

Abstract
Noonan syndrome is a relatively common and heterogeneous genetic disorder, associated with congenital heart defect in about 50% of the cases. If the defect is not severe, life expectancy is normal. We report a case of Noonan syndrome in a preterm infant with hypertrophic cardiomyopathy and lethal outcome associated to acute respiratory distress syndrome caused by Adenovirus pneumonia. A novel mutation in the RAF1 gene was identified: c.782C>G (p.Pro261Arg) in heterozygosity, not described previously in the literature. Consequently, the common clinical course in this mutation and its respective contribution to the early fatal outcome is unknown. No conclusion can be established regarding genotype/phenotype correlation.
Key words: RASopathies, RAF1 gene, prematurity, hypertrophic cardiomyopathy, death
Introduction
Hypertrophic cardiomyopathy (HCM) is defined by the presence of increased left ventricular wall thickness that is not solely explained by abnormal loading conditions. This definition applies to children and adults and makes no a priori assumptions about etiology or myocardial pathology.1 In newborns, HCM may be associated with prenatal transient causes including maternal diabetes, fetal hypoxia with impaired intrauterine growth and administration of corticosteroids. It can also occur as a secondary effect to polymalformative syndromes such as Noonan, LEOPARD, Beckwith-Weidemann, or Barth, inborn errors of metabolism like deposit diseases (glycogenosis, mucopolysaccharidosis, oligosaccharidosis, sphingolipidosis), disorders of energy metabolism (fatty acid oxidation defects and carnitine deficiency) and neuromuscular syndromes (Friedrich ataxia and Duchenne muscular dystrophy).2,3 The current case describes a preterm with Noonan syndrome due to a novel RAF1 mutation, who developed a rapidly progressive hypertrophic cardiomyopathy with fatal outcome, in the context of cardiorespiratory failure associated with Adenovirus pneumonia.
Case Report
This male infant was born to healthy non-consanguineous parents. It was the mother’s first pregnancy, complicated at 28 weeks of gestation by polyhydramnios. The fetal ultrasounds revealed bilateral hydronephrosis (right 14 mm, left 10 mm), bladder wall thickening and shortened long bones. He was delivered at the 30th week of gestation by cesarean section due to fetal distress by placental abruption. Apgar scores were 3/3/4 at one, five and ten minutes respectively, with sustained bradycardia. The bodyweight was 1840 g (P75-90), height 38 cm (P10) and head circumference 32 cm (P90). The newborn was intubated and ventilated in the first minutes and one dose of exogenous surfactant was given due to respiratory distress syndrome. Physical examination revealed marked hypotonia, generalized edema and shortened limbs. The echocardiogram showed poor contractility, low cardiac output, myocardial hypertrophy and mild pericardial effusion. Hence, the newborn was submitted to invasive ventilation, fluid replacement, albumin and inotropic support. He developed metabolic acidosis requiring correction and antibiotic therapy was performed due to sepsis. In the first days of life, cerebral ultrasound was normal. In the second day, a weight loss of 7.6% was confirmed, along with redundant skin, course facies, hepatomegaly and palpable kidneys. The echocardiogram showed improved function and vascular filling, left myocardial hypertrophy and signs of pulmonary hypertension. Clinical and electric seizures were observed, requiring treatment with phenobarbital. On day four, dopamine was suspended but invasive ventilation and dobutamine were maintained. Five days later ventilatory and hemodynamic status were stable on non-invasive ventilation. At this time a significant reduction of edema (weight loss of 400 g: –22% of weight birth) was observed. On day ten, dobutamine was suspended and the echocardiogram confirmed hypertrophic cardiomyopathy with left ventricular outflow tract obstruction (Figure 1). Propranolol was initiated (maximum dose of 4 mg/kg/day) with the heart rate around 150 bpm. Seizures were controlled and cerebral ultrasound on day 15 revealed a grade II intraperiventricular hemorrhage. Investigation included karyotype (46,XY), plasma amino acids, serum lactate, ammonia, total and esterified carnitine, urine metabolic screen and renal ultrasound, all with normal results. TORCH screen was negative.
Figure 1.
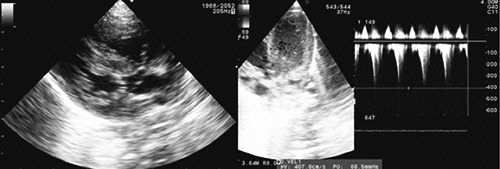
Echocardiogram: hypertrophic cardiomyopathy with left ventricular outflow tract obstruction.
A molecular study for RASopathies, testing for specific mutations in PTPN11, BRAF, SOS1, HRAS, KRAS, RAF1, MAP2K1, and MAP2K2, revealed a novel RAF1 gene mutation: c.782C>G (p.Pro261Arg) in heterozygosity, forecasting a diagnosis of probable Noonan syndrome. The genetic parents’ study was negative. On day 31, a cerebral magnetic resonance showed intraventricular and parenchymal venous infarction and relatively poor gyration, with the presence of shallow grooves especially in the frontal regions (Figure 2).
Figure 2.
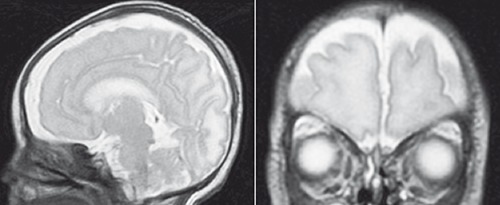
Cerebral magnetic resonance images: poor gyration, with the presence of shallow grooves especially in the frontal regions.
There was some decrease of the left ventricular outflow tract obstruction, under high propranolol doses and the newborn remained clinically stable in dependence on noninvasive ventilation and oxygen therapy. On day 60 (postmenstrual age 39th+4 weeks), he developed respiratory failure requiring invasive ventilation and antibiotics due to pneumonia. Adenovirus was isolated in respiratory secretions. It progressed to Acute Respiratory Distress Syndrome, requiring high-frequency ventilation and pulmonary vasodilators (nitric oxide and sildenafil) for refractory hypoxemia. On day 79, severe pulmonary hypertension and congestive heart failure with low cardiac output were registered. Despite the therapeutic optimization, cardiorespiratory status worsened and he died in the 80th day.
Discussion
The RASopathies are a clinically defined group of medical genetic syndromes caused by germline mutations in genes that encode components or regulators of the RAS/mitogen-activated protein kinase (MAPK) pathway. These disorders include neurofibromatosis type 1, Noonan syndrome, LEOPARD syndrome, capillary malformation-arteriovenous malformation syndrome, Costello syndrome, cardio-faciocutaneous syndrome, and Legius syndrome. Given the common underlying RAS/MAPK pathway dysregulation, the RASopathies exhibit numerous overlapping phenotypic features.4-6
The combination of prenatal features like polyhydramnios, hydrops, shortened long bones and somatic characteristics such as excessive neck skin, widely spaced nipples and low-set ears is suggestive of Noonan syndrome. In the present case, the diagnosis of Noonan syndrome was made based on HCM associated with clinical characteristics and normal karyotype.7
Noonan syndrome is a relatively common genetic condition with an incidence of one in 1,000 to 2,000 live births.6,7 Mutations have been described in 7 genes: PTPN11 (50%), SOS1 (20%), RAF1 (15%), KRAS (5%), and NRAS, BRAF, MAP2K1 (in fewer than 2%), causing gain-of-function and up-regulation of the RAS-MAPK pathway. The condition is often inherited as an autosomal dominant trait, but at least 50% of these mutations occur de novo, as observed in this patient.1,6,8 In the current case, we identified a novel mutation in the RAF1 gene (c.782C>G (p.Pro261Arg) in heterozygosity. Two different mutations have been described in this position, c.782C>A(p.Pro261His) and c.782C>T (p.Pro261Leu), and three additional ones also affecting the same residue, c.781C>T (p.Pro261Ser), c.781C>G(p.Pro261Ala) and c.781C>A(p.Pro261Thr). There is a submission of this variant in ClinVar.9 It was classified by the submitting user as an uncertain significance germline variant, but it was not provided the status (affected/unaffected) of that individual. It is in dbSNP (rs397516828) as a not validated entry.
Bioinformatic software analysis with Align GVGD (class C65), MutationTaster (disease causing), SIFT (deleterious) and PolyPhen-2 (probably damaging), were consistent predicting that the variant found is most likely pathogenic. Additionally, the mutation was not present in both unaffected parents, and, therefore, is most likely de novo. This, along with the presented data, indicates that this variant is very likely pathogenic and the cause of Noonan syndrome in this patient.
The syndrome is associated with a wide variety of signs and symptoms due to multiorgan involvement. It is a very heterogeneous disorder, predominantly characterized by dysmorphic facial features, post-natal short stature, webbed neck, chest deformity, cryptorchidism in men, lymphatic dysplasia, variable bleeding disorders, and intellectual disability.2,6,10
Rare structural cerebral anomalies are described and we speculate if the cerebral alterations found in this case were related to the pathology.
Noonan syndrome is also linked to congenital cardiac defects, with an estimated rate of 35-50%. The predominant lesions are pulmonary valve stenosis and HCM, followed by atrial and ventricular septal defects. HCM is present in about 20% of NS patients and in 95% of mutations in the RAF1 gene.2,10
The HCM in our patient was not detected on prenatal ultrasound but rather in the first days of life. The natural course of HCM is markedly variable. While some patients may become symptomatic and succumb in early childhood, others may remain asymptomatic until late childhood. Rapidly progressive HCM, resulting in an early death or a need for heart transplantation, occurs only sporadically in Noonan Syndrome and other RASopathies.2,7 Hirsch et al reported two children with Noonan syndrome and hypertrophic cardiomyopathy who developed progressive left ventricular outflow tract obstruction over the first year of life.11 They died at 9 and 23 months of age, respectively. In our patient, HCM conducted to cardiorespiratory failure but prematurity and respiratory infection have also contributed to the early death. However, since the common clinical course in this mutation is unknown, it is impossible to assess its contribution to the early fatal outcome.
Acknowledgements
The authors would like to gratefully acknowledge Jorge Pinto Basto, Clinical Director at CGC Genetics/CGC Centro Genética Clínica, Porto, Portugal for his critical reading of the paper.
References
- 1.Elliott PM, Anastasakis A, Borger MA, et al. ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy: the Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC). Eur Heart J 2014;35:2733-79. [DOI] [PubMed] [Google Scholar]
- 2.Andre s AS, Gutiérrez AM, Moreno J. [Prenatal hypertrophic cardiomyopathy and neonatal Noonan syndrome: an association to remember]. Rev Esp Cardiol 2011;64:535-43. [Article in Spanish]. [DOI] [PubMed] [Google Scholar]
- 3.Faienza MF, Giordani L, Ferraris M, et al. PTPN11 gene mutation and severe neonatal hypertrophic cardiomyopathy: what is the link? Pediatr Cardiol 2009;30:1012-5. [DOI] [PubMed] [Google Scholar]
- 4.Rauen KA. The RASopathies. Annu Rev Genomics Hum Genet 2013;14:355-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tidyman WE, Rauen KA. The RASopathies: developmental syndromes of Ras/MAPK pathway dysregulation. Curr Opin Genet Dev 2009;19:230-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nosan G, Bertok S, Vesel S, et al. A lethal course of hypertrophic cardiomyopathy in Noonan syndrome due to a novel germline mutation in the KRAS gene: case study. Croat Med J 2013;54:574-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gibson W, Trevenen C, Giuffre M, et al. Noonan syndrome in a premature infant with hypertrophic cardiomyopathy and death in infancy. J Natl Med Assoc 2005;97:805-7. [PMC free article] [PubMed] [Google Scholar]
- 8.Allanson JE, Roberts AE. Noonan syndrome. Pagon RA, Adam MP, Ardinger HH, et al. GeneReviews® [Internet]. Seattle: University of Washington, Seattle; 1993-2015. [PubMed] [Google Scholar]
- 9.ClinVar, accession n. RCV000037706. Available from: http://www.ncbi.nlm.nih.gov/clinvar /5359551/. [Google Scholar]
- 10.Romano AA, Allanson JE, Dahlgren J, et al. Noonan syndrome: clinical features, diagnosis, and management guidelines. Pediatrics 2010;126:746-59. [DOI] [PubMed] [Google Scholar]
- 11.Nishikawa T, lshiyama S, Shimojo T, et al. Hypertrophic cardiomyopathy in Noonan syndrome. Acta Paediatr Jpn 1996;38:91-8 [DOI] [PubMed] [Google Scholar]