

Abstract
Despite advances in surgical and medical therapy, glioblastoma multiforme (GBM) remains a fatal disease. There has been no significant increase in survival for patients with this disease over the last 20 years. Tumor vasculature formation and glioma cell invasion along the white matter tracts both play a pivotal role in glioma development. Angiogenesis and invasion are the major factors believed to be responsible for treatment resistance in tumors, and a better understanding of the glioma invasion and angiogenesis mechanisms will lead to the development of potential new treatments. In this review, we focus on the molecular characteristics of angiogenesis and invasion in human malignant glioma. We discuss bevacizumab and cilengitide, which are used to inhibit angiogenesis in GBM.
Keywords: cilengitide, bevacizumab, angiogenesis, invasion
Introduction
Despite advances in surgical and medical therapy, glioblastoma multiforme (GBM) remains a fatal disease. The pathophysiological processes of angiogenesis and tumor cell invasion play pivotal roles in glioma development and growth, beginning in the earliest phase of tumor growth.4) The main reasons for the resistance of treatment in these tumors were the formation of abnormal dysfunctional tumor vasculature and glioma cell invasion along white matter tracts. Recent insight into the glioma angiogenesis and invasion mechanisms have provided renewed hope for developing novel strategies aimed at reducing morbidity due to this fatal disease. However, glioma angiogenesis and invasion are challenging to investigate in experimental settings because most of the animal models fail to mimic the unique angiogenesis and invasiveness of human glioma cells.
In this article, we review histopathological studies that focus on invasion and angiogenesis of human malignant gliomas. We also focus on the molecular aspects of glioma angiogenesis and invasion and the key mediators of these processes. In addition, we consider several animal glioma models that are available for studying invasion and angiogenesis, including our novel animal models. Finally, we discuss bevacizumab (a recombinant humanized monoclonal antibody targeting vascular endothelial growth factor [VEGF]) and cilengitide (an inhibitor of αvβ3 and αvβ5 integrins).
Histopathological Analysis of Angiogenesis and Invasion
GBM is known to have blood vessels of increased diameter with high permeability, thickened basement membranes, and highly proliferative endothelial cells.41) The histopathological hallmark of GBM is the presence of microvascular proliferation with the formation of glomerular capillary loops in a garland-like formation.54) One of the malignancy evaluation criteria is increased neoplastic proliferation of glial cells running parallel to endothelial vascular proliferation.40) Vascular density in GBM is markedly higher than that in glioma of a lower histological grade.63) An increase in vascularization significantly worsens the disease’s prognosis.40)
Histopathological studies have given some insights into tumor invasion. We showed previously that there are at least two invasive and angiogenic glioma phenotypes. Clusters of glioma cells were seen around newly developed vessels in the normal parenchyma adjacent to the tumor margins. Single cell infiltrations were also seen in normal brain parenchyma independent of the vasculature (Fig. 1). These different invasive and angiogenic phenotypes are either angiogenesis-dependent or angiogenesis-independent. GBM consists of a mixture of subclones with both angiogenesis-dependent and angiogenesis-independent invasion phenotypes present in various proportions.27,46,49)
Fig. 1.
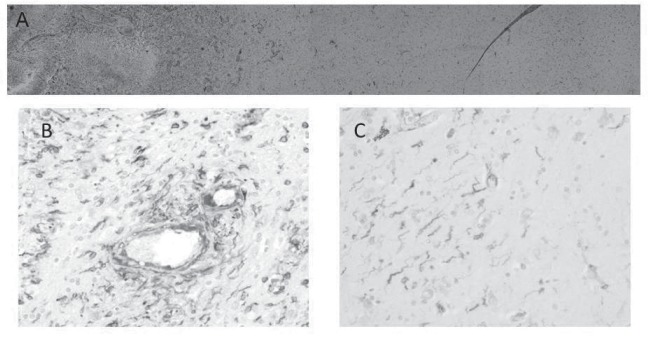
Microtubule-associated protein (MAP) 2e and von Willebrand factor (vWF) immunohistochemical staining of human GBM samples. A: MAP2e, a splice variant of MAP2, was a candidate glioma-specific antigen. Tumor cells diffusely infiltrated from the tumor center to normal brain tissue; there is no border between them. B: At the tumor border, MAP2e-positive tumor cells clustered around dilated vessels. C: Single MAP2e-positive tumor cell infiltration into normal brain parenchyma that are independent of vasculature were also seen. MAP2e: diaminobenzidine (DAB), vWf: DAB-Ni, Counterstain: hematoxylin.
Molecular Biology of Angiogenesisin GBM
Angiogenesis is one of the key events in GBM development, and the histological diagnosis of GBM was led by the presence of microvascular proliferation.65) Among all solid tumors, GBM has been reported to be the most angiogenic because it displays the highest degree of endothelial cell hyperplasia and vascular proliferation.9) The peritumoral edema resulting from a defective blood brain barrier (BBB) in the newly formed tumor vasculature is a pathological feature of GBM.17,67) Vascular homeostasis is maintained by a balance between pro-angiogenic and anti-angiogenic stimuli.29) Angiogenesis is activated in developing GBM when the pro-angiogenic stimuli outweigh the anti-angiogenic stimuli. Tissue hypoxia is the most potent activator of angiogenic mechanisms in brain tumors. The hypoxia-inducible factor (HIF) -1/VEGF-A pathway is one of the well-studied pathways. The HIF-1/VEGF-A pathway leads to endothelial cell proliferation and migration.30) HIF-1 activates deoxyribonucleic acid (DNA) promoter regions, which are known as hypoxia response elements (HREs). HREs induce transcription of > 100 genes that help the cell to adapt to low O2 conditions.8,62) VEGF is an example of a gene that is regulated by an HIF-1 through an HRE. VEGF regulates brain edema surrounding brain tumors and blood vessel formation; specifically, VEGF-A is known to be upregulated in GBM.24) VEGF-A regulates endothelial cell survival, permeability, proliferation, and migration primarily via the VEGF-receptor 2 (VEGFR2).13) VEGF promotes endothelial proliferation by activation of the mitogen-activated protein kinase (MAPK) pathway.65) VEGF also enhances vascular permeability through the MAPK signaling cascade. VEGF rearranges adherin/catenin complexes and loosens adhering junctions between endothelial cells.19,31) VEGF stimulates endothelial production of urokinase-type plasminogen activator (uPA).45) uPA induces the conversion of plasminogen to plasmin and the breakdown of extracellular matrix (ECM) components, and leads to ECM remodeling.65) Immature, highly permeable blood vessels with subsequent poor maintenance of BBB and parenchymal edema are produced as a result of VEGF signaling in tumors.28,29)
Angiogenesis is the formation of new blood vessels by rerouting or remodeling of existing vessels. It is believed to be the primary method of vessel formation in gliomas. Angiogenesis requires three distinct steps: (1) blood vessel breakdown, (2) degradation of the vessel basement membrane and surrounding ECM, and (3) migration of endothelial cells and the formation of new blood vessels.49)
Blood vessel breakdown
The first step of angiogenesis is the dissolution of native vessels aspects. Glioma cells first accumulate around the existing cerebral blood vessels and lift off the astrocytic foot processes, which leads to the disruption of the normal contact between endothelial cells and the basement membrane.66) The affected endothelial cells express angiopoietin-2 (Ang-2) resulting in destabilization of the vessel wall and decreased pericyte coverage.26,66,68) Ang-1 and -2 are important endothelial growth factors that signal via the Tie2 receptor tyrosine kinase (RTK) expressed on endothelial cells. In the normal brain, Ang-1 binds to Tie2, inducing association between pericytes and endothelial cells and resulting in stabilization of the vasculature.6,55) Conversely, Ang-2 may act as an antagonist to Tie2 phosphorylation, and lead to destabilization of blood vessels. Therefore, Ang-2 represents a checkpoint for Ang-1/Tie2-mediated angiogenesis.44,66)
Degradation of the vessel basement membrane and surrounding ECM
Degradation of the vessel basement membrane and surrounding ECM facilitates the invasion of endothelial cells. This step is an integral part of the ongoing angiogenic process.56) Gelatinase-A (MMP-2) and gelatinase-B (MMP-9) are highly expressed in astrocytomas.21,52) MMP-2 and MMP-9 expression is strongly induced by hypoxia, and these two molecules appear to have a synergistic effect on basement membrane degradation.36)
Migration of endothelial cells and formation of new blood vessels
After regression of existing vessels and breakdown of the basement membrane, endothelial cells proliferate and migrate toward the tumor cells expressing pro-angiogenic compounds. Integrin αvβ3 and α5β1 are upregulated in endothelial cells during angiogenesis, enhancing endothelial cell adhesion and migration.10,32) Migration of pericytes is an important part of tumor vessel formation. Platelet-derived growth factor (PDGF) that is secreted by activated endothelial cells recruits pericytes to the site of newly sprouting vessels and aids in establishing a new basement membrane.20,41)
Molecular Biology of GBM Invasion
One of the insidious biological features of gliomas is the potential of single cells to invade normal brain tissue. Details of the glioma invasion mechanisms are only beginning to be determined.
Tumor cell invasion requires four distinct steps: (1) detachment of invading cells from the primary tumor mass, (2) adhesion to ECM, (3) degradation of ECM, and (4) cell motility and contractility.49)
Detachment of invading cells from the primary tumor mass
The detachment of invading glioma cells from the primary tumor mass involves several events. The first event is destabilization and disorganization of cadherin-mediated junctions that hold the primary mass together. The second event is a decline in the expression of connexin 43. The reduction in connexion 43 leads to the reduction in gap junction formation.23) The third event is cleavage of CD44. CD44 anchors the primary mass to ECM, by the metalloproteinase A Disintegrin and Metalloproteinase (ADAM).48)
Adhesion to ECM
Integrins are the most common molecules that allow glioma cells to adhere to ECM. In particular, the integrin αvβ3 is thought to play a central role in glioma invasion. Integrin αvβ3 binds to fibronectin, vitronectin, and tenascin-C in ECM.39)
ECM degradation
MMPs are the most common proteases that degrade the ECM and create space for the invading glioma cells. MMP-2 and MMP-3 levels and MMP-2/MMP-9 activity correlate with glioma cell migration and invasion.64)
Cell motility and contractility
Cell motility requires cytoplasmic contractile force. Myosin II allows glioma cells to squeeze through pores smaller than their nuclear diameter, which is important because the brain has particularly narrow extracellular spaces.3) Small GTPases, such as RhoA, Rac, cdc42, as well as RLC-interacting protein are also involved in this process in glioma cells.7,57)
Animal Models for Studying Glioma Invasion and Angiogenesis
Studying glioma invasion and angiogenesis are challenging, because most animal models fail to mimic the unique features of human glioma cell invasiveness and angiogenesis16). Typically, such transplantable tumors in mice or rats form solid nodules at the injection site and compress rather than invade the surrounding brain regions.
We have established two novel animal models with different invasive and angiogenic phenotypes. Two subclones, J3T-1 and J3T-2, were established by passage of the parental canine glioma cell line, J3T, in immunocompromised animals. These cells established tumors following intracerebral inoculation in athymic mouse and rat.27) J3T-1 cells clustered around dilated blood vessels at tumor borders whereas J3T-2 cells showed diffuse single cell infiltration into surrounding normal parenchyma. Marked angiogenesis was seen only in J3T-1 gliomas (Fig. 2). The described animal models histologically imitated two invasive and angiogenic phenotypes, namely angiogenesis-dependent and -independent invasion, which is also observed in human GBM. Angiogenesis-dependent invasion is that tumor cells coopt around pre-existing vessels and secrete angiogenic factors to develop neovasculature and tumor cells proliferate around newly developed vessels and migrate along them, resulting in the formation of perivascular cuffing. Angiogenesis-independent invasion is that tumor cells migrate as single cells along myelinated axons without inducing angiogenesis. These novel models would be particularly beneficial for analyzing the molecular mechanisms of glioma invasion and angiogenesis and investigating new glioma therapies.27,46,49,50) We performed proteomic analysis using J3T-1 (angiogenesis-dependent invasion phenotype) and J3T-2 (angiogenesis-independent invasion phenotype) to investigate the molecular basis of invasion and angiogenesis by malignant gliomas.46) One of the proteins identified was annexin A2, which was expressed at higher levels in J3T-1 than in J3T-2. Moreover, immunohistochemical analysis of human GBM specimens showed that annexin A2 was expressed at high levels in the tumor cells that formed clusters around dilated vessels. Thus, annexin A2 may be related to angiogenesis-dependent invasion.46)
Fig. 2.
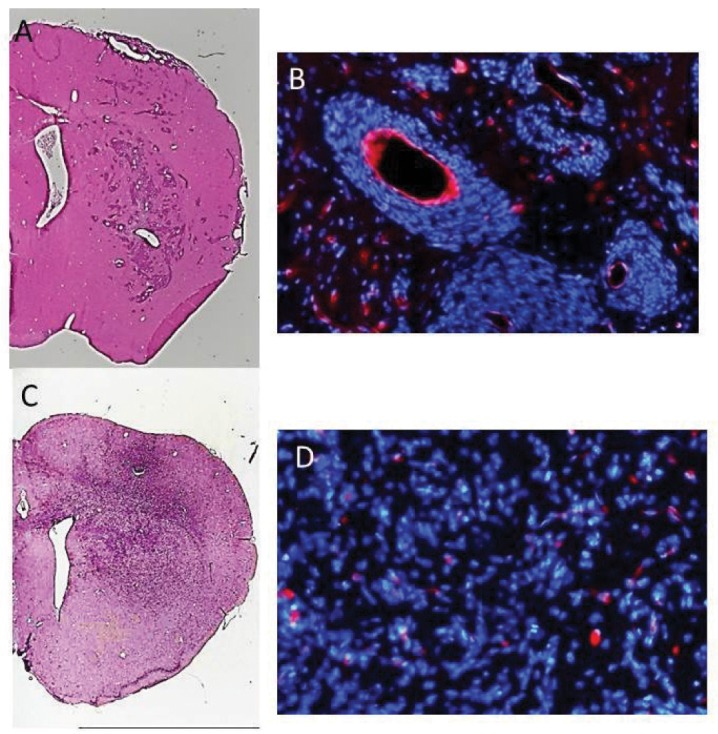
Two distinct invasion phenotypes in animals harboring J3T-1 and J3T-2 brain tumors. A, B: A J3T-1 brain tumor was established in athymic rat brains. C, D: J3T-1 cells formed well demarcated and highly angiogenic tumors in the rat brain. Multiple small satellite tumors are also seen at tumor borders. A J3T-2 brain tumor was established in athymic rat brains. J3T-2 cells gradually dispersed from the tumor center to the surrounding normal brain tissue. A and C: hematoxylin-eosin staining. B and D: vascular stain (RECA-1, red) and nuclear stain (DAPI, blue). DAPI: 4’,6-diamidino-2-phenylindole, RECA: rat endothelial cell antigen.
Bevacizumab
Bevacizumab (Avastin) is a recombinant humanized monoclonal antibody that targets VEGF; it was the first anti-angiogenesis agent to be approved by the United States Food and Drug Administration (FDA) in 2004. Several Phase II clinical trials have studied the therapeutic efficacy of bevacizumab as a single-agent or in combination with chemotherapy or radiation for recurrent GBM. The results support the conclusion that bevasizumab is effective for recurrent GBM. Bevacizumab has demonstrated encouraging radiographic response in patients with recurrent malignant gliomas.58) Retrospective studies have also supported the same conclusion.2,14) In 2013, during the American Society of Clinical Oncology (ASCO) Annual Meeting, the results of the positive phase III AVAglio study was presented by Roger Henriksson et al.25) The study showed bevacizumab in combination with radiation and temozolomide chemotherapy reduced the risk of cancer progression or death (progression-free survival; PFS) by 36% in people with newly diagnosed GBM. The results for overall survival (OS) did not reach statistical significance. The result of the phase III RTOG 0825 study was presented at the 2013 ASNO Annual Meeting by Gilbert et al. The RTOG 0825 study was the double-blind placebo-controlled trial evaluating bevacizumab in patients with newly diagnosed GBM. The addition of bevacizumab for newly diagnosed GBM did not improve OS, but improved PFS, but did not reach the significance criterion. De Groot et al.15) showed that patients with glioma developed an apparent phenotypic shift to a predominantly infiltrative pattern of tumor progression after treatment with bevacizumab. Indeed, a preclinical study showed that anti-angiogenic therapy of murine gliomas with an antibody against VEGF-R2 caused small satellite tumors to arise near the primary mass, centered around core vessels, which is similar to the perivascular invasion found more recently in the VEGF knockout glioma cell lines that were described previously.37) Further research is needed to identify mediators of this invasion and to determine whether the invasion seen after bevacizumab treatment of human GBM is the perivascular invasion seen in the murine cell lines or the parenchymal type of invasion along white matter typically seen in GBM.4) It has been proposed that hypoxia caused by vessel regression during the course of anti-angiogenic therapy leads to up-regulation of proangiogenic factors and recruitment of bone marrow-derived cells (BMDCs) that have the capacity to increase tumor growth by means of new blood vessel growth.5) Glioma cells evade antiangiogenic therapies by up-regulating alternative proangiogenic signal circuits, including those utilizing fibroblast growth factor, ephrin A1, and angiopoietin 1. Another adaptive measure is the hypoxia-regulated recruitment of vascular progenitor cells and proangiogenic monocytes from the bone marrow to tumors.5)
Cilengitide
Cilengitide (EMD121974), an inhibitor of αvβ3 and αvβ5 integrins, demonstrated preclinical efficacy against malignant glioma.43) It is speculated that cilengitide can inhibit tumor growth, invasion, and angiogenesis. However, the effects of cilengitide on these processes have not been sufficiently examined.
We investigated the anti-glioma effect of cilengitide using DNA microarray analysis. U87ΔEGFR cells (human malignant glioma cell line) were used for this experiment. The cells were harvested after 16 h of cilengitide treatment, and mRNA was extracted. Gene expression and pathway analyses were performed using a DNA microarray (CodeLinkTM human whole Genome Bioarray, [Applied Microarrays, Inc., Tempe, AZ, USA]). The expression of 264 genes was changed with cilengitide treatment. The expression of 214 genes was up-regulated and that of 50 genes was down-regulated compared to the controls. In pathway analysis, “apoptotic cleavage of cellular proteins” and “TNF receptor signaling pathway” were over-represented. Apoptotic-associated genes such as caspase 8 were up-regulated. We revealed that cilengitide activated caspase-8 and induced apoptosis-related pathways (Fig. 3).51)
Fig. 3.

In vivo immunohistochemical analysis of caspase 8 expression in U87ΔEGFR brain tumors. U87ΔEGFR cells were injected into the right frontal lobe of athymic rats. Cilengitide or phosphate buffered saline (PBS) was administered 3 times/week intraperitoneally starting on day 5 after tumor cell implantation. To assess the expression of caspase 8, athymic rats harboring U87ΔEGFR brain tumors were sacrificed at 14 days after tumor implantation. A subpopulation of caspase 8-positive cells was visualized using immunostaining (caspase 8-positive cells: caspase 8, red; nuclei: DAPI, blue) of U87ΔEGFR control xenografts (A) and U87ΔEGFR cilengitide-treated xenografts (B). The control sections exhibited scattered red fluorescence (A), whereas more punctate red fluorescence was observed in the cilengitide-treated xenografts (B). DAPI: 4’,6-diamidino-2-phenylindole.
In our previous study, we investigated the anti-glioma mechanisms of cilengitide utilizing the novel invasive glioma models, J3T-1 (angiogenesis-dependent invasion phenotype) and J3T-2 (angiogenesis-independent invasion phenotype). Cilengitide treatment resulted in a significantly decreased diameter of the J3T-1 tumor vessel clusters and its core vessels when compared to controls, while an anti-invasive effect was shown in the J3T-2 brain tumor with a significant reduction of diffuse cell infiltration around the tumor center (Fig. 4). Our results indicate that cilengitide exerts a phenotypic anti-tumor effect by inhibiting angiogenesis and glioma cell invasion in vivo.50) Angiogenesis requires three distinct steps: (1) blood vessel breakdown, (2) degradation of the vessel basement membrane and surrounding ECM, and (3) migration of endothelial cells and the formation of new blood vessels.60) During the third step of angiogenesis, integrin αvβ3 is upregulated in endothelial cells, enhancing endothelial cell adhesion and migration.10,11) The final product of glioma angiogenesis is a vasculature with highly tortuous dilated vessels.60) Cilengitide prevents the third step of angiogenesis and reduces the size of tumor vessels. Glioma cell invasion requires four distinct steps: (1) detachment of invading cells from the primary tumor mass, (2) adhesion to the ECM, (3) degradation of the ECM, and (4) cell motility and contractility.49) Integrins are the molecules that allow glioma cells to adhere to the ECM during the second step.60) Cilengitide might inhibit the second step, thereby suppressing the invasion of glioma.
Fig. 4.

Bimodal anti-glioma mechanisms of cilengitide demonstrated by novel invasive glioma models. Anti-angiogenic effects of cilengitide on the J3T-1 brain tumor in rats. Examined with immunofluorescence staining (vascular: RECA-1, red; nuclei: DAPI, blue), the diameter of the J3T-1 tumor clusters’ core vessels in cilengitide-treated animals (B) was smaller than that in untreated animals (A). We observed the anti-invasive effects of cilengitide on the J3T-2 gliomas in rats. Immunofluorescence staining (vascular endothelial cells: RECA-1, red; nuclei: DAPI, blue) of J3T-2 control tumors (C) and cilengitide-treated J3T-2 tumors (D) revealed that the tumor borders in treated animals were more evident. The cell density of control tumors was gradually reduced from the tumor center toward the normal brain parenchyma, while the cell density of treated tumors dropped steeply at the tumor border.DAPI: 4’,6-diamidino-2-phenylindole, RECA: rat endothelial cell antigen.
Several preclinical studies have shown an enhanced antitumor effect of cilengitide when administered in combination therapeutic regimens.1,12,53,61) Mikkelsen et al. demonstrated that cilengitide dramatically amplified the efficacy of radiation therapy in an animal glioma model.47) We demonstrated the enhanced therapeutic efficacy of an oncolytic virus on experimental glioma following pretreatment with cilengitide.35) This research showed that pretreatment of gliomas with the angiogenesis inhibitor cilengitide reduced inflammation, vascular hyperpermeability, and leukocyte infiltration of tumor tissue upon treatment with oncolytic virus. Reduction of host immune responses by cilengitide treatment enhanced the anticancer efficacy of oncolytic virus treatment by increasing oncolytic virus propagation in tumors. We also reported that oncolytic HSV-1 infection of tumors induces angiogenesis and upregulates cysteine-rich protein 61 (CYR61).34) CYR61 was identified as a member of the CCN (CYR61/CTGF/NOV) family of matricelluar proteins regulating cell growth, differentiation, survival, angiogenesis, and migration in development, tissue remodeling, and repair.38)
In order to test the role of CYR61-mediated integrin activation in Oncolytic Virus (OV)-induced angiogenesis, the impact of cilengitide on OV treatment-induced angiogenesis was investigated.33,38)
Clinical Trials
A phase I clinical trial of cilengitide in recurrent malignant gliomas by the New Approaches to Brain Tumor Therapy (NABTT) has been completed with no dose limiting toxicities.18) The most notable trial to date was a randomized phase II study of cilengitide, which was associated with a median survival of 10 months in recurrent glioma patients.53) The North American Brain Tumor Consortium (NABTC) study22) was designed to determine whether cilengitide effectively penetrates into GBM in human patients. This study confirmed that cilengitide is effectively delivered into primary human GBM tumors with good retention. The result of the CENTRIC study (phase III), which compares the efficacy and tolerability of cilengitide in patients with newly diagnosed GBM and a methylated O6-methylguanine-DNA methyltransferase (MGMT) gene promoter status,59) was presented at 2013 ASCO Annual Meeting by Roger Stupp et al.59) Cilengitide failed to prolong PFS or OS in patients with newly diagnosed GBM and methylated MGMT gene promoter. The previously reported safety profile of cilengitide in addition to standard therapy was confirmed. The phase II CORE trial, which included only patients with an unmethylated MGMT gene promoter status, are currently ongoing.
Conclusions
A better understanding of the molecular components responsible for glioma angiogenesis and invasion will hopefully lead to the development of new treatment methods. Antiangiogenic therapy is dramatically altering the treatment landscape for patients with GBM.
References
- 1). Abdollahi A, Griggs DW, Zieher H, Roth A, Lipson KE, Saffrich R, Gröne HJ, Hallahan DE, Reisfeld RA, Debus J, Niethammer AG, Huber PE: Inhibition of alpha(v)beta3 integrin survival signaling enhances antiangiogenic and antitumor effects of radiotherapy. Clin Cancer Res 11: 6270– 6279, 2005. [DOI] [PubMed] [Google Scholar]
- 2). Agha CA, Ibrahim S, Hassan A, Elias DA, Fathallah-Shaykh HM: Bevacizumab is active as a single agent against recurrent malignant gliomas. Anticancer Res 30: 609– 611, 2010. [PubMed] [Google Scholar]
- 3). Beadle C, Assanah MC, Monzo P, Vallee R, Rosenfeld SS, Canoll P: The role of myosin II in glioma invasion of the brain. Mol Biol Cell 19: 3357– 3368, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4). Bello L, Giussani C, Carrabba G, Pluderi M, Costa F, Bikfalvi A: Angiogenesis and invasion in gliomas. Cancer Treat Res 117: 263– 284, 2004. [DOI] [PubMed] [Google Scholar]
- 5). Bergers G, Hanahan D: Modes of resistance to anti-angiogenic therapy. Nat Rev Cancer 8: 592– 603, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6). Bergers G, Song S: The role of pericytes in blood-vessel formation and maintenance. Neuro-oncology 7: 452– 464, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7). Bornhauser BC, Lindholm D: MSAP enhances migration of C6 glioma cells through phosphorylation of the myosin regulatory light chain. Cell Mol Life Sci 62: 1260– 1266, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8). Brahimi-Horn C, Berra E, Pouysségur J: Hypoxia: the tumor's gateway to progression along the angiogenic pathway. Trends Cell Biol 11: S32– 36, 2001. [DOI] [PubMed] [Google Scholar]
- 9). Brem S, Cotran R, Folkman J: Tumor angiogenesis: a quantitative method for histologic grading. J Natl Cancer Inst 48: 347– 356, 1972. [PubMed] [Google Scholar]
- 10). Brooks PC, Clark RA, Cheresh DA: Requirement of vascular integrin alpha v beta 3 for angiogenesis. Science 264: 569– 571, 1994. [DOI] [PubMed] [Google Scholar]
- 11). Brooks PC, Montgomery AM, Rosenfeld M, Reisfeld RA, Hu T, Klier G, Cheresh DA: Integrin alpha v beta 3 antagonists promote tumor regression by inducing apoptosis of angiogenic blood vessels. Cell 79: 1157– 1164, 1994. [DOI] [PubMed] [Google Scholar]
- 12). Burke PA, DeNardo SJ, Miers LA, Lamborn KR, Matzku S, DeNardo GL: Cilengitide targeting of alpha(v)beta(3) integrin receptor synergizes with radioimmunotherapy to increase efficacy and apoptosis in breast cancer xenografts. Cancer Res 62: 4263– 4272, 2002. [PubMed] [Google Scholar]
- 13). Carmeliet P, Jain RK: Angiogenesis in cancer and other diseases. Nature 407: 249– 257, 2000. [DOI] [PubMed] [Google Scholar]
- 14). Chamberlain MC, Johnston SK: Salvage therapy with single agent bevacizumab for recurrent glioblastoma. J Neurooncol 96: 259– 269, 2010. [DOI] [PubMed] [Google Scholar]
- 15). de Groot JF, Fuller G, Kumar AJ, Piao Y, Eterovic K, Ji Y, Conrad CA: Tumor invasion after treatment of glioblastoma with bevacizumab: radiographic and pathologic correlation in humans and mice. Neuro-oncology 12: 233– 242, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16). de Vries NA, Beijnen JH, van Tellingen O: High-grade glioma mouse models and their applicability for preclinical testing. Cancer Treat Rev 35: 714– 723, 2009. [DOI] [PubMed] [Google Scholar]
- 17). Del Maestro RF, Megyesi JF, Farrell CL: Mechanisms of tumor-associated edema: a review. Can J Neurol Sci 17: 177– 183, 1990. [DOI] [PubMed] [Google Scholar]
- 18). Eskens FA, Dumez H, Hoekstra R, Perschl A, Brindley C, Böttcher S, Wynendaele W, Drevs J, Verweij J, van Oosterom AT: Phase I and pharmacokinetic study of continuous twice weekly intravenous administration of Cilengitide (EMD 121974), a novel inhibitor of the integrins alphavbeta3 and alphavbeta5 in patients with advanced solid tumours. Eur J Cancer 39: 917– 926, 2003. [DOI] [PubMed] [Google Scholar]
- 19). Esser S, Lampugnani MG, Corada M, Dejana E, Risau W: Vascular endothelial growth factor induces VE-cadherin tyrosine phosphorylation in endothelial cells. J Cell Sci 111 ( Pt 13): 1853– 1865, 1998. [DOI] [PubMed] [Google Scholar]
- 20). Ferrara N, Kerbel RS: Angiogenesis as a therapeutic target. Nature 438: 967– 974, 2005. [DOI] [PubMed] [Google Scholar]
- 21). Forsyth PA, Wong H, Laing TD, Rewcastle NB, Morris DG, Muzik H, Leco KJ, Johnston RN, Brasher PM, Sutherland G, Edwards DR: Gelatinase-A (MMP-2), gelatinase-B (MMP-9) and membrane type matrix metalloproteinase-1 (MT1-MMP) are involved in different aspects of the pathophysiology of malignant gliomas. Br J Cancer 79: 1828– 1835, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22). Gilbert MR, Kuhn J, Lamborn KR, Lieberman F, Wen PY, Mehta M, Cloughesy T, Lassman AB, Deangelis LM, Chang S, Prados M: Cilengitide in patients with recurrent glioblastoma: the results of NABTC 03-02, a phase II trial with measures of treatment delivery. J Neurooncol 106: 147– 153, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23). Goodenough DA, Goliger JA, Paul DL: Connexins, connexons, and intercellular communication. Annu Rev Biochem 65: 475– 502, 1996. [DOI] [PubMed] [Google Scholar]
- 24). Hanahan D, Folkman J: Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell 86: 353– 364, 1996. [DOI] [PubMed] [Google Scholar]
- 25). Henriksson R, Bottomley A, Mason W, et al. : Progression-free survival (PFS) and health-related quality of life (HRQoL) in AVAglio, a phase III study of bevacizumab (Bv), temozolomide (T), and radiotherapy (RT) in newly diagnosed glioblastoma (GBM). 2013 ASCO Annual Meeting Abstract 2005. Presented June 1, 2013 [Google Scholar]
- 26). Holash J, Maisonpierre PC, Compton D, Boland P, Alexander CR, Zagzag D, Yancopoulos GD, Wiegand SJ: Vessel cooption, regression, and growth in tumors mediated by angiopoietins and VEGF. Science 284: 1994– 1998, 1999. [DOI] [PubMed] [Google Scholar]
- 27). Inoue S, Ichikawa T, Kurozumi K, Maruo T, Onishi M, Yoshida K, Fujii K, Kambara H, Chiocca EA, Date I: Novel animal glioma models that separately exhibit two different invasive and angiogenic phenotypes of human glioblastomas. World Neurosurg 78: 670– 682, 2012. [DOI] [PubMed] [Google Scholar]
- 28). Jain RK: Molecular regulation of vessel maturation. Nat Med 9: 685– 693, 2003. [DOI] [PubMed] [Google Scholar]
- 29). Jain RK, di Tomaso E, Duda DG, Loeffler JS, Sorensen AG, Batchelor TT: Angiogenesis in brain tumours. Nat Rev Neurosci 8: 610– 622, 2007. [DOI] [PubMed] [Google Scholar]
- 30). Jiang BH, Rue E, Wang GL, Roe R, Semenza GL: Dimerization, DNA binding, and transactivation properties of hypoxia-inducible factor 1. J Biol Chem 271: 17771– 17778, 1996. [DOI] [PubMed] [Google Scholar]
- 31). Kevil CG, Payne DK, Mire E, Alexander JS: Vascular permeability factor/vascular endothelial cell growth factor-mediated permeability occurs through disorganization of endothelial junctional proteins. J Biol Chem 273: 15099– 15103, 1998. [DOI] [PubMed] [Google Scholar]
- 32). Kim S, Bell K, Mousa SA, Varner JA: Regulation of angiogenesis in vivo by ligation of integrin alpha5beta1 with the central cell-binding domain of fibronectin. Am J Pathol 156: 1345– 1362, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33). Kireeva ML, Lam SC, Lau LF: Adhesion of human umbilical vein endothelial cells to the immediate-early gene product Cyr61 is mediated through integrin alphavbeta3. J Biol Chem 273: 3090– 3096, 1998. [DOI] [PubMed] [Google Scholar]
- 34). Kurozumi K, Hardcastle J, Thakur R, Shroll J, Nowicki M, Otsuki A, Chiocca EA, Kaur B: Oncolytic HSV-1 infection of tumors induces angiogenesis and upregulates CYR61. Mol Ther 16: 1382– 1391, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35). Kurozumi K, Hardcastle J, Thakur R, Yang M, Christoforidis G, Fulci G, Hochberg FH, Weissleder R, Carson W, Chiocca EA, Kaur B: Effect of tumor microenvironment modulation on the efficacy of oncolytic virus therapy. J Natl Cancer Inst 99: 1768– 1781, 2007. [DOI] [PubMed] [Google Scholar]
- 36). Lakka SS, Gondi CS, Rao JS: Proteases and glioma angiogenesis. Brain Pathol 15: 327– 341, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37). Lamszus K, Kunkel P, Westphal M: Invasion as limitation to anti-angiogenic glioma therapy. Acta Neurochir Suppl 88: 169– 177, 2003. [DOI] [PubMed] [Google Scholar]
- 38). Leask A, Abraham DJ: All in the CCN family: essential matricellular signaling modulators emerge from the bunker. J Cell Sci 119: 4803– 4810, 2006. [DOI] [PubMed] [Google Scholar]
- 39). Leavesley DI, Ferguson GD, Wayner EA, Cheresh DA: Requirement of the integrin beta 3 subunit for carcinoma cell spreading or migration on vitronectin and fibrinogen. J Cell Biol 117: 1101– 1107, 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40). Lebelt A, Dziecioł J, Guzińska-Ustymowicz K, Lemancewicz D, Zimnoch L, Czykier E: Angiogenesis in gliomas. Folia Histochem Cytobiol 46: 69– 72, 2008. [DOI] [PubMed] [Google Scholar]
- 41). Lindahl P, Johansson BR, Levéen P, Betsholtz C: Pericyte loss and microaneurysm formation in PDGF-B-deficient mice. Science 277: 242– 245, 1997. [DOI] [PubMed] [Google Scholar]
- 42). Lopes MB: Angiogenesis in brain tumors. Microsc Res Tech 60: 225– 230, 2003. [DOI] [PubMed] [Google Scholar]
- 43). MacDonald TJ, Taga T, Shimada H, Tabrizi P, Zlokovic BV, Cheresh DA, Laug WE: Preferential susceptibility of brain tumors to the antiangiogenic effects of an alpha(v) integrin antagonist. Neurosurgery 48: 151– 157, 2001. [DOI] [PubMed] [Google Scholar]
- 44). Maisonpierre PC, Suri C, Jones PF, Bartunkova S, Wiegand SJ, Radziejewski C, Compton D, McClain J, Aldrich TH, Papadopoulos N, Daly TJ, Davis S, Sato TN, Yancopoulos GD: Angiopoietin-2, a natural antagonist for Tie2 that disrupts in vivo angiogenesis. Science 277: 55– 60, 1997. [DOI] [PubMed] [Google Scholar]
- 45). Mandriota SJ, Seghezzi G, Vassalli JD, Ferrara N, Wasi S, Mazzieri R, Mignatti P, Pepper MS: Vascular endothelial growth factor increases urokinase receptor expression in vascular endothelial cells. J Biol Chem 270: 9709– 9716, 1995. [DOI] [PubMed] [Google Scholar]
- 46). Maruo T, Ichikawa T, Kanzaki H, Inoue S, Kurozumi K, Onishi M, Yoshida K, Kambara H, Ouchida M, Shimizu K, Tamaru S, Chiocca EA, Date I: Proteomics-based analysis of invasion-related proteins in malignant gliomas. Neuropathology 33: 264– 275, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47). Mikkelsen T, Brodie C, Finniss S, Berens ME, Rennert JL, Nelson K, Lemke N, Brown SL, Hahn D, Neuteboom B, Goodman SL: Radiation sensitization of glioblastoma by cilengitide has unanticipated schedule-dependency. Int J Cancer 124: 2719– 2727, 2009. [DOI] [PubMed] [Google Scholar]
- 48). Nagano O, Saya H: Mechanism and biological significance of CD44 cleavage. Cancer Sci 95: 930– 935, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49). Onishi M, Ichikawa T, Kurozumi K, Date I: Angio-genesis and invasion in glioma. Brain Tumor Pathol 28: 13– 24, 2011. [DOI] [PubMed] [Google Scholar]
- 50). Onishi M, Ichikawa T, Kurozumi K, Fujii K, Yoshida K, Inoue S, Michiue H, Chiocca EA, Kaur B, Date I: Bimodal anti-glioma mechanisms of cilengitide demonstrated by novel invasive glioma models. Neuropathology 33: 162– 174, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51). Onishi M, Kurozumi K, Ichikawa T, Michiue H, Fujii K, Ishida J, Shimazu Y, Chiocca EA, Kaur B, Date I: Gene expression profiling of the anti-glioma effect of Cilengitide. Springerplus 2: 160, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52). Rao JS, Yamamoto M, Mohaman S, Gokaslan ZL, Fuller GN, Stetler-Stevenson WG, Rao VH, Liotta LA, Nicolson GL, Sawaya RE: Expression and localization of 92 kDa type IV collagenase/gelatinase B (MMP-9) in human gliomas. Clin Exp Metastasis 14: 12– 18, 1996. [DOI] [PubMed] [Google Scholar]
- 53). Reardon DA, Fink KL, Mikkelsen T, Cloughesy TF, O'Neill A, Plotkin S, Glantz M, Ravin P, Raizer JJ, Rich KM, Schiff D, Shapiro WR, Burdette-Radoux S, Dropcho EJ, Wittemer SM, Nippgen J, Picard M, Nabors LB: Randomized phase II study of cilengitide, an integrin-targeting arginine-glycine-aspartic acid peptide, in recurrent glioblastoma multiforme. J Clin Oncol 26: 5610– 5617, 2008. [DOI] [PubMed] [Google Scholar]
- 54). Reifenberger G, Collins VP: Pathology and molecular genetics of astrocytic gliomas. J Mol Med 82: 656– 670, 2004. [DOI] [PubMed] [Google Scholar]
- 55). Reiss Y, Machein MR, Plate KH: The role of angiopoietins during angiogenesis in gliomas. Brain Pathol 15: 311– 317, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56). Rooprai HK, McCormick D: Proteases and their inhibitors in human brain tumours: a review. Anti-cancer Res 17: 4151– 4162, 1997. [PubMed] [Google Scholar]
- 57). Salhia B, Rutten F, Nakada M, Beaudry C, Berens M, Kwan A, Rutka JT: Inhibition of Rho-kinase affects astrocytoma morphology, motility, and invasion through activation of Rac1. Cancer Res 65: 8792– 8800, 2005. [DOI] [PubMed] [Google Scholar]
- 58). Sathornsumetee S, Reardon DA, Desjardins A, Quinn JA, Vredenburgh JJ, Rich JN: Molecularly targeted therapy for malignant glioma. Cancer 110: 13– 24, 2007. [DOI] [PubMed] [Google Scholar]
- 59). Stupp R, Van Den Bent MJ, Erridge SC, Reardon DA, Hong Y, Wheeler H, Hegi M, Perry JR, Picard M, Weller M: Cilengitide in newly diagnosed glioblastoma with MGMT promoter methylation: protocol of a multicenter, randomized, open-label, controlled phase III trial (CENTRIC). J Clin Oncol 28: 15s, 2010. [Google Scholar]
- 60). Tate MC, Aghi MK: Biology of angiogenesis and invasion in glioma. Neurotherapeutics 6: 447– 457, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61). Tentori L, Dorio AS, Muzi A, Lacal PM, Ruffini F, Navarra P, Graziani G: The integrin antagonist cilengitide increases the antitumor activity of temozolomide against malignant melanoma. Oncol Rep 19: 1039– 1043, 2008. [PubMed] [Google Scholar]
- 62). Wenger RH, Stiehl DP, Camenisch G: Integration of oxygen signaling at the consensus HRE. Sci STKE 2005: re12, 2005. [DOI] [PubMed] [Google Scholar]
- 63). Wesseling P, Ruiter DJ, Burger PC: Angiogenesis in brain tumors; pathobiological and clinical aspects. J Neurooncol 32: 253– 265, 1997. [DOI] [PubMed] [Google Scholar]
- 64). Wild-Bode C, Weller M, Wick W: Molecular determinants of glioma cell migration and invasion. J Neurosurg 94: 978– 984, 2001. [DOI] [PubMed] [Google Scholar]
- 65). Wong ML, Prawira A, Kaye AH, Hovens CM: Tumour angiogenesis: its mechanism and therapeutic implications in malignant gliomas. J Clin Neurosci 16: 1119– 1130, 2009. [DOI] [PubMed] [Google Scholar]
- 66). Zagzag D, Amirnovin R, Greco MA, Yee H, Holash J, Wiegand SJ, Zabski S, Yancopoulos GD, Grumet M: Vascular apoptosis and involution in gliomas precede neovascularization: a novel concept for glioma growth and angiogenesis. Lab Invest 80: 837– 849, 2000. [DOI] [PubMed] [Google Scholar]
- 67). Zagzag D, Goldenberg M, Brem S: Angiogenesis and blood-brain barrier breakdown modulate CT contrast enhancement: an experimental study in a rabbit brain-tumor model. AJR Am J Roentgenol 153: 141– 146, 1989. [DOI] [PubMed] [Google Scholar]
- 68). Zagzag D, Hooper A, Friedlander DR, Chan W, Holash J, Wiegand SJ, Yancopoulos GD, Grumet M: In situ expression of angiopoietins in astrocytomas identifies angiopoietin-2 as an early marker of tumor angiogenesis. Exp Neurol 159: 391– 400, 1999. [DOI] [PubMed] [Google Scholar]