

SUMMARY
Acute myeloid leukemia (AML) manifests as phenotypically and functionally diverse cells, often within the same patient. Intratumor phenotypic and functional heterogeneity have been linked primarily by physical sorting experiments, which assume that functionally distinct subpopulations can be prospectively isolated by surface phenotypes. This assumption has proven problematic and we therefore developed a data-driven approach. Using mass cytometry, we profiled surface and intracellular signaling proteins simultaneously in millions of healthy and leukemic cells. We developed PhenoGraph, which algorithmically defines phenotypes in high-dimensional single-cell data. PhenoGraph revealed that the surface phenotypes of leukemic blasts do not necessarily reflect their intracellular state. Using hematopoietic progenitors, we defined a signaling-based measure of cellular phenotype, which led to isolation of a gene expression signature that was predictive of survival in independent cohorts. This study presents new methods for large-scale analysis of single-cell heterogeneity and demonstrates their utility, yielding insights into AML pathophysiology.
INTRODUCTION
Intratumor heterogeneity is accepted to be functionally and clinically significant (Marusyk et al., 2012). Recent evidence implies that the pathobiology of cancer results from the actions and interactions of diverse subpopulations within the tumor. Thus, it is necessary to study tumors with methods that preserve single-cell resolution. Emerging technologies such as mass cytometry (Bendall et al., 2011) and single-cell RNAseq (Patel et al., 2014) have attained dramatic increases in dimensionality and throughput, bringing unprecedented resolution to the diversity of cellular states detectable in a given tissue. Yet, to take advantage of these technological gains, computational methods are required to robustly identify high-dimensional phenotypes and compare them within and between individuals. Data-driven phenotypic dissection may then form the basis for downstream analyses in which subpopulations are isolated and compared, revealing the role of complex population structure in uncharacterized systems such as malignancies.
Intratumor heterogeneity is pervasive in acute myeloid leukemia (AML), an aggressive liquid tumor of the bone marrow characterized by an overwhelming abundance of poorly differentiated myeloid cells (‘blasts’). Arising from the disruption of regulated myeloid differentiation (Tenen, 2003), AML results in a disordered developmental hierarchy wherein leukemic stem cells (LSCs) are capable of re-establishing the disease in immunodeficient mice (Bonnet and Dick, 1997). LSCs were first thought to be restricted to the same CD34+/CD38− cellular compartment as normal hematopoietic stem cells (HSCs). Subsequent studies have demonstrated increased plasticity in AML where both CD38+ (Taussig et al., 2008) and CD34− (Taussig et al., 2010) cells have LSC capacity, indicating that AML does not follow the hierarchy of normal hematopoiesis. While AML exhibits a differentiated hierarchy, no uniform phenotypic identifier for LSCs has been found across patients (Eppert et al., 2011).
Recognizing a disconnect between functionally primitive (e.g., tumor-initiating) cells associated with cancer persistence and their surface phenotype, we simultaneously examined surface antigen expression and regulatory signaling in individual AML cells. We reasoned that intracellular signaling rather than antigen profile more accurately represents the functional state of a diseased cell. We used mass cytometry to measure protein expression and activation state in millions of cells from AML patients and healthy bone marrow donors in 31 simultaneous dimensions. By measuring cells after ex vivo perturbations we further expanded the dimensionality of the data by revealing functional responses to environmental cues reflecting the broader cellular network beyond what can be inferred from the unperturbed state (Irish et al., 2004). To avoid the pitfalls of manual gating, we developed PhenoGraph, a robust computational method that partitions high-dimensional single-cell data into subpopulations. Building on these subpopulations we developed additional methods to extract high-dimensional signaling phenotypes and infer differences in functional potential between subpopulations.
Our data-driven approach revealed two new perspectives on the pathobiology of AML. First, we found that pediatric AML draws from a surprisingly limited repertoire of surface phenotypes, indicating some memory of normal myelopoiesis. Despite genetic diversity, patterns of surface antigen expression followed trends in myeloid development, indicating limits in the ability of leukemic cells to phenotypically diverge from normal antigen profiles. Second, we found that the signaling pattern of undifferentiated hematopoietic progenitors defined a primitive signaling phenotype that was recapitulated in a majority of AML samples at varying frequencies. Functionally primitive leukemic cells—defined by signaling—were not linked to a consistent surface phenotype, including the standard HSC/LSC antigen profile (i.e., CD34+/CD38−), demonstrating that surface antigens are decoupled from regulatory networks in leukemia. The frequency of these functionally primitive cells enabled isolation of a gene expression signature that was enriched for stem cell annotations and formed a significant predictor of overall survival in independent AML clinical cohorts.
Taken together, we provide an alternative paradigm for identifying primitive cancer cells that complements the immunophenotypic definitions of cancer stem cells traditionally used in both AML and other systems. Moreover, this analysis framework is robust and broadly applicable to the characterization of subpopulation structure and function from single-cell data in a wide range of systems.
RESULTS
High-dimensional single-cell profiling of pediatric AML by mass cytometry
We used mass cytometry to obtain single-cell proteomic profiles of cryopreserved bone marrow aspirates from pediatric AML patients obtained at diagnosis (n = 16) and from healthy adult donors (n = 5). We performed preliminary analysis to select 16 highly informative surface markers that efficiently captured the intra- and intertumor heterogeneity in our cohort (see Extended Experimental Procedures). We added 14 antibody probes against intracellular phosphorylation, thus allowing simultaneous measurement of surface phenotype and signaling behavior in single cells. Each sample was subjected ex vivo to a battery of short-term molecular perturbations (cytokines and chemical inhibitors; Table S1) to elicit functionally relevant signaling responses (Bendall et al., 2011; Irish et al., 2004). The complete data set contained over 15 million single cells from 21 individuals measured in 31 simultaneous protein epitope dimensions following exposure to one of 18 conditions (Fig. 1A).
Fig. 1. Mass cytometry analysis of signaling responses in pediatric acute myeloid leukemia.

(A) Summary of experimental design. (B) PhenoGraph method for clustering high-dimensional single-cell data. Each node in the neighbor graph represents one of 500 random cells from healthy donor H1 colored by CD34 expression. CD34+ HSPCs form a dense subgraph and are automatically assigned to a single subpopulation. See Figure S1 and Experimental Procedures for more details on the PhenoGraph algorithm. (C) HSPCs identified by PhenoGraph from donor H1. This subpopulation (red histograms) had a CD34+/CD45low phenotype relative to the other cells in the sample (gray histograms). Each PhenoGraph subpopulation contained cells from all perturbations, permitting analysis of 224 signaling responses.
PhenoGraph dissects population structure in high-dimensional single-cell data
Complex tissues such as bone marrow are composed of biologically meaningful subpopulations that are phenotypically coherent despite the intrinsic variability that makes each cell unique. A fundamental challenge is to establish the major phenotypes present, enabling an efficient and meaningful profile of the tissue. While normal immune cells are typically binned into predefined “landmark” cell subsets, this strategy is unsuitable for less predictable or under-studied tissues such as cancer, where new phenotypes have been shown to occur. Thus a data-driven, unsupervised approach is needed that takes single-cell measurements and returns a grouping of cells into distinct subpopulations (i.e., clusters).
Dimensionality reduction techniques such as stochastic neighbor embedding (SNE) (Amir et al., 2013; Maaten and Hinton, 2008) help visualize the data, but do not explicitly identify and partition cells into subpopulations. Moreover, not all subpopulations are visually distinct when rendering high-dimensional data in only two dimensions. We evaluated a number of leading methods for clustering fluorescence cytometry data and found that these did not perform well for mass cytometry data (Aghaeepour et al., 2013). Parametric methods (Pyne et al., 2009) require strong assumptions about the high-dimensional shape of cellular populations (e.g., ellipsoid, convex), which are violated in single-cell data (Amir et al., 2013). Therefore a non-parametric approach is needed, yet these currently use unstable heuristics or suffer from computational inefficiency and do not scale well to higher dimensions. We found that as the number of dimensions increased, available methods routinely failed to correctly identify known subsets, gave inconsistent results and were prohibitively slow (see Extended Experimental Procedures).
To robustly discover subpopulations in high-dimensional single-cell data we developed PhenoGraph. The parameters measured for each cell define a point in high-dimensional space wherein clustering is tantamount to finding dense regions in this space. The difficulty is that density detection in high dimensions is both computationally hard and statistically unstable. Following our previous work (Bendall et al., 2014), we model this high-dimensional space using a nearest-neighbor graph. In this graph, each cell is represented by a node and connected by a set of edges to a neighborhood of its most similar cells. The graph distills the high-dimensional distribution of single cells into a compact, information-rich data structure that captures phenotypic relatedness and overcomes many of the pitfalls of standard geometries.
After the nearest-neighbor graph is constructed, the problem of density detection corresponds to the task of finding sets of highly interconnected nodes (Fig. 1B). To this end we borrow from the social network field, which has developed powerful algorithms to partition large social networks into communities (Girvan and Newman, 2002). In our setting, communities represent an accumulation of phenotypically similar cells that likely reflects biologically meaningful phenotypic stability, thus revealing stable cellular states in the population. Partitioning the graph into these communities produces a dissection of the population into phenotypically coherent subpopulations. Community detection algorithms make no assumption about the size, number, or form of subpopulations (Fortunato, 2010). Importantly, communities need not be convex, symmetric, or ellipsoid—assumptions that are questionable for complex cellular populations. Efficient implementations can partition large graphs in minimal computation time (Blondel et al., 2008).
A key step in the PhenoGraph method is converting the single-cell data to a graph that faithfully represents the phenotypic relationships between cells. Without a carefully constructed graph, large populations can obscure rare ones (which may be outnumbered by orders of magnitude). This problem is further exacerbated by measurement noise that can spuriously link unrelated parts of the graph. We addressed both problems by constructing the graph in two iterations, using the Jaccard similarity coefficient in the second iteration. Thus, the similarity between cells is redefined by the number of shared neighbors following the first iteration (see Experimental Procedures and Fig. S1). The Jaccard metric exploits the local density at each data point, removing spurious edges and strengthening well-supported ones. The co-occurrence of rare cells in the same phenotypic vicinity produces strongly interconnected modules that distinguish these rare cells from noise. Overall, the modular nature of the population is better revealed in the resulting graph.
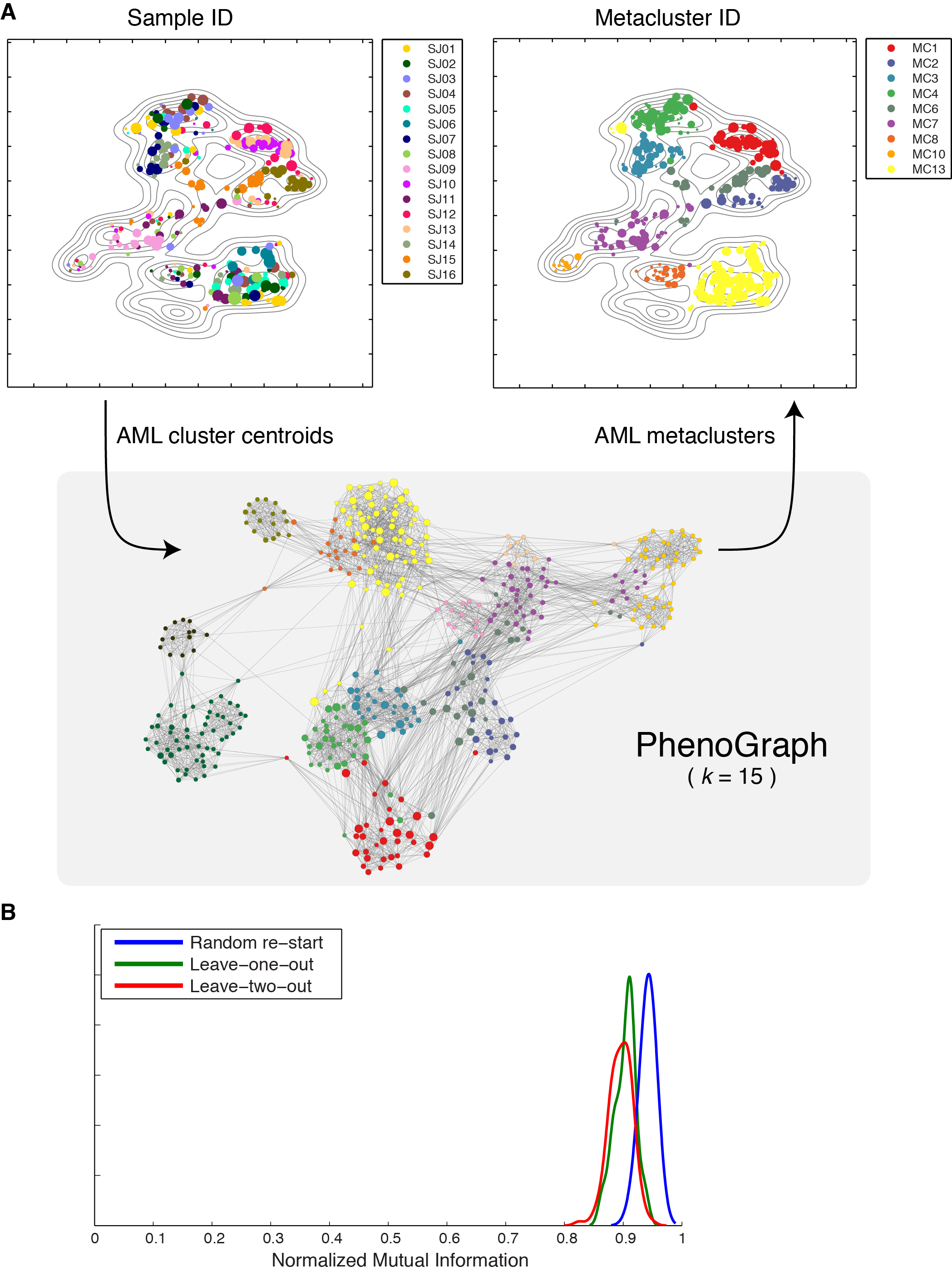
Healthy human bone marrow, which is rich in distinct and well-characterized immunological cell types, presents a benchmark case for phenotypic dissection. We tested PhenoGraph on three different mass cytometry data sets of healthy human bone marrow (Bendall et al., 2011) and PhenoGraph correctly identified labeled immune cell types, displaying superior precision, recall and robustness against leading methods (Aghaeepour et al., 2013) (see Extended Experimental Procedures and Figs. 2, S2A–C & Data S1). PhenoGraph runs efficiently on large data sets with substantially better scaling than other methods (Fig. S2D) and can process millions of cells with modest computational resources. PhenoGraph is able to resolve subpopulations as rare as 1/2000 cells, and is robust to random subsampling and to the choice of the single user-defined parameter (Figs. 2, S2A–C & Data S1).
Fig. 2. PhenoGraph clustering recapitulates manual assignments of healthy immune cells.

(A) viSNE (Amir et al., 2013) display of 30,000 cells from healthy BPMC benchmark data (Bendall et al., 2011). Cells are colored by cell type assignments established by manual gating (left panel) or subpopulations detected by PhenoGraph (right panel). (B) Comparison PhenoGraph to other methods on the benchmark data set, assessed for ability to recover the manual cell type assignments shown in (A, left panel), quantified using the F-measure statistic (Aghaeepour et al., 2013) and normalized mutual information (Fig. S2C). Box plots show the distributions of F-measure computed from 50 random samples of 20,000 cells from the full data set. PhenoGraph was tested with 4 different settings of its single parameter k, small interquartile ranges demonstrate that PhenoGraph accurately identifies the structure of the original population and is robust to random subsampling and its single parameter k. Comparison on additional benchmark datasets is provided in Data S1G–I.
Conformity of phenotypes in the landscape of AML
After validating PhenoGraph on healthy cells, we applied it to our pediatric AML cohort. We ran PhenoGraph on each sample individually, defining subpopulations based on the 16 measured surface markers. This yielded an average of 28 subpopulations per sample (ranging between 17 and 48), totaling 616 subpopulations across the entire cohort. Subpopulation size varied by orders of magnitude, from 7×102 to 2×105 cells (or .06% to 20% of a sample). For each sample, we pooled data from all conditions, enabling characterization of subpopulation-specific signaling patterns. Each resulting subpopulation was a multifaceted data object, containing information about surface phenotypes, as well as the response of each signaling marker to each molecular perturbation (Fig. 1C).
Each leukemia presented a diversity of surface phenotypes defined by distinct combinations of marker expression (Data S2A). We sought an overview of the similarities and differences between detected subpopulations across patients that could reveal larger trends and enable direct comparison of all subpopulations simultaneously. To do so, we began by representing each PhenoGraph subpopulation by its surface marker centroid. We then used t-SNE (Maaten and Hinton, 2008), to reduce the 16-dimensional data to 2 dimensions, following an approach previously taken with cytometry data (Amir et al., 2013). The resulting 2D landscape provided an intuitive and comprehensive overview of the major phenotypes present in the cohort and also demonstrated the extent of intra- and inter-tumoral heterogeneity or similarity (Fig. 3A). Subpopulations from healthy and leukemic samples were mapped simultaneously so the healthy cell types could act as “landmarks” to aid interpretation of the leukemic subpopulations. Normal lymphoid cell types were excluded from the landscape (see Extended Experimental Procedures) to focus on primitive and myeloid phenotypes, “zooming in” on the myeloid lineages relevant to AML.
Fig. 3. Intra- and intertumor heterogeneity is visible across the phenotypic landscape of pediatric AML.

(A) t-SNE landscape of average surface marker expression of non-lymphoid PhenoGraph clusters from the AML cohort. Each cluster is represented by a single point scaled to represent its sample proportion and in the main panel colored by patient identity. Normal bone marrow cell types (H1–H5; blue) provide landmarks for interpreting the phenotypes of the leukemic bone marrow samples (SJ01–SJ16). In additional panels each subpopulation is colored by median expression of indicated surface markers. (B) PhenoGraph applied to cluster centroids consolidated the 616 patient-level subpopulations into 14 cohort-level metaclusters (MCs). Stacked columns indicate the contribution made by each patient to each MC. (C) Average surface marker expression in each MC, summarizing the major phenotypes observed across the cohort. Columns match those represented in B. (D) Intrapatient heterogeneity for each patient is represented graphically by a horizontal bar in which segment lengths represent the proportion of the patient assigned to each MC, colored according to the accompanying legend (bottom right). Hierarchical clustering of these patient descriptions revealed that some patterns of intrapatient heterogeneity were significantly correlated with genetic biomarkers. (CBF, core binding transcription factor translocation: P=0.0014; NPM1: P=0.0083, nucleophosmin mutation; CN, cytogenetically normal: P=0.018).
The AML cohort landscape organized the subpopulations into regions of phenotypic similarity, distinguished by particular marker combinations. Inspecting the structure of this landscape, we found that the vertical axis largely mimicked trends in normal myeloid development with primitive markers expressed toward the top and more mature markers toward the bottom (Fig. 3A & Data S2B–C). Healthy CD34+/CD38mid hematopoietic stem and progenitor cells (HSPCs) provided the most primitive landmark, located at the top of the landscape plot. AML subpopulations in this region displayed surface profiles that resembled the HSPC phenotype. At the bottom of the landscape, the CD11b+ healthy monocytes served as a landmark for differentiated myeloid cells, representing full maturation not observed in the leukemic samples. Between these two poles, other developing myeloid antigens—CD38, CD117, CD123, CD33—peaked and subsided, thus the vertical axis of the landscape resembled normal myeloid development (Fig. Data S2B–C). The adherence of AML phenotypes to this axis suggests that myeloid developmental programs continue to influence the phenotypic diversity of leukemic cells even after malignant transformation. The patterns of intratumor heterogeneity support this view, as most patients contained a mixture of ‘primitive’ and ‘mature’ surface phenotypes (Fig. Data S2D).
Metaclusters highlight inter-patient similarity
Despite the widespread phenotypic diversity observed within patients (Data S2E), the cohort landscape revealed a surprising conformity when comparing AML subpopulations across different patients. Multiple patients occupied each phenotypic region in the landscape and no patient presented a substantially unique phenotype, suggesting that subpopulations could be matched across patients, cohort-wide. To examine these cohort-level phenotypes further, we pursued a metaclustering approach in which subpopulations from each patient were merged by a secondary clustering analysis (Pyne et al., 2009). We represented each AML subpopulation by its centroid and used PhenoGraph to group centroids into metaclusters (MCs; see Experimental Procedures and Fig. S3A), identifying 14 MCs that delineated the major cohort phenotypes (Fig. 3B–C). Each MC had a mixed patient composition, containing subpopulations from at least 2 patients and a median of 11 patients.
To evaluate the robustness of these MCs we performed cross-validation and observed high reproducibility (see Fig. S3B and Extended Experimental Procedures). Subsequently, we used the healthy samples (H1–H5) to interpret the MCs by systematically matching cells from healthy bone marrow with the MC surface marker profiles (see Extended Experimental Procedures). Several MCs corresponded clearly to non-malignant cell types (constituting a small proportion of each leukemic sample), while the remaining MCs represented presumptive blast phenotypes. We determined that 7/14 MCs represented malignant expansions (MC 1–4, 6, 7, 13), based on the relative frequency of healthy cognates (Fig. 3B) and surface marker profiles (Fig. 3C). As expected from the histopathology of AML, the blast phenotypes resembled normal primitive and progenitor phenotypes with a myeloid bias. Each malignant phenotype was detected in multiple patients, but only MC13 was detected in all patients. The CD64+/HLA-DR+ expression profile of MC13 indicates an immature monocytic phenotype that was often drastically more abundant in AML than in healthy samples. Occupancy in MC13 varied substantially between patients (0.8%–77%), consistent with a model of AML as a block in myeloid differentiation with variable severity (Tenen, 2003).
Samples were evaluated quantitatively in terms of their proportional occupancies of the 14 MCs (Fig. 3D). As expected, the 5 healthy samples were similar to each other and distinct from AML. Interestingly, MC occupancies organized the AML samples into subgroups that were significantly correlated with other molecular biomarkers (Fig. 3D). For example, patients with core binding factor translocation [t(8;21) or inv(16)] had large numbers of cells in MC4 and MC13, placing them in a group enriched for this clinical annotation (P = 0.0014, hypergeometric test). Patients with nucleophosmin mutations displayed a different phenotypic distribution—occupancy of MC2, MC7 and MC13—forming another distinct patient group (P = 0.0083). Finally, the 3 patients characterized by large occupancies of MC1 were all cytogenetically normal (P = 0.018). Taken together, each leukemia, although unique, appears to be formed from a limited palette of possible phenotypes. Remarkably, the specific composition and relative proportion of MCs was determined in part by genetic background, demonstrating a genetic influence on the distribution of phenotypes observed in each patient.
Signaling phenotypes define functionally distinct subpopulations
Surface markers have become standard tools for clinical diagnosis and monitoring of blood neoplasia (Craig and Foon, 2008). In normal bone marrow, cell surface markers identify stem and progenitor cell populations with distinct lineage potential and intracellular signaling behaviors (Bendall et al., 2011). However, in AML, no surface marker phenotype has been established that consistently distinguishes the more primitive blasts universally across patients (Eppert et al., 2011; Taussig et al., 2008; 2010).
We hypothesized that intracellular signaling might be a better surrogate of the underlying functional potential and therefore included molecular perturbations known to elicit signaling responses that are functionally relevant to normal and malignant hematopoiesis (Table S1). Intracellular signaling markers were selected to represent pathways known to be functionally and clinically relevant in AML, including JAK/STAT, PI3K/AKT and MAPK. The response of each of 14 signaling proteins to each of 16 perturbations revealed a facet of the underlying network that controls cellular function, resulting in 224 signaling responses per subpopulation. We used these data to build a quantitative signaling phenotype representing the structure and function of the intracellular signaling network in each subpopulation.
To fully harness the single-cell nature of our data, we developed SARA (Statistical Analysis of Perturbation Response; see Experimental Procedures and Fig. 4A). SARA examines the entire single-cell distribution of phosphoprotein intensities to detect meaningful changes between two conditions. SARA incorporates a measure of statistical significance through permutation testing, producing estimates that are sensitive to small responsive subsets yet robust to sampling error and noise. Together, PhenoGraph and SARA distilled high-dimensional data for 15 million cells into a single matrix of subpopulations and their signaling phenotypes (Fig. 4B), revealing a rich variety of signaling potential across subpopulations.
Fig. 4. Analysis of perturbation response generates signaling phenotypes.

(A) A cartoon depicting how SARA uses the single-cell distributions together with permutation testing to score signaling response. (B) SARA, applied to every signaling molecule for every perturbation in every subpopulation, produced ~138,000 responses, which were compiled into 224-dimensional signaling phenotypes for each subpopulation (columns) for each of 616 subpopulations (rows). Rows and columns ordered by agglomerative linkage. (C) Hierarchical clustering of 4 developmentally-relevant signaling responses in the healthy samples (top panel) identified patterns of primitive signaling (PS) and mature signaling (MS) correlated with expression of CD34 and CD45, in the healthy samples. Hierarchical clustering of the same signaling responses in the AML samples (bottom panel) identified a cluster of subpopulations that recapitulated the primitive signaling pattern, but lacked a consistent surface phenotype. Color scales are as in Figures 3A and 4A. (D) Box plots comparing CD34 expression between signaling clusters identified in (C). CD34 expression was significantly associated with primitive signaling only in the healthy samples.
Within the healthy samples, surface and signaling phenotypes were tightly coupled, consistent with previous reports (Bendall et al., 2011; Gibbs et al., 2011). Hierarchical clustering of a curated set of progenitor- and lineage-associated signaling features produced a complete separation of primitive (CD34+) and mature (CD34−) cell types among the healthy samples (Fig. 4C–D; P = 2.0×10−52, Student’s t test). In the leukemic samples, the same procedure produced a similar stratification of signaling phenotypes, including a set of subpopulations that recapitulate the signaling profile of healthy primitive cells. However, this stratification of primitive (PS) and mature (MS) signaling had no association with CD34 expression (P = 0.83, Student’s t test; Fig. 4D). Decoupling of surface and signaling phenotypes in the leukemic samples is consistent with evidence that surface markers are unreliable proxies of cellular function in AML (Eppert et al., 2011; Gibbs et al., 2011; Taussig et al., 2008; 2010). We therefore sought to use signaling phenotypes rather than surface phenotypes as alternative proxies for functional state.
Classification of leukemic maturity by signaling phenotype
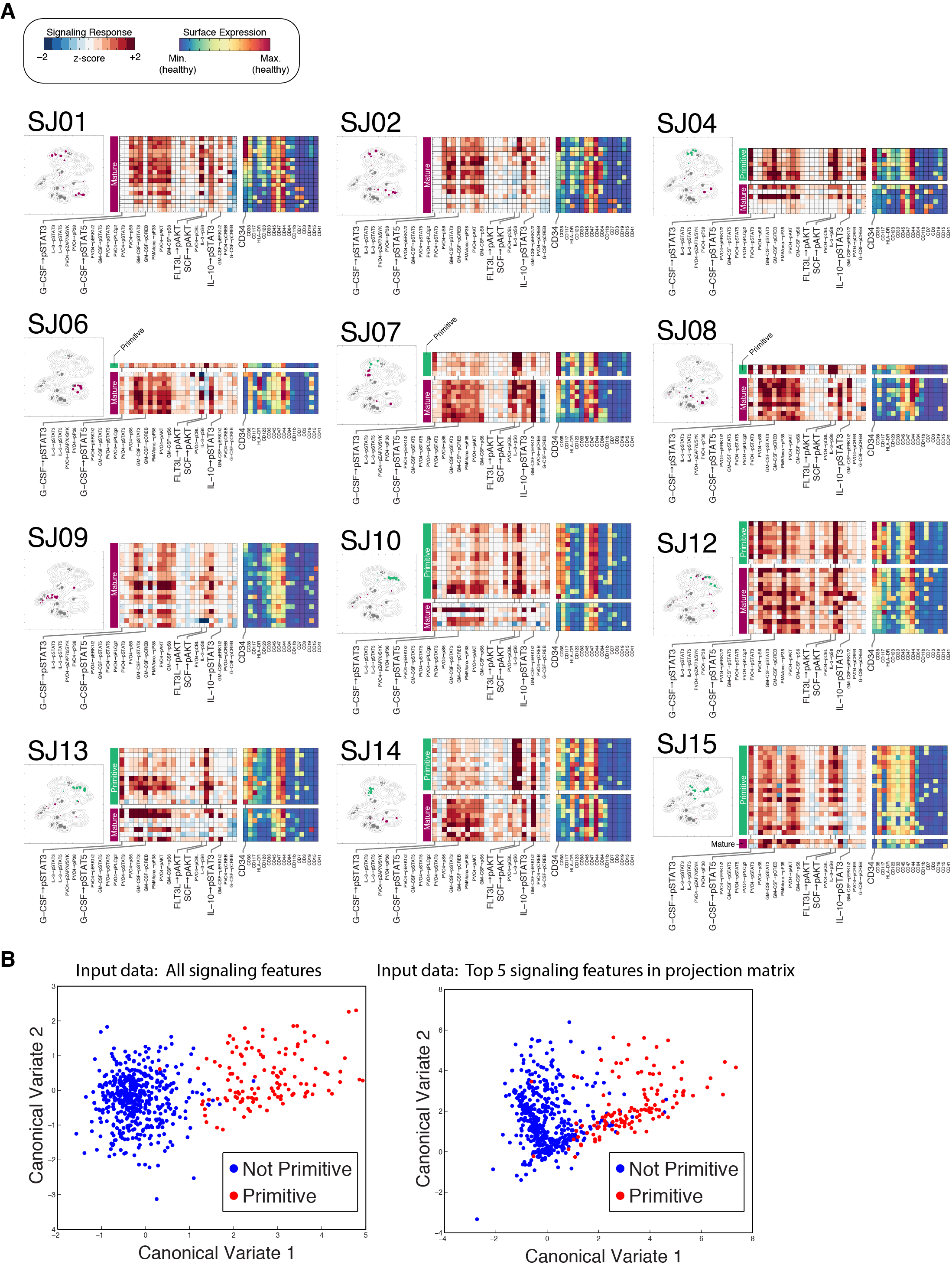
PhenoGraph and SARA yielded two alternative representations for each subpopulation: a 16-dimensional surface phenotype, and a 224-dimensional signaling phenotype (Fig. 5A). We asked if there was a characteristic signaling phenotype of undifferentiated healthy cells that could act as a high-dimensional generalization of the CD34/CD38 surface phenotype, which more faithfully captures the functional aspect of the primitive state.
Fig. 5. Data-driven scoring of leukemic maturity by either surface or signaling phenotype.

(A) Each PhenoGraph subpopulation has two alternative phenotypes: surface and signaling (B) Normal cell types identified in healthy samples display characteristic surface and signaling phenotypes, represented by heat maps. Each row represents the indicated cell type. Surface markers (left) and signaling responses (right) are colored as in (A). Signaling responses are ordered from left to right by decreasing significance of association with cell type (Table S2). (C) The same t-SNE map presented in Fig. 3A, labeled by results of PhenoGraph classification. Colors depict whether a subpopulation was assigned to either, both, or neither primitive class as determined IFPC or SDPC; (see Fig. S4A–B). (D) Frequencies of primitive cells: %IFPC or %SDPC for each patient sample.
Harnessing the tight coupling between surface and signaling in the healthy system, we grounded our analysis in a characterization of healthy subpopulations. PhenoGraph metaclustering of the 5 normal marrow samples identified 20 healthy cell types (Fig. 5B and Data S3A). Using ANOVA, we examined their signaling profiles for consistent responses that were associated with particular cell types and found that a large number of signaling responses had significant associations with cell type (Table S2). Many of these were induction responses specific to undifferentiated cells, including G-CSF→pSTAT3 (Q = 6.4 × 10−42) and SCF→pAKT (Q = 1.0 × 10−9), as previously reported (Gibbs et al., 2011).
We then asked whether cell types could be distinguished entirely by their signaling phenotypes, rendering surface phenotypes dispensable for characterizing the subpopulations. To test this, we developed a framework for classifying subpopulations based on either their surface or signaling phenotypes. We derived an extension of PhenoGraph that uses the same graph-based model but assigns observations to classes according to user-defined training examples (‘PhenoGraph classification’; see Experimental Procedures and Fig. S4A). First, we verified that PhenoGraph was capable of recovering “held out” healthy cell type labels using a graph derived from surface phenotypes. Performance was evaluated using the cross-validated correct classification rate (CCR) and indeed, PhenoGraph correctly recovered 99.42% of the cell type labels in this test. Next, we constructed a graph based only on similarity among signaling phenotypes, withholding all surface phenotype information. Using this graph, PhenoGraph’s ability to recover the surface-defined labels was modestly diminished (CCR = 94%), due to errors distinguishing mature cell types for which characteristic signaling phenotypes had not been measured. Focusing on the task of distinguishing the most primitive cells (i.e., HSPCs) from the mature cell types, we found that signaling phenotypes performed equivalently to surface phenotypes (CCR = 99.85%; see Experimental Procedures).
Considering that signaling phenotypes were sufficient to distinguish healthy primitive cells, we hypothesized that the functional state of AML subpopulations could be inferred by direct examination of their signaling phenotypes. With the healthy subpopulations as training examples, we used PhenoGraph to classify the AML subpopulations, producing an estimate of functional state for each subpopulation (e.g., HSPC-like or monocyte-like). Because there were two alternative phenotypes for each subpopulation—surface and signaling—we performed two separate classifications (Fig. S4B). The result was a data-driven assessment of each AML subpopulation, indicating which healthy cell type it resembled in its surface marker expression on one hand and in its high-dimensional signaling phenotype on the other.
Inferred functional maturity diverges from surface phenotype in AML
The classifiers identified primitive subpopulations within each patient sample, reflecting the heterogeneous nature of the samples. At the cohort level, each classifier labeled ~25% of subpopulations as primitive, but only 16% were identified as primitive by both classifiers simultaneously (Fig. 5C). In many cases (32/99), subpopulations with primitive surface marker phenotypes exhibited signaling that resembled mature cells. Conversely, many subpopulations displayed primitive signaling in the absence of primitive surface marker expression (51/118).
We denote cells labeled primitive by the surface phenotype classifier as Surface-Defined Primitive Cells (SDPCs) and cells labeled primitive by the signaling classifier as Inferred Functionally Primitive Cell (IFPC). For each patient, the sample proportion assigned to each of these labels produced two alternative measures of maturity (%SDPC or %IFPC; Fig. 5D and Table S2). This is similar to summarizing the degree of maturation by the enumeration of CD34+/CD45low blasts, a practice often used in the clinical diagnosis and classification of leukemias (Craig and Foon, 2008). Indeed, we found that %SDPC was highly correlated with this standard manual gating procedure (Pearson’s r = 0.96, P = 4.4 × 10−9; Fig. S4C & Data S3B). Conversely, %SDPC was only weakly correlated with %IFPC (Pearson’s r = 0.5; P = 0.05), demonstrating that these two metrics are not redundant in AML. Instead, examination of signaling phenotypes in AML often revealed a different degree of maturation than was indicated by the surface phenotype. We noted that the degree of discordance between IFPC and SDPC assignments was not constant across patients, indicating that the tendency of IFPCs to express canonical LSC markers was itself a variable patient feature. For example, the IFPCs in patient SJ05 were well represented by the CD34+/CD38mid phenotype (Fig. 6, left column). In other cases, IFPCs were found exclusively in the CD34− fraction, even when CD34+ blasts were abundant (e.g., SJ16).
Fig. 6. Leukemic subpopulations with primitive signaling exhibit diverse surface phenotypes.

Detailed surface and signaling phenotypes of IFPC subpopulations in 4 representative samples. Each row represents a particular patient using a number of visuals. Biaxial dot plots (left) show the CD34/CD38 phenotype of IFPCs (red) in each sample. IFPCs displayed the canonical primitive CD34+/CD38mid phenotype in only a subset of samples. The IFPCs displayed using the t-SNE landscape of Fig. 3A (center; IFPCs in green, non-IFPCs in maroon, healthy cells in gray). Heat maps (right) display the signaling and surface phenotypes of all non-lymphoid subpopulations of each sample, stratified by IFPC classification (indicated by green and maroon bars). Signaling responses are ordered as in Fig. 5B. Signaling responses marked in bold with vertical lines were especially distinctive of IFPCs (see Main Text and Extended Experimental Procedures). See Figure S5A for all patients not shown here.
Differences in signaling patterns between primitive and mature leukemic subpopulations reveal the responses most important for these classifications (Fig. 6; see Fig. S5A for all patients). We used canonical variates analysis (see Extended Experimental Procedures for details) to quantify this importance, finding that the majority of discriminative power could be attributed to 5 responses: G-CSF→pSTAT3, SCF→pAkt, G-CSF→pSTAT5, Flt3-L→pAkt, and IL-10→pSTAT3 (Fig. S5B and Table S2). Primitive subpopulations displayed strong activation in the first four of these responses, which have all been previously implicated in the biology of HSPCs (Gibbs et al., 2011) and in the pathobiology of AML (Irish et al., 2004). Additionally, attenuation of the IL-10→pSTAT3 response—a response exhibited by mature immune cells—was also a distinctive feature of IFPCs. Other signaling responses were strongly associated with primitive subpopulations despite being less powerful for classification (Table S2).
Evaluating the ability of surface markers to identify IFPCs, it was clear that no surface phenotype could be applied universally across patients (Fig. 6 and Fig. S5A). CD34 was often an important label for IFPCs, but in a subset of cases. For example, CD34 marked both primitive and mature subpopulations in patient SJ03, where HLA-DR was a more specific marker of IFPCs (P = 0.0007 vs. P = 0.003, Student’s t test). In SJ05, where CD34 expression was tightly associated with IFPCs (P = 7.4 × 10−8), the multiparameter surface measurements revealed that CD123 was also an important marker (P = 4.4 × 10−6), whereas CD123 did not identify IFPCs in SJ03. Patient SJ11 lacked CD34 expression almost entirely, as expected for this nucleophosmin-mutated case (Taussig et al., 2010). In this patient, IFPCs were distinctly labeled by elevated expression of CD47 (P = 7.1 × 10−6) and CD123 (P = 3.4 × 10−5). Surprisingly, we found that CD34 expression can be strongly anti-correlated with primitive signaling, as in patient SJ16, where CD34 expression was higher in mature cells (P = 0.0027) and IFPCs were marked instead by elevated expression of CD117 (P = 0.0026). Complete median surface marker profiles for IFPC and non-IFPC subpopulations are displayed in heat maps for each patient in Figs. 6 and S5A.
Primitive signaling phenotype identifies clinically prognostic gene expression signature
Ultimately, the importance of intratumor heterogeneity depends on whether functionally distinct subpopulations influence clinical outcomes, especially patient survival (Pearce et al., 2006). While our cohort was too small for survival analysis, genome-wide expression arrays for 15 of our 16 patients were available from a previous study (Radtke et al., 2009), providing a link to larger cohorts for which gene expression and survival data were available. Because our samples displayed a wide range of IFPC frequencies (Fig. 5D), we reasoned that this variance could be exploited to identify genes whose expression covaried with these frequencies by in silico expression deconvolution (Lu et al., 2003). As IFPC frequency varies across samples, genes expressed specifically by these cells should be detectably more or less abundant in the bulk gene expression measurements, thereby providing an estimate of %IFPC in independent samples from the level of this gene signature, measured in bulk.
We developed a deconvolution method based on linear regression and cross-validation and used both %IFPC and %SDPC to produce two gene expression signatures, containing 42 and 49 genes, respectively (see Experimental Procedures, Fig. 7A and Table S3). To characterize these signatures, we queried the Molecular Signatures Database (Subramanian et al., 2005) for significant annotations overlapping with each. The SDPC signature—which contained CD34 among its top-ranked genes—was highly enriched for gene sets associated specifically with CD34+ AML (Table S3). Alternatively, the most significant annotation for the IFPC signature was a set of genes upregulated in CD133+ hematopoietic stem cells (Jaatinen et al., 2006) (Q = 5.5 × 10−8; Table S3). CD133 marks healthy stem cells that are possibly more primitive than CD34+ HSCs (Gallacher et al., 2000) and has been linked to cancer stem cells in multiple cancer types (Collins et al., 2005; O’Brien et al., 2007). The mean expression of each signature was highly correlated with its corresponding subpopulation frequency (Fig. S6A), indicating that the signature mean was an appropriate proxy for these frequencies in independent cohorts.
Fig. 7. Frequency of IFPCs identifies a gene expression signature that predicts clinical outcome.

(A) IFPC gene signature identified by deconvolution of bulk expression data using IFPC frequency. The heat map displays expression of each gene in the bulk measurements. Rows are alphabetically ordered; columns are ordered by the mean expression of the genes in the signature. (B) The mean of the IFPC signature forms a clinically significant prognostic indicator of overall survival in 2 independent cohorts of adult AML (Metzeler et al., 2008). Patients were assigned to groups for Kaplan-Meier analysis based on whether their IFPC expression score was below or above the cohort median. P values obtained from log-rank test.
We tested our signatures in two independent cohorts of adult AML for which both gene expression and survival data were available (Metzeler et al., 2008). While the SDPC signature was associated with survival in one cohort, this was not replicated in the other (Fig. S6B). Alternatively, the IFPC signature was predictive of poor survival in both cohorts (Fig. 7B). Combining the data into a single, large cohort (n = 242), the IFPC signature was highly predictive of poor survival (P = 4.8 × 10−6, Hazard Ratio [HR] = 3.4), while the SDPC signature formed a less significant predictor (P = 0.005, HR = 1.6). To test these signatures against each other, we placed them together in a bivariate Cox regression model. In this setting, the IFPC signature retained its predictive power (P = 8.2 × 10−5, HR = 3.0), while the SDPC signature became completely uninformative for survival (P = 0.29, HR = 1.2).
We examined the relationship between the IFPC signature and three signatures reported by (Eppert et al., 2011), which were also developed to capture primitive gene expression programs in AML. For each Eppert signature, we were able to reproduce the significant correlation with survival in the data from (Metzeler et al., 2008). To assess the prognostic value of the IFPC signature when these other signatures were known, we tested three bivariate Cox regression models in which each of the Eppert signatures was used as a predictor alongside the IFPC signature (see Extended Experimental Procedures). The IFPC signature proved to be a stronger predictor of survival than any of the Eppert signatures (Table S3). In each model, the IFPC signature retained significance (P < 0.005), while each Eppert signature became statistically insignificant (P > 0.07). In a multivariate Cox regression model containing all signatures (IFPC, SDPC and the Eppert signatures), only the IFPC signature retained significance (P = 0.012, HR = 2.4; Table S3).
DISCUSSION
Tissues are complex populations of cells residing in phenotypically and functionally diverse states. A key challenge is to dissect the high-dimensional structure of these complex populations into components that can be studied individually and collectively. In AML, where the relationship between phenotypic and functional heterogeneity has been elusive, we used mass cytometry to profile both surface and signaling features simultaneously in millions of leukemic cells.
PhenoGraph revealed a phenotypic landscape of a pediatric AML cohort, providing a comprehensive overview of the major phenotypes and an explicit characterization of intra- and intertumor heterogeneity. The landscape resembled normal myeloid development, but with aberrations resulting from malignant accumulation of cells and neoplastic divergence from normal phenotypes. However, this AML landscape was surprisingly restricted to a limited repertoire of 14 MCs, each defined by distinct surface marker patterns. Importantly, these MCs were shared among a wide variety of AML genetic subtypes, yet genetics had a detectable influence on the phenotypic composition of each patient. Together these observations suggest the persistence of developmental mechanisms that control the available repertoire of phenotypes even in the context of genetic dysregulation associated with cancer.
We used mass cytometry in conjunction with molecular interrogation to construct signaling phenotypes that reflect differences in functional potential between subpopulations. Surface and signaling phenotypes displayed tight coregulation in healthy samples, whereas this coregulation was broken in AML. This substantial decoupling of surface and signaling phenotypes in the leukemic cells renders the surface markers typically used in diagnostics unreliable proxies of cellular state and function in AML.
Our demonstration that surface markers are unreliable reporters of signaling state in AML sheds light on the controversies surrounding the LSC model, which rely on manual gating and surface marker expression to define subpopulations. To avoid the assumption that surface markers indicate the functional state of leukemic cells, we used healthy HSPCs to define a primitive signaling phenotype, reflecting the functional state of undifferentiated hematopoietic cells. We found that the primitive signaling phenotype was present in most AML samples and could be used to identify intratumor functional heterogeneity. Leukemic cells displaying primitive signaling (Inferred Functionally Primitive Cells [IFPCs]), were thereby identified using datadriven techniques and without reference to surface phenotypes.
The IFPC phenotype was found to occur in most AML samples at varying frequencies and with variable surface phenotypes, often with low or absent CD34 expression. While no universal surface phenotype captured IFPCs across patients, within each patient IFPCs displayed homogeneous expression in certain markers—markers whose importance was neither universal nor unique. Our results suggest that a subset of leukemic cells maintains a conserved, progenitor-like signaling program that phenocopies the regulatory state of normal HSPCs, regardless of surface marker expression and underlying genetic mutations.
Deconvolution analysis of microarray data identified a gene expression signature associated with the IFPC phenotype that can serve as a proxy for the frequency of this phenotype in a given sample. This gene expression signature was enriched for annotations related to primitive hematopoietic cells and included genes—such as PROM1, SOCS2, and CD96—that have been previously associated with healthy and/or leukemic stem cells (Toren et al., 2005). Importantly, this gene expression signature predicted survival in multiple independent AML patient cohorts, suggesting that this signaling-based definition describes a clinically relevant cellular phenotype.
It was previously demonstrated (Eppert et al., 2011) that functional characterization by physical sorting and xenotransplantation could be used to identify genes correlated with patient survival. Our analysis is conceptually related, but instead of differential expression between sorted cells, we used in silico deconvolution to identify genes, based on the measured cellular frequencies of the IFPC phenotype. Ultimately, both approaches seek to identify primitive cells by means that emphasize functional over surface phenotypes, and to test whether the predominance of primitive cells—approximated by expression of a gene signature—is associated with poor survival.
Our findings were enabled by computational dissection of intratumor heterogeneity. PhenoGraph creates a graph-based model of cellular phenotypes, similar to that used previously to identify developmental trajectories (Bendall et al., 2014) and in this case defining phenotypes as communities of densely interconnected nodes. PhenoGraph is general and highly scalable both in terms of dimensionality and sample size, making it suitable in a wide range of settings for which single-cell population structure is of interest, including other cancers or healthy tissues, and for use with other emerging single-cell technologies such as single-cell RNAseq. Many such cases are presented by the tumor microenvironment, including drug-resistant tumor subpopulations, infiltrating immune cells, and reactive stromal components. These methods are also applicable to healthy tissues, within which a large diversity of cell types remains uncharted.
Our signaling-based definition of primitive cells warrants further investigation as it may indicate pathways that influence the maturation of leukemic cells and could be leveraged therapeutically to block survival or direct differentiation. More broadly, this molecular interrogation approach could be used to characterize primitive cells in any cancer where a cognate healthy primitive cell type is available to serve as a reference point. This study provides a framework for interrogating and discovering other features of cell biology that define network response states and their associated mechanistic or clinical outcomes.
EXPERIMENTAL PROCEDURES
Patient samples
Sixteen (16) cryopreserved diagnostic bone marrow mononuclear cells (BMMC) of pediatric AML patients were supplied by St. Jude Children’s Hospital (Memphis, TN) (Table S1). For healthy adult controls, cryopreserved healthy BMMCs were purchased from AllCells, Inc. (Emeryville, CA). All human samples were obtained with informed consent in compliance with IRB-approved protocols.
Mass Cytometry Analysis
Mass cytometry analysis including data pre-processing is as previously described (Bendall et al., 2011; Finck et al., 2013; Zunder et al., 2015). Surface marker expression was normalized based on the maximum intensity observed in healthy samples, determined as the 99.5th percentile of the ~3M healthy bone marrow cells. Data from all samples were divided by these maximum values, yielding expression values that can be interpreted as x-fold of the maximum observed in healthy. Mass cytometry data are publicly available at http://cytobank.org/nolanlab/reports. See Extended Experimental Procedures for full details.
Microarray data and normalization
Matched Affymetrix U133A gene expression arrays for the 16 pediatric AML patients (Radtke et al., 2009) were downloaded from the Gene Expression Omnibus (GSE14471). Gene expression and survival data for 242 cytogenetically normal adult AML patients from two independent cohorts (Metzeler et al., 2008) were downloaded from the Gene Expression Omnibus (GSE12417). All microarray data were processed and normalized as described previously (Akavia et al., 2010).
The PhenoGraph algorithm
PhenoGraph takes as input a matrix of N single-cell measurements and partitions them into subpopulations by clustering a graph that represents their phenotypic similarity. PhenoGraph builds this graph in two steps. First, it finds the k nearest neighbors for each cell (using Euclidean distance), resulting in N sets of k-neighborhoods. Second, it operates on these sets to build a weighted graph such that the weight between nodes scales with the number of neighbors they share. The Louvain community detection method (Blondel et al., 2008) is then used to find a partition of the graph that maximizes modularity. See Extended Experimental Procedures for full details on the method and an assessment of its accuracy, efficiency, and robustness compared to other methods. Source code for PhenoGraph is available online for MATLAB and Python (www.c2b2.columbia.edu/danapeerlab/html/software.html).
PhenoGraph classification
Given a dataset of N d-dimensional vectors, M distinct classes and a vector providing the class labels for the first L samples, the PhenoGraph classifier assigns labels to the remaining N–L unlabeled vectors. First, a graph is constructed as described above. The classification problem then corresponds to the probability that a random walk originating at unlabeled node x will first reach a labeled node from each of the M classes. This defines an M-dimensional probability distribution for each node x that records its affinity for each class. See Extended Experimental Procedures for full details on this method, as well as an evaluation of its performance on benchmark data.
Applying PhenoGraph and SARA to AML cohort
We ran PhenoGraph on each sample individually, defining subpopulations based on expression of the 16 surface markers. For each sample, all ex vivo conditions were pooled as we previously demonstrated that surface marker distributions are not altered by these short-term perturbations (Bendall et al., 2011). PhenoGraph was run on the normalized surface phenotype matrices for each sample, with the parameter k=50.
Subpopulation signaling phenotypes were computed for each cluster using SARA, followed by z-score standardization. See Extended Experimental Procedures for full details.
Defining AML metaclusters
Each AML subpopulation was represented by its centroid, resulting in a 425 × 16 matrix. PhenoGraph was run on 425 subpopulations centroids with the parameter k=15, resulting in 14 metaclusters (MCs) delineating the major cohort phenotypes. These MCs are a robust feature of the data and remained consistent when the metaclustering was performed on subsets of patients (see Extended Experimental Protocols and Fig. S3B). To characterize these MCs, we systematically matched cells from healthy bone marrow (H1–H5) with the MC surface marker profiles using linear discriminant analysis. See Extended Experimental Procedures for full details.
PhenoGraph classification of leukemic subpopulations
We used the PhenoGraph classifier to classify leukemic subpopulations based on training examples provided by the healthy subpopulations. For each, k-neighbor graphs (k = 15) were constructed over 616 subpopulations (healthy and leukemic) using similarities derived either from surface or signaling phenotypes. Specifically, we used a weighted Euclidean distance in which each phenotypic feature was weighted according to its statistical association with known cell types in the healthy samples. Each AML subpopulation was classified based on its phenotypic proximity to the healthy training examples. Classification was performed using surface and signaling classifiers separately, resulting in two alternative classifications per AML subpopulation (Figs. 6 and S4B). See Extended Experimental Procedures for full details.
Gene expression signatures and survival analysis
For each score, %SDPC or %IFPC, a set of associated genes was defined based on correlation with the expression patterns across patients, using linear regression. This in silico gene expression deconvolution (Lu et al., 2003), assumes that changes in bulk expression of certain genes will track with changes in subpopulation size. We used leave-two-out cross-validation across 15 patients to select genes that placed in the top one percentile and had a standard deviation across subsets < 5%.
We used gene expression and survival data for 242 cytogenetically normal adult AML patients from two independent cohorts (Metzeler et al., 2008). For each patient, the frequency of a cell type (%IFPC or %SDPC) was estimated as the mean expression intensity of the associated gene signature. For Kaplan-Meier analysis, patients were stratified into two groups based on the median expression value of the signature of interest. See Extended Experimental Procedures.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
We thank G. Behbehani, W. Fantl, B.J. Chen and L. Zelnik for helpful discussion. E.F.S. and S.C.B. are supported by DRCF Fellowships (DRG 2190-14 &DRG-2017-09) and NIH 1R00-GM104148 to S.C.B. Grants NIH (DP1-HD084071, DP2-OD002414, R01-CA164729 U54-CA121852), Stand Up To Cancer Phillip A. Sharp Award SU2C-AACR-PS04 and Packard Fellowship for Science and Engineering supported D.P. Grants NIH (1R01CA130826, 5U54CA143907, HHSN272200700038C, N01-HV-00242, P01 CA034233, U19 AI057229 and U54CA149145), CIRM (DR1-01477 and RB2-01592), EC (HEALTH.2010.1.2-1), US FDA (HHSF223201210194C),US DOD (W81XWH-12-1-0591), the Entertainment Industry Foundation, and the Rachford and Carlota Harris Endowed Professorship supported G.P.N.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author contributions
J.H.L., E.F.S., S.C.B., D.P., G.P.N. conceived the study. J.H.L., E.F.S., S.C.B., E.D.A., D.P., G.P.N designed experiments. J.H.L., E.D.A., D.P. designed and developed PhenoGraph. A.L.G., I.R., J.R.D. provided clinical samples. E.F.S., S.C.B., K.L.D., H.F., A.J. performed all data acquisition experiments. E.Z., R.F. provided barcoding methods. J.H.L., D.P. developed new analysis algorithms. J.H.L., E.D.A., M.T., O.L. implemented analysis tools. J.H.L., E.F.S., D.P. analyzed and interpreted the data. J.H.L., E.F.S., S.C.B., D.P., G.P.N. wrote the manuscript.
REFERENCES
- Aghaeepour N, Finak G, Hoos H, Mosmann TR, Brinkman R, Gottardo R, Scheuermann RH FlowCAP Consortium, DREAM Consortium. Critical assessment of automated flow cytometry data analysis techniques. Nat Methods. 2013;10:228–238. doi: 10.1038/nmeth.2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akavia U-D, Litvin O, Kim J, Sanchez-Garcia F, Kotliar D, Causton HC, Pochanard P, Mozes E, Garraway LA, Pe'er D. An Integrated Approach to Uncover Drivers of Cancer. Cell. 2010;143:1005–1017. doi: 10.1016/j.cell.2010.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amir E-AD, Davis KL, Tadmor MD, Simonds EF, Levine JH, Bendall SC, Shenfeld DK, Krishnaswamy S, Nolan GP, Pe'er D. viSNE enables visualization of high dimensional single-cell data and reveals phenotypic heterogeneity of leukemia. Nat Biotechnol. 2013 doi: 10.1038/nbt.2594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendall SC, Davis KL, Amir E-AD, Tadmor MD, Simonds EF, Chen TJ, Shenfeld DK, Nolan GP, Pe'er D. Single-Cell Trajectory Detection Uncovers Progression and Regulatory Coordination in Human B Cell Development. Cell. 2014 doi: 10.1016/j.cell.2014.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendall SC, Simonds EF, Qiu P, Amir E-AD, Krutzik PO, Finck R, Bruggner RV, Melamed R, Trejo A, Ornatsky OI, et al. Single-cell mass cytometry of differential immune and drug responses across a human hematopoietic continuum. Science (New York, NY) 2011;332:687–696. doi: 10.1126/science.1198704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blondel VD, Guillaume J-L, Lambiotte R, Lefebvre E. Fast unfolding of communities in large networks. Journal of Statistical Mechanics: Theory and Experiment 2008. 2008 [Google Scholar]
- Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. 1997;3:730. doi: 10.1038/nm0797-730. [DOI] [PubMed] [Google Scholar]
- Collins AT, Berry PA, Hyde C, Stower MJ, Maitland NJ. Prospective Identification of Tumorigenic Prostate Cancer Stem Cells. Cancer Res. 2005;65:10946–10951. doi: 10.1158/0008-5472.CAN-05-2018. [DOI] [PubMed] [Google Scholar]
- Craig FE, Foon KA. Flow cytometric immunophenotyping for hematologic neoplasms. Blood. 2008;111:3941–3967. doi: 10.1182/blood-2007-11-120535. [DOI] [PubMed] [Google Scholar]
- Eppert K, Takenaka K, Lechman ER, Waldron L, Nilsson B, van Galen P, Metzeler KH, Poeppl A, Ling V, Beyene J, et al. Stem cell gene expression programs influence clinical outcome in human leukemia. Nat Med. 2011;17:1086–1093. doi: 10.1038/nm.2415. [DOI] [PubMed] [Google Scholar]
- Finck R, Simonds EF, Jager A, Krishnaswamy S, Sachs K, Fantl W, Pe'er D, Nolan GP, Bendall SC. Normalization of mass cytometry data with bead standards. Cytometry A. 2013;83:483–494. doi: 10.1002/cyto.a.22271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortunato S. Community detection in graphs. Physics Reports. 2010;486:75–174. [Google Scholar]
- Gallacher L, Murdoch B, Wu DM, Karanu FN, Keeney M, Bhatia M. Isolation and characterization of human CD34−Lin− and CD34+Lin− hematopoietic stem cells using cell surface markers AC133 and CD7. Blood. 2000;95:2813–2820. [PubMed] [Google Scholar]
- Gibbs KD, Gilbert PM, Sachs K, Zhao F, Blau HM, Weissman IL, Nolan GP, Majeti R. Single-cell phospho-specific flow cytometric analysis demonstrates biochemical and functional heterogeneity in human hematopoietic stem and progenitor compartments. Blood. 2011;117:4226–4233. doi: 10.1182/blood-2010-07-298232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girvan M, Newman MEJ. Community structure in social and biological networks. Proc Natl Acad Sci USA. 2002;99:7821–7826. doi: 10.1073/pnas.122653799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irish JM, Hovland R, Krutzik PO, Perez OD, Bruserud Ø, Gjertsen BT, Nolan GP. Single Cell Profiling of Potentiated Phospho-Protein Networks in Cancer Cells. Cell. 2004;118:217–228. doi: 10.1016/j.cell.2004.06.028. [DOI] [PubMed] [Google Scholar]
- Jaatinen T, Hemmoranta H, Hautaniemi S, Niemi J, Nicorici D, Laine J, Yli-Harja O, Partanen J. Global Gene Expression Profile of Human Cord Blood–Derived CD133+ Cells. Stem Cells. 2006;24:631–641. doi: 10.1634/stemcells.2005-0185. [DOI] [PubMed] [Google Scholar]
- Lu P, Nakorchevskiy A, Marcotte EM. Expression deconvolution: A reinterpretation of DNA microarray data reveals dynamic changes in cell populations. Proc Natl Acad Sci USA. 2003;100:10370–10375. doi: 10.1073/pnas.1832361100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maaten LVD, Hinton G. Visualizing Data using t-SNE. Journal of Machine Learning Research. 2008;9:2579–2605. [Google Scholar]
- Marusyk A, Almendro V, Polyak K. Intra-tumour heterogeneity: a looking glass for cancer? Nat Rev Cancer. 2012;12:323–334. doi: 10.1038/nrc3261. [DOI] [PubMed] [Google Scholar]
- Metzeler KH, Hummel M, Bloomfield CD, Spiekermann K, Braess J, Sauerland M-C, Heinecke A, Radmacher M, Marcucci G, Whitman SP, et al. An 86-probe-set gene-expression signature predicts survival in cytogenetically normal acute myeloid leukemia. Blood. 2008;112:4193–4201. doi: 10.1182/blood-2008-02-134411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien CA, Pollett A, Gallinger S, Dick JE. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature. 2007;445:106–110. doi: 10.1038/nature05372. [DOI] [PubMed] [Google Scholar]
- Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, Cahill DP, Nahed BV, Curry WT, Martuza RL, et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science (New York, NY) 2014;344:1396–1401. doi: 10.1126/science.1254257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearce DJ, Taussig D, Zibara K, Smith L-L, Ridler CM, Preudhomme C, Young BD, Rohatiner AZ, Lister TA, Bonnet D. AML engraftment in the NOD/SCID assay reflects the outcome of AML: implications for our understanding of the heterogeneity of AML. Blood. 2006;107:1166–1173. doi: 10.1182/blood-2005-06-2325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyne S, Hu X, Wang K, Rossin E, Lin T-I, Maier LM, Baecher-Allan C, McLachlan GJ, Tamayo P, Hafler DA, et al. Automated high-dimensional flow cytometric data analysis. Proceedings of the National Academy of Sciences. 2009;106:8519–8524. doi: 10.1073/pnas.0903028106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radtke I, Mullighan CG, Ishii M, Su X, Cheng J, Ma J, Ganti R, Cai Z, Goorha S, Pounds SB, et al. Genomic analysis reveals few genetic alterations in pediatric acute myeloid leukemia. Proceedings of the National Academy of Sciences. 2009;106:12944–12949. doi: 10.1073/pnas.0903142106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taussig DC, Miraki-Moud F, Anjos-Afonso F, Pearce DJ, Allen K, Ridler C, Lillington D, Oakervee H, Cavenagh J, Agrawal SG, et al. Anti-CD38 antibody-mediated clearance of human repopulating cells masks the heterogeneity of leukemia-initiating cells. Blood. 2008;112:568–575. doi: 10.1182/blood-2007-10-118331. [DOI] [PubMed] [Google Scholar]
- Taussig D, Vargaftig J, Miraki-Moud F, Griessinger E, Sharrock K, Luke T, Lillington D, Oakervee H, Cavenagh J, Agrawal S, et al. Leukemia-initiating cells from some acute myeloid leukemia patients with mutated nucleophosmin reside in the CD34- fraction. Blood. 2010;115:1976. doi: 10.1182/blood-2009-02-206565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tenen DG. Disruption of differentiation in human cancer: AML shows the way. Nat Rev Cancer. 2003;3:89–101. doi: 10.1038/nrc989. [DOI] [PubMed] [Google Scholar]
- Toren A, Bielorai B, Jacob-Hirsch J, Fisher T, Kreiser D, Moran O, Zeligson S, Givol D, Yitzhaky A, Itskovitz-Eldor J, et al. CD133-positive hematopoietic stem cell “stemness” genes contain many genes mutated or abnormally expressed in leukemia. Stem Cells. 2005;23:1142–1153. doi: 10.1634/stemcells.2004-0317. [DOI] [PubMed] [Google Scholar]
- Zunder ER, Finck R, Behbehani GK, El-ad DA. Palladium-based mass tag cell barcoding with a doublet-filtering scheme and single-cell deconvolution algorithm. Nature. 2015 doi: 10.1038/nprot.2015.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.