The multivariate Cox regression analysis showed that age at onset was a significant predictor of death (hazard ratio, 1.22; 95%CI, 1.02-1.44; P=0.027) (Table 3). Similar to the univariate analysis, the other baseline characteristics were not associated with prognosis. Figure 2 shows distribution of ages according to the outcomes of the patients. The median age of the 11 fatal patients was 75 (range 53-83). On the other hand, the median age of the 23 patients who survived was 39 (range 15-86). The survivors constituted a younger populations among all cases.
Figure 2.
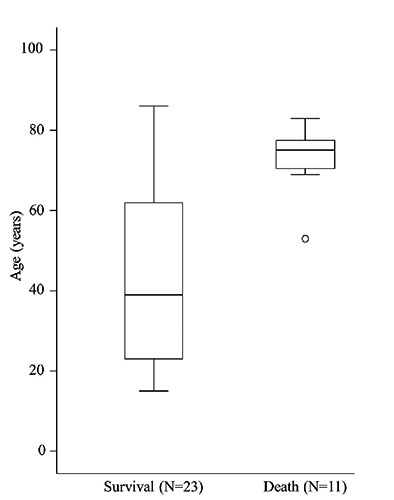
Distribution of ages according to patient outcomes. Boxes contain 50% of data with the inside horizontal line representing the median value.
Results of autopsy
Autopsy was performed in 4 of the 11 fatal cases. The results of the autopsies are shown in Table 4. Hemophagocytosis in at least three organs was seen in all 4 cases. The 69 yo female (case 1) developed HLH secondary to granulomatosis with polyangitis. A systemic steroid was given but she died of respiratory failure. The autopsy revealed aspergillus pneumonia. The remaining 3 cases underwent autopsy to investigate the underlying cause of HLH (cases 2-4). All 3 patients underwent biopsy of bone marrow and skin with a suspicion of IVL before death, but the results of the biopsies were negative. The clinical causes of death were multi-organ failure (cases 2 and 3) and respiratory failure (case 4). The major findings of the autopsies were myelodysplastic syndrome (MDS) (cases 2 and 3) and severe pneumonia (case 4). The subtype of MDS in two cases was refractory anemia with excess of blast-2 (RAEB-2). Postmortem pathological examination did not reveal the underlying disease of HLH in the 3 cases.
Table 4.
Results of four autopsy cases.
| No. | Age (y) | Sex | Underlying disease of HLH | Cause of death | Findings |
|---|---|---|---|---|---|
| 1 | 69 | F | GPA | Respiratory failure | Aspergillus pneumonia; lymphohistyocytosis (spleen, lymph node, bone marrow) |
| 2 | 73 | F | Unknown | MOF | Lymphohistyocytosis (liver, lymph node, bone marrow) |
| 3 | 79 | M | Unknown | MOF | MDS (RAEB-2); Pseudomenbranous colitis; lymphohistyocytosis (liver, spleen, lymph node) |
| 4 | 71 | F | Unknown | Respiratory failure | MDS (RAEB-2); severe pneumonia; lymphohistyocytosis (liver, spleen, lymph node) |
HLH, hemophagocytic lymphohistiocytosis; GPA, granulomatosis with polyangitis; MOF, multi-organ failure.
Discussion
The results of this study indicate that the major etiology of HLH was neoplastic disease including lymphoma, followed by infection, collagen vascular disease, Kikuchi’s disease and drugs. The underlying disease was not diagnosed in about half of the cases. We have shown that age at onset is the strongest prognostic factor regardless of the etiology of HLH. This study is a relatively large retrospective study that explored the etiology and prognosis of rare adult-onset HLH.
A previous Japanese survey showed that the major etiology of adult-onset HLH is malignant lymphoma (42%) and infection (43%). They report that 7.2% of cases developed from an unknown etiology.6 A report from China showed similar results.8 These findings are inconsistent with the results of this study concerning the low proportion of viral infection and malignant lymphoma. It is necessary to consider the possibility of undiagnosed viral infection and malignant lymphoma.
Among infection-associated HLH, the major pathogen is EBV, CMV, HSV-1, adenovirus, parvovirus B19, influenza virus and parainfluenza virus.6 Patients in our study underwent examination for influenza virus, EBV, CMV, and HSV-1 only. It is possible that patients with HLH of an unknown primary in our study might have an unexamined viral infection.
HLH in adults is often associated with malignant disease, typically malignant lymphoma. The major etiology of lymphoma-associated HLH is B cell or NK/T cell lymphoma. The predominant histology of the B-cell tumors was DLBCL and IVL.6 IVL is a rare disease form of non-Hodgkin lymphoma which is characterized by the absence of peripheral lymphadenopathy and specific laboratory abnormalities.9 HLH is a major clinical manifestation in Asian patients with IVL and biopsy of the bone marrow or skin seems to be a high-yield diagnostic procedure.9,10 Therefore, we carefully performed examinations to look for underlying IVL but the results were non-contributory. Moreover, postmortem autopsy of the three fatal cases did not reveal IVL either. We believe that patients with HLH of an unknown primary disease had a low probability of malignant lymphoma. It remains unknown why this study includes a lower proportion of malignant lymphoma.
We did not perform genetic testing for familial HLH in this study. However, patients with HLH of an unknown primary disease might have undiagnosed late-onset familial HLH. Recently Zhan et al. reported that about 14% of patients with adult-onset HLH have hypomorphic mutations in PRF1, MUNC13-4, and STXBP2.11 They proposed that mutations cause adult-onset HLH when affected patients experience stresses such as viral infection. These gene mutations are observed in familial HLH and inherited in the autosomal recessive pattern. Patients with gene abnormalities have abnormally activated cytotoxic lymphocytes and generally develop HLH within the first year of life. To date, some reports have been published about adult-onset familial HLH.12,13 Patients with HLH of an unknown primary disease in our study may have undiagnosed gene mutations associated with familial HLH. Therefore, we should consider genetic testing when we see patients with HLH of an unknown origin and who are suspected of having lateonset familial HLH.
It is noteworthy that autopsy of the 4 fatal cases found MDS (RAEB-2) in 2 cases. So far a few reports have been published on the association of MDS and HLH. MDS can directly cause HLH due to increased CD8+ lymphocytes, or increased levels of cytokine cause morphologic changes in bone marrow mimicking MDS.14,15
However, a definite association between MDS and HLH is not unknown and further studies are required.
Without therapy, the mortality of patients with HLH is high. In contrast, patients treated on the HLH-94 protocol had a median survival of 54 percent at 6.2 years.1 The results of our study shows a comparable survival outcome. There are no standard outcomes predictors for HLH. However, recent investigations have demonstrated that patients with high serum ferritin level or malignant disease have a worse prognosis.16,17 Our study indicates that the age of patients is the strongest predictive factor for death and this is a new finding in adult-onset HLH. The other major finding is the disparity between early deaths and long survivors. Most of the fatal cases in this study showed primary resistance to the initial treatment, while some cases recovered without treatment. This result means that adult-onset HLH consists of a diverse population.
The main limitation of this study is related to its retrospective design. It is possible that some cases of HLH may not have been recorded, and that not all patients with HLH may have been accurately identified.
Conclusions
Adult-onset HLH has high diversity and various outcomes. This study is a relatively large retrospective study that explored the etiology and prognosis of adult-onset HLH. We have shown that the age at onset is the strongest prognostic factor regardless of the etiology of HLH, and that the underlying disease was not diagnosed in about half of the cases. The pathogenesis of adult-onset HLH is not fully understood and further research is required.
References
- 1.Trottestam H, Horne A, Aricò M, et al. Chemoimmunotherapy for hemophagocytic lymphohistiocytosis: long-term results of the HLH-94 treatment protocol. Blood 2011;118:4577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Okamoto M, Yamaguchi H, Isobe Y, et al. Analysis of triglyceride value in the diagnosis and treatment response of secondary hemophagocytic syndrome. Intern Med 2009;48:775. [DOI] [PubMed] [Google Scholar]
- 3.Fukaya S, Yasuda S, Hashimoto T, et al. Clinical features of haemophagocytic syndrome in patients with systemic autoimmune diseases: analysis of 30 cases. Rheumatology (Oxford) 2008;47:1686. [DOI] [PubMed] [Google Scholar]
- 4.Jordan MB, Allen CE, Weitzman S, et al. How I treat hemophagocytic lymphohistiocytosis. Blood 2011;118:4041-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Göransdotter Ericson K, Fadeel B, Nilsson-Ardnor S, et al. Spectrum of perforin gene mutations in familial hemophagocytic lymphohistiocytosis. Am J Hum Genet 2001;68:590-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ishii E, Ohga S, Imashuku S, et al. Nationwide survey of hemophagocytic lymphohistiocytosis in Japan. Int J Hematol 2007;86:58-65. [DOI] [PubMed] [Google Scholar]
- 7.Dobke J. Treatment Protocol of the Second International HLH study 2004. Available from: http://www.kinderkrebsinfo.de/health_professionals/clinical_trials/pohkinderkre bsinfotherapiestudien/hlh_2004/index_eng.html [Google Scholar]
- 8.Li J, Wang Q, Zheng W, et al. Hemophagocytic lymphohistiocytosis: clinical analysis of 103 adult patients. Medicine 2014;93:100-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shimada K, Kinoshita T, Naoe T, Nakamura S. Presentation and management of intravascular large B-cell lymphoma. Lancet Oncol 2009;10:895-902. [DOI] [PubMed] [Google Scholar]
- 10.Murase T, Yamaguchi M, Suzuki R, et al. Intravascular large B-cell lymphoma (IVL-BCL): a clinicopathologic study of 96 cases with special reference to the immunophenotypic heterogeneity of CD5. Blood 2007; 109:478-85. [DOI] [PubMed] [Google Scholar]
- 11.Zhang K, Jordan MB, Marsh RA, et al. Hypomorphic mutations in PRF1, MUNC13-4, and STXBP2 are associated with adult-onset familial HLH. Blood 2011;118:5794-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nagafuji K, Nonami A, Kumano T, et al. Perforin gene mutations in adult-onset hemophagocytic lymphohistiocytosis. Haematologica 2007;92:978-81. [DOI] [PubMed] [Google Scholar]
- 13.Ueda I, Kurokawa Y, Koike K, et al. Late-onset cases of familial hemophagocytic lymphohistiocytosis with missense perforin gene mutations. Am J Hematol 2007; 82:427-32. [DOI] [PubMed] [Google Scholar]
- 14.Tsuji T, Yamasaki H, Arima N, Tsuda H. Hemophagocytic lymphohistiocytosis associated with myelodysplastic syndromes. Int J Hematol 2010;92:547-9. [DOI] [PubMed] [Google Scholar]
- 15.Imashuku S, Kitazawa K, Ishii M, et al. Bone marrow changes mimicking myelodysplasia in patients with hemophagocytic lymphohistiocytosis. Int J Hematol 2000;72:353-7. [PubMed] [Google Scholar]
- 16.Lin TF, Ferlic-Stark LL, Allen CE, et al. Rate of decline of ferritin in patients with hemophagocytic lymphohistiocytosis as a prognostic variable for mortality. Pediatr Blood Cancer 2011;56:154-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Parikh SA, Kapoor P, Letendre L, et al. Prognostic factors and outcomes of adults with hemophagocytic lymphohistiocytosis. Mayo Clin Proc 2014;89:484-92. [DOI] [PubMed] [Google Scholar]