

Abstract
Piperlongumine (PPLGM), an alkaloid isolated from the long pepper (Piper longum L.), can selectively trigger cancer cell death in colorectal cancer cells. The present study investigated whether the c-Jun NH2-terminal kinase (JNK) signaling pathway is involved in PPLGM-induced apoptosis in the human colorectal cancer HCT116 cell line. The results demonstrated that PPLGM reduced the cell viability and induced cell apoptosis in a time- and concentration-dependent manner, without a significant effect on cell cycle distribution. Meanwhile, treatment with 10 µM PPLGM resulted in JNK activation within 1 h, and a marked and sustained increase in c-Jun phosphorylation in the HCT116 cells. In addition, SP600125, a general inhibitor of JNK, inhibited PPLGM-induced apoptosis in the HCT116 cells by inhibiting PPLGM-induced c-Jun phosphorylation. Altogether, it can be concluded that the JNK signaling pathway, at least in part, is involved in PPLGM-mediated apoptosis in HCT116 cells.
Keywords: piperlongumine, colorectal cancer, c-Jun NH2-terminal kinases, apoptosis, phosphorylation, SP600125
Introduction
Colorectal cancer (CRC) is one of the leading causes of cancer-related mortality worldwide (1). Nearly half of patients newly diagnosed with colorectal cancer will develop metastases and require systemic chemotherapy (2). As current chemotherapeutic drugs for colorectal cancer have clinically significant toxicities, it is a challenging endeavor to develop novel chemotherapeutic agents with good efficacy and selectivity (3).
Piperlongumine (PPLGM) is a bioactive component that was first isolated from Piper longum L., commonly referred to as the long pepper (4). Traditionally, PPLGM has been used for treating gastrointestinal and respiratory diseases (5). Notably, PPLGM has been shown to selectively target a wide spectrum of cancer cells (6–9). PPLGM induces cancer cell death by triggering different pathways, including apoptosis, necrosis and autophagy (10–13). The elevation of reactive oxygen species (ROS), characteristic of oxidative stress, is an important mechanism by which PPLGM induces cancer-selective cell death (7,8). In addition, ROS-independent mechanisms, such as cellular cross-linking events, may also contribute to the induction of apoptosis by PPLGM (14). Extensive research has documented the fact that PPLGM induces apoptosis in cancer cells by interfering with redox and ROS homeostatic regulators (8,15). The outstanding efficiency of PPLGM in inducing cancer cell death and its low toxicity favor the potential development of this compound as a chemotherapeutic agent against cancer (8).
The c-Jun NH2-terminal kinases (JNKs) are protein kinases that can phosphorylate the c-Jun transcription factor at serine (Ser)63 and −73, resulting in the robust induction of c-Jun transactivation (16). Evidence has accumulated demonstrating that JNKs are involved in a variety of cell activities, including cell apoptosis, which is an important process for tumor suppression (17,18). Further studies have revealed that JNKs play an essential role in cancer cell death induced by redox chemotherapeutic agents (19,20). In particular, PPLGM-mediated oxidative stress has been shown to be partially induced by inhibiting glutathione S-transferase π-1 (GSTP1), which is a direct negative regulator of JNK, resulting in cancer cell death (8,21). We thus hypothesize that PPLGM induces cell death in colorectal cancer cells via activation of the JNK signaling pathway. The present study investigates the effects of PPLGM on JNK signal transduction and the role of the JNK signaling pathway on PPLGM-mediated cell apoptosis in HCT116 cells.
Materials and methods
Reagents
PPLGM and SP600125 (Sigma-Aldrich, St. Louis, MO, USA) were dissolved in dimethyl sulphoxide (DMSO) to a 50 mM solution and stored at −20°C. Rabbit monoclonal antibodies (Abs) against c-Jun and phospho-c-Jun at Ser63 were purchased from Epitomics (Burlingame, CA, USA). Rabbit monoclonal Abs against cysteinyl aspartate-specific proteinase-3 (caspase-3) and JNK, and mouse monoclonal Abs against phospho-JNK at Thr183/Tyr185 were obtained from Cell Signaling Technology, Inc., (Beverly, MA, USA). Mouse monoclonal Abs against poly(adenosine diphosphate-ribose) polymerase (PARP) were obtained from BD Biosciences (San Jose, CA, USA). Mouse monoclonal Abs against β-actin, and anti-mouse immunoglobulin G and anti-rabbit immunoglobulin G horseradish peroxidase-conjugated secondary antibodies were purchased from Proteintech Group, Inc. (Chicago, IL, USA).
Cell culture
Human epithelial colorectal adenocarcinoma HCT116 cells were purchased from Culture Collection of Chinese Academy of Science (Shanghai, China) and cultured in RPMI 1640 medium (Gibco Life Technologies, Carlsbad, CA, USA) supplemented with 10% inactivated fetal bovine serum, 100 IU/ml penicillin and 100 µg/ml streptomycin in a humidified atmosphere of 5% CO2 at 37°C until confluence.
Cell viability assay
An MTS assay (CellTiter 96® AQueous One Solution Cell Proliferation Assay; Promega Corporation, Madison, WI, USA) was used to test cell viability. A total of 3×104/ml cells in 100 µl cell culture medium were incubated with increasing concentrations of PPLGM for 72 h. Detailed description of the indicated duration periods and doses is provided in Fig. 1. Control cells received DMSO for a final concentration that was the identical to the highest concentration of PPLGM, but less than 0.1% (v/v). At 4 h prior to culture termination, 20 µl MTS was added to the wells. The absorbance density was read on a 96-well plate reader (Mithras LB 940 Multimode Microplate Reader; Berthold Technologies GmbH & Co. KG, Bad Wildbad, Baden-Württemberg, Germany). at wavelength of 490 nm.
Figure 1.

Piperlongumine (PPLGM) reduces the cell viability of HCT116 cells. The HCT116 cells were exposed to the indicated concentrations of PPLGM for 72 h and then subjected to an MTS proliferation assay. Data are presented as the mean ± standard deviation from three experiments.
Trypan blue dye exclusion assay
The HCT116 cells were seeded into 24-well plates to 20–30% confluency, and then treated with PPLGM in various concentrations for the indicated duration. Detailed description of the indicated duration periods and doses is provided in Fig. 2. The cells were then trypsinized and stained with 0.4% (w/v) trypan blue (Sigma-Aldrich) and the viable cells were counted using a hematocytometer (Qiujing Biology Company Limited, Shanghai, China).
Figure 2.

Piperlongumine (PPLGM) inhibits HCT116 cell proliferation in a dose- and time-dependent manner. An equal number of HCT116 cells were plated and cultured in 24-well plates for 24 h, treated with different concentrations of PPLGM and incubated for the indicated time intervals. Cells were then stained with trypan blue and the viable cells were counted. Each experiment was performed in triplicate and the number of viable cells was plotted graphically with the standard deviation.
Cell cycle analysis by flow cytometry
Subsequent to being exposed to a fixed dose of PPLGM for various periods of time, the HCT116 cells were collected and fixed overnight in 66% cold ethanol at 4°C. A detailed description of the indicated duration periods and doses is provided in Fig. 3. The cells were then washed twice in cold phosphate-buffered saline (PBS) and labeled with propidium iodide (PI; BD Biosciences, Franklin Lakes, NJ, USA). Cell cycle distribution was determined using a BD FACScanto II flow cytometry analyzer equipped with BD FACSDiva software, version 6.1.3 (BD Biosciences, Franklin Lakes, NJ, USA).
Figure 3.
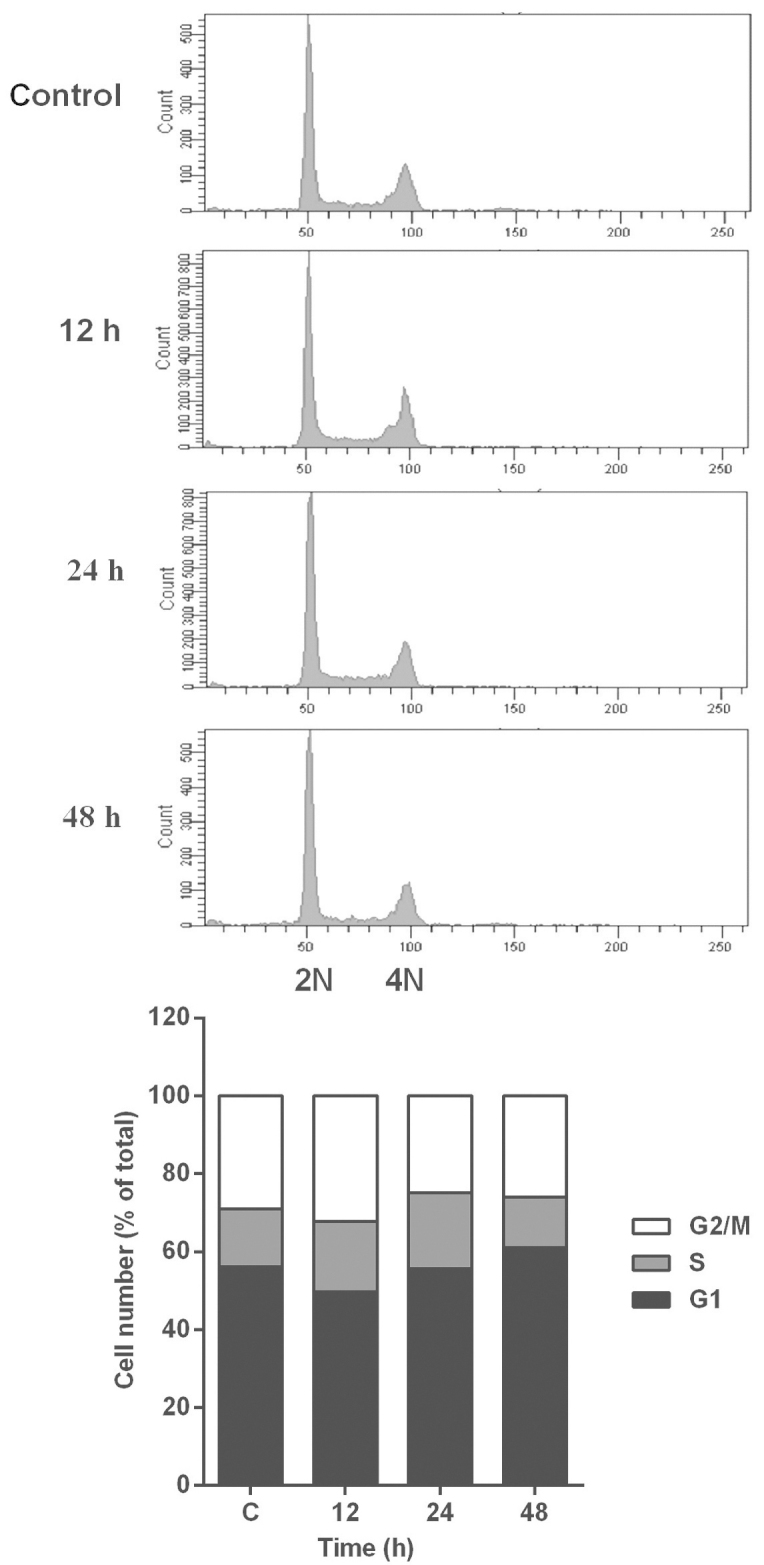
Effect of piperlongumine (PPLGM) on the distribution of cell cycle phases in HCT116 cells. The HCT116 cells were exposed to 5 µM PPLGM for the indicated time intervals and analyzed by flow cytometry following staining with propidium iodide. Figures were selected as representative data from three independent experiments. 2N, diploid cells; 4N, tetraploid cells.
Analysis of cell apoptosis by flow cytometry
Apoptosis was determined by flow cytometry using an Annexin V-fluoroisothiocyanate (FITC)/PI double-staining kit (Nanjing KeyGen Biotechnology, Co., Ltd., Nanjing, Jiangsu, China). The HCT116 cells were incubated with increasing concentration of drugs for the indicated times. A detailed description of the indicated duration periods and doses is provided in Fig. 4. Following the drug treatment, the cells were collected, washed and stained in working solution (500 µl binding buffer with 5 µl Annexin V-FITC and 5 µl PI) for 15 min in the dark. Apoptotic cells were then determined by flow cytometry, and the results were analyzed by BD FACSDiva software version 6.1.3.
Figure 4.

Piperlongumine (PPLGM) triggers apoptosis in HCT116 cells. (A) The cells were treated with PPLGM at increasing concentrations for 48 h and then underwent Annexin V/propidium iodide (PI) staining for flow cytometric analysis of cell apoptosis. (B) The cells were exposed to PPLGM at increasing concentrations for 48 h or were treated with 10 µM PPLGM for the indicated times. PARP and caspase-3 protein levels were determined by western blotting. β-actin was used to verify equal loading. Figures were selected as representative data from three independent experiments. PARP, poly(adenosine diphosphate-ribose) polymerase.
Western blotting
The procedures for western blotting were performed as described previously (22). Briefly, equal amounts of protein were separated by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to a polyvinylidene difluoride membrane (Millipore, Billerica, MA, USA). The membranes were then blocked with 5% skimmed-milk, 20 mM PBS and 0.1% Tween-20 buffer for 1 h at room temperature, and incubated with rabbit anti-c-Jun (dilution, 1:1,000), rabbit anti-phospho-c-Jun at Ser63 (dilution, 1:1,000), rabbit anti-caspase-3 (dilution, 1:1,000), mouse anti-PARP (dilution, 1:1,000), mouse anti-β-actin (dilution, 1:10,000) and rabbit anti-JNK (dilution, 1:1,000), mouse anti-phospho-JNK at Thr183/Tyr185 (dilution, 1:1,000) overnight at 4°C. The next day, following incubation with horseradish peroxidase-conjugated secondary antibodies (dilution, 1:10,000) for 1 h at room temperature, the signals were detected using an enhanced chemiluminescence detection kit (Santa Cruz Biotechnology, Inc., Dallas, TX, USA).
Treatment with JNK inhibitors
The cells were preincubated with the JNK inhibitor, SP600125 (10 µM), for 2 h and then treated with PPLGM (10 µM) for 24 h. Cell apoptosis was analyzed by flow cytometry and the protein levels were measured by western blotting.
Statistical analysis
All experiments were performed at least three times, and results are expressed as the mean ± standard deviation. Statistical analysis was performed by one-way analysis of variance followed by Tukey's test using GraphPad Prism 6.02 software (GraphPad Software, Inc., La Jolla, CA, USA). P<0.05 was considered to indicate a statistically significant difference.
Results
PPLGM causes concentration- and time-dependent growth inhibition of HCT116 cells
The dose-response effects of PPLGM on HCT116 were evaluated by MTS assay (Fig. 1). The half maximal inhibitory concentration value for PPLGM was 4.6 µM in the HCT116 cells at 72 h. Next, the cytotoxic effects of PPLGM on the HCT116 cells were tested by trypan-blue dye exclusion assay (Fig. 2). PPLGM at different concentrations inhibited the cell viability in the HCT116 cells, and the cellular response to the drug increased markedly as the drug concentration was raised from 2.5 to 10 µM (Fig. 2). The result showed that the inhibition of cell proliferation by PPLGM in the HCT116 cells was concentration- and time-dependent. After the cells had been treated with 5 µM PPLGM for 24 h, the cell viability decreased sharply compared with the control (Fig. 2). In addition, flow cytometric analysis revealed no significant alteration in cell-cycle distribution in the HCT116 cells incubated with 5 µM PPLGM (Fig. 3). Thus, PPLGM inhibited the growth of HCT116 cells without significantly affecting the cell cycle, suggesting that the loss of viability in the HCT116 cells may be attributable to cell death, but not to cell cycle withdrawal.
PPLGM induces apoptosis in HCT116 cells
To confirm the capability of PPLGM in inducing apoptosis, the control and PPLGM-treated HCT116 cells were assessed by flow cytometry subsequent to staining with Annexin V and PI. After 48 h of treatment with 10 µM PPLGM, the population of apoptotic HCT116 cells reached 67.3% (Fig. 4A). These data supported the occurrence of apoptosis in the HCT116 cells following PPLGM treatment. Next, to further verify apoptotic induction, cleaved PARP and caspase-3 in the PPLGM-treated HCT116 cells were monitored by western blotting. As illustrated in Fig. 4B, PARP cleavage and the reduction of caspase-3 protein levels, known hallmarks of apoptosis, appeared after 24 h of treatment with 10 µM PPLGM. Taken together, these results show that PPLGM induces a substantial level of apoptosis in HCT116 cells.
PPLGM activates the JNK signaling pathway in HCT116 cells
As JNK is the protein kinase that can phosphorylate c-Jun at Ser63 and −73, c-Jun phosphorylation at site Ser63 was selected for evaluation of the JNK signaling pathway. When the cells were incubated with increasing concentrations of PPLGM for 48 h, a moderate increase of c-Jun phosphorylation was detected at 5 µM only (Fig. 5A). According to the results of previous studies (8,9) and the current results (Fig. 4), 10 µM PPLGM induced cancer cell death more significantly than 5 µM PPLGM. Therefore, the concentration of 10 µM PPLGM was used in subsequent experiments. Treatment with 10 µM PPLGM for 12 h resulted in a marked increase in c-Jun phosphorylation in the HCT116 cells when the cell lysates were immunoblotted with the appropriate phospho-specific antibodies (Fig. 5B). The 10-µM PPLGM treatment resulted in sustained c-Jun phosphorylation, which was still apparent after 24 h in the HCT116 cells (Fig. 5B). In addition, the c-Jun expression level was elevated in parallel with increased c-Jun phosphorylation in the PPLGM-treated HCT116 cells (Fig. 5). The 10-µM PPLGM treatment induced c-Jun phosphorylation prior to PARP cleavage and loss of cell viability, suggesting that the JNK signal pathway may mediate PPLGM-induced apoptosis.
Figure 5.

Piperlongumine (PPLGM) promotes c-Jun phosphorylation at Ser63. Protein expression levels were determined by western blotting. β-actin was used to verify equal loading. (A) HCT116 cells were exposed to PPLGM at increasing concentrations for 48h. (B) HCT116 cells were treated with 10 µM PPLGM for the indicated times. Figures were selected as representative data from three independent experiments. p-, phosphorylated; t-, total.
To further examine whether the JNK signaling pathway is activated by PPLGM in HCT116 cells, the cells were exposed to 10 µM PPLGM for various periods of time, and analyzed for PPLGM-induced changes in JNK phosphorylation by western blot analysis using dual phospho-specific JNK antibodies. As shown in Fig. 6, JNK phosphorylation levels increased in the HCT116 cells within 1 h of PPLGM incubation and then decreased again from 3 h onwards.
Figure 6.

Piperlongumine (PPLGM) induces c-Jun NH2-terminal kinase (JNK) phosphorylation. Protein expression levels were determined by western blotting. β-actin was used to verify equal loading. HCT116 cells were exposed to 10 µM PPLGM for various periods of time. Figures were selected as representative data from three independent experiments. p-, phosphorylated; t-, total.
SP600125 inhibits PPLGM-mediated apoptosis through restraining JNK signal pathway activation in HCT116 cells
To identify whether the JNK signaling pathway was involved in PPLGM-mediated apoptosis, SP600125, a general inhibitor of JNK, was co-incubated with PPLGM in the HCT116 cells and cell apoptosis was then determined using flow cytometry. Pre-treatment with the SP600125 JNK inhibitor completely blocked the PPLGM-induced phosphorylation of c-Jun (Fig. 7) and significantly inhibited PPLGM-induced cell apoptosis in the HCT116 cells (Fig. 8). The percentage of apoptotic cells following treatment with 20 µM SP600125, 10 µM PPLGM, or 20 µM SP600125 plus 10 µM PPLGM were 7.2, 35.7 and 12.0%, respectively. The difference between PPLGM alone and PPLGM plus SP600125 was significant (P<0.0001).
Figure 7.

SP600125 inhibits piperlongumine (PPLGM)-induced c-Jun phosphorylation. For the inhibition experiments, the cells were incubated for 2 h in the presence or absence of 20 µM SP600125, then 10 µM PPLGM was added and incubated for 24 h. p-c-Jun and t-c-Jun was determined by western blotting. β-actin was used to verify equal loading. Figures were selected as representative data from three independent experiments. p-, phosphorylated; t-, total.
Figure 8.

SP600125 rescues HCT116 cells from apoptosis induced by piperlongumine (PPLGM). For the inhibition experiments, the cells were incubated for 2 h in the presence or absence of 20 µM SP600125, then 10 µM PPLGM was added and incubated for 24 h. The induction of apoptosis was estimated by flow cytometric analysis. Each value is the mean ± standard deviation of three determinations. ***P<0.0001. PI, propidium iodide.
Discussion
Recent data has showed that PPLGM can selectively kill various cancer cells, including human colorectal cancer cells (8,9). To the best of our knowledge, the present study is the first to indicate that the JNK signaling pathway is involved in PPLGM-induced cell apoptosis in human colorectal cancer cells. The results showed that PPLGM reduced cell viability independently of cell cycle withdrawal and induced cell death in a time- and concentration-dependent manner. At the same time, JNK signaling was activated during PPLGM treatment in the HCT116 cells. In addition, SP600125 inhibited PPLGM-induced JNK signaling and apoptosis in the HCT116 cells, suggesting that PPLGM-mediated apoptosis was at least partially dependent on the activation of the JNK signal pathway in the HCT116 cells (23).
Accumulated data have shown that PPLGM induces cell death through different pathways in multiple types of cancer cells (7–12). Previous studies have demonstrated that caspase-3-mediated PARP cleavage and cell cycle arrest at the G2/M phase are involved in PPLGM-induced cell apoptosis in human prostate cancer PC-3 cells (11). In the present study, HCT116 cells treated with PPLGM demonstrated likewise up-regulation of PARP/procaspase-3 cleavage (Fig. 4B), but not cell cycle arrest (Fig. 3); this discrepancy in the effect of PPLGM on cell cycle distribution may be a result of the different responses to PPLGM in different cell lines.
c-Jun, a cognate substrate for JNK, is a labile protein that is degraded by JNK under non-stressed conditions (24). However, a variety of cellular stresses, such as oxidative stress, can strongly activate the JNKs, which inhibit c-Jun ubiquitination and promote c-Jun transcription through c-Jun phosphorylation (24,25). Inhibition of the ubiquitin-proteasome system induced by PPLGM in cancer cells may also reduce the ubiquitin-dependent degradation of c-Jun (26). Collectively, a steady elevation of c-Jun expression is associated with the expression and stabilization of c-Jun. Hence, in the present study, it followed that sustained c-Jun phosphorylation was concomitant with the c-Jun overexpression observed during PPLGM treatment (Fig. 5).
The positive association between c-Jun activation and cell apoptosis has been well documented in neurons, endothelial and myeloma cells, fibroblasts and colorectal cancer cells (3,27–30). Similarly, the activation of JNK and the subsequent phosphorylation of c-Jun have been linked with apoptotic cell death induced by PPLGM in HCT116 cells (Figs. 7 and 8). Conversely, other studies have shown that the inhibition of JNK by SP600125 sensitizes tumor cells to CD95-induced apoptosis in HCT116 cells and that this effect is cell line-specific (31). Whether c-Jun activation leads to the inhibition or promotion of apoptosis should be dependent on the stimuli and the cell type.
Recent data have indicated that the mitogen-activated protein kinase (MAPK) core pathways are involved in PPLGM-induced cancer cell death. Notably, p38 MAPK activation leads to cell death through autophagy in human osteosarcoma U2OS cells (12). The present results showed that JNK signaling is involved in PPLGM-induced apoptosis in HCT116 cells (Figs. 7 and 8). As reported earlier in the colorectal cancer HT-29 cell line (9), the extracellular signal-regulated kinase (ERK) signaling pathway is also activated in PPLGM-treated HCT116 cells (data not shown), strongly arguing that a decrease in ERK and an increase in JNK are required for the induction of apoptosis (17). A previous study in hamster fibroblast CC139 cells demonstrated that phosphoinositide 3′-kinase (PI3K) inhibition is necessary for JNK-mediated cell death (32). In addition, PPLGM causes PI3K inhibition to induce caspase-dependent apoptosis in human triple-negative breast cancer cells (13). Altogether, the mechanisms of signal transduction are complicated during the course of PPLGM-induced cancer cell death and the cross-talk between different signaling pathways should be further elucidated.
In summary, the present results suggested that in the HCT116 cells, PPLGM reduced cell viability and triggered cell apoptosis through the JNK signal pathway in a concentration- and time-dependent manner. A clear understanding of the molecular mechanisms of PPLGM-mediated cell apoptosis may shed light on the further clinical development of PPLGM for chemotherapy.
Acknowledgements
The authors would like to thank Dr Xinhui Fu, Dr Zihuan Yang, Dr Jun Hu and Ms. Zhiting Chen for providing technical assistance. This study was supported by Sun Yat-sen University ‘100 Talents Program’, the Guangdong Innovative Research Team Program (grant no. 2009010058), the Science and Information Technology Bureau of Guangzhou, Guangdong (grant no. 2011J5200009), the Guangdong Provincial Department of Science and Technology (grant no. 2012B050500004), Overseas Excellent Professor Project, and the Japanese Ministry of Education, Culture, Sports, Science and Technology, Program of Japanese Initiative for Global Research Network on Infectious Diseases.
References
- 1.Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014;64:9–29. doi: 10.3322/caac.21208. [DOI] [PubMed] [Google Scholar]
- 2.Van Cutsem E, Köhne CH, Hitre E, et al. Cetuximab and chemotherapy as initial treatment for metastatic colorectal cance. N Engl J Med. 2009;360:1408–1417. doi: 10.1056/NEJMoa0805019. [DOI] [PubMed] [Google Scholar]
- 3.Ham J, Babij C, Whitfield J, et al. A c-Jun dominant negative mutant protects sympathetic neurons against programmed cell death. Neuron. 1995;14:927–939. doi: 10.1016/0896-6273(95)90331-3. [DOI] [PubMed] [Google Scholar]
- 4.Chatterjee A, Dutta CP. Alkaloids of Piper longum Linn. I. Structure and synthesis of piperlongumine and piperlonguminine. Tetrahedron. 1967;23:1769–1781. doi: 10.1016/S0040-4020(01)82575-8. [DOI] [PubMed] [Google Scholar]
- 5.Bezerra DP, Pessoa C, de Moraes MO, Saker-Neto N, Silveira ER, Costa-Lotufo LV. Overview of the therapeutic potential of piplartine (piperlongumine) Eur J Pharm Sci. 2013;48:453–463. doi: 10.1016/j.ejps.2012.12.003. [DOI] [PubMed] [Google Scholar]
- 6.Han SS, Son DJ, Yun H, Kamberos NL, Janz S. Piperlongumine inhibits proliferation and survival of Burkitt lymphoma in vitro. Leuk Res. 2013;37:146–154. doi: 10.1016/j.leukres.2012.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu JM, Pan F, Li L, et al. Piperlongumine selectively kills glioblastoma multiforme cells via reactive oxygen species accumulation dependent JNK and p38 activation. Biochem Biophys Res Commun. 2013;437:87–93. doi: 10.1016/j.bbrc.2013.06.042. [DOI] [PubMed] [Google Scholar]
- 8.Raj L, Ide T, Gurkar AU, et al. Selective killing of cancer cells by a small molecule targeting the stress response to ROS. Nature. 2011;475:231–234. doi: 10.1038/nature10167. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 9.Randhawa H, Kibble K, Zeng H, Moyer MP, Reindl KM. Activation of ERK signaling and induction of colon cancer cell death by piperlongumine. Toxicol In Vitro. 2013;27:1626–1633. doi: 10.1016/j.tiv.2013.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bezerra DP, Militão GC, de Castro FO, et al. Piplartine induces inhibition of leukemia cell proliferation triggering both apoptosis and necrosis pathways. Toxicol In Vitro. 2007;21:1–8. doi: 10.1016/j.tiv.2006.07.007. [DOI] [PubMed] [Google Scholar]
- 11.Kong EH, Kim YJ, Cho HJ, et al. Piplartine induces caspase-mediated apoptosis in PC-3 human prostate cancer cells. Oncol Rep. 2008;20:785–792. [PubMed] [Google Scholar]
- 12.Wang Y, Wang JW, Xiao X, et al. Piperlongumine induces autophagy by targeting p38 signaling. Cell Death Dis. 2013;4:e824. doi: 10.1038/cddis.2013.358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shrivastava S, Kulkarni P, Thummuri D, et al. Piperlongumine, an alkaloid causes inhibition of PI3K/Akt/mTOR signaling axis to induce caspase-dependent apoptosis in human triple-negative breast cancer cells. A Internat J Program Cell Death. 2014;19:1148–1164. doi: 10.1007/s10495-014-0991-2. [DOI] [PubMed] [Google Scholar]
- 14.Adams DJ, Dai M, Pellegrino G, et al. Synthesis, cellular evaluation, and mechanism of action of piperlongumine analogs. Proc Natl Acad Sci USA. 2012;109:15115–15120. doi: 10.1073/pnas.1212802109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim TH, Song J, Kim SH, et al. Piperlongumine treatment inactivates peroxiredoxin 4, exacerbates endoplasmic reticulum stress, and preferentially kills high-grade glioma cells. Neuro Oncol. 2014;16:1354–1364. doi: 10.1093/neuonc/nou088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kyriakis JM, Avruch J. Mammalian MAPK signal transduction pathways activated by stress and inflammation: a 10-year update. Physiol Rev. 2012;92:689–737. doi: 10.1152/physrev.00028.2011. [DOI] [PubMed] [Google Scholar]
- 17.Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science. 1995;270:1326–1331. doi: 10.1126/science.270.5240.1326. [DOI] [PubMed] [Google Scholar]
- 18.Chang L, Karin M. Mammalian MAP kinase signalling cascades. Nature. 2001;410:37–40. doi: 10.1038/35065000. [DOI] [PubMed] [Google Scholar]
- 19.Hu R, Kim BR, Chen C, Hebbar V, Kong AN. The roles of JNK and apoptotic signaling pathways in PEITC-mediated responses in human HT-29 colon adenocarcinoma cells. Carcinogenesis. 2003;24:1361–1367. doi: 10.1093/carcin/bgg092. [DOI] [PubMed] [Google Scholar]
- 20.Xu C, Shen G, Yuan X, et al. ERK and JNK signaling pathways are involved in the regulation of activator protein 1 and cell death elicited by three isothiocyanates in human prostate cancer PC-3 cells. Carcinogenesis. 2006;27:437–445. doi: 10.1093/carcin/bgi251. [DOI] [PubMed] [Google Scholar]
- 21.Wang T, Arifoglu P, Ronai Z, Tew KD. Glutathione S-transferase P1-1 (GSTP1-1) inhibits c-Jun N-terminal kinase (JNK1) signaling through interaction with the C terminus. J Biol Chem. 2001;276:20999–21003. doi: 10.1074/jbc.M101355200. [DOI] [PubMed] [Google Scholar]
- 22.Song M, Chen D, Lu B, et al. PTEN loss increases PD-L1 protein expression and affects the correlation between PD-L1 expression and clinical parameters in colorectal cancer. PLoS One. 2013;8:e65821. doi: 10.1371/journal.pone.0065821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bennett BL, Sasaki DT, Murray BW, et al. SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase. Proc Natl Acad Sci USA. 2001;98:13681–13686. doi: 10.1073/pnas.251194298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fuchs SY, Dolan L, Davis RJ, Ronai Z. Phosphorylation-dependent targeting of c-Jun ubiquitination by Jun N-kinase. Oncogene. 1996;13:1531–1535. [PubMed] [Google Scholar]
- 25.Musti AM, Treier M, Bohmann D. Reduced ubiquitin-dependent degradation of c-Jun after phosphorylation by MAP kinases. Science. 1997;275:400–402. doi: 10.1126/science.275.5298.400. [DOI] [PubMed] [Google Scholar]
- 26.Jarvius M, Fryknäs M, D'Arcy P, Sun C, Rickardson L, Gullbo J, Haglund C, Nygren P, Linder S, Larsson R. Piperlongumine induces inhibition of the ubiquitin-proteasome system in cancer cells. Biochem Biophys Res Commun. 2013;431:117–123. doi: 10.1016/j.bbrc.2013.01.017. [DOI] [PubMed] [Google Scholar]
- 27.Wang N, Verna L, Hardy S, et al. c-Jun triggers apoptosis in human vascular endothelial cells. Circ Res. 1999;85:387–393. doi: 10.1161/01.RES.85.5.387. [DOI] [PubMed] [Google Scholar]
- 28.Podar K, Raab MS, Tonon G, et al. Up-regulation of c-Jun inhibits proliferation and induces apoptosis via caspase-triggered c-Abl cleavage in human multiple myeloma. Cancer Res. 2007;67:1680–1688. doi: 10.1158/0008-5472.CAN-06-1863. [DOI] [PubMed] [Google Scholar]
- 29.Bossy-Wetzel E. Bakiri L and Yaniv M: Induction of apoptosis by the transcription factor c-Jun. EMBO J. 1997;16:1695–1709. doi: 10.1093/emboj/16.7.1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Teraishi F, Wu S, Zhang L, et al. Identification of a novel synthetic thiazolidin compound capable of inducing c-Jun NH2-terminal kinase-dependent apoptosis in human colon cancer cells. Cancer Res. 2005;65:6380–6387. doi: 10.1158/0008-5472.CAN-05-0575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kuntzen C, Sonuc N, De Toni EN, et al. Inhibition of c-Jun-N-terminal-kinase sensitizes tumor cells to CD95-induced apoptosis and induces G2/M cell cycle arrest. Cancer Res. 2005;65:6780–6788. doi: 10.1158/0008-5472.CAN-04-2618. [DOI] [PubMed] [Google Scholar]
- 32.Molton SA, Todd DE, Cook SJ. Selective activation of the c-Jun N-terminal kinase (JNK) pathway fails to elicit Bax activation or apoptosis unless the phosphoinositide 3-kinase (PI3K) pathway is inhibited. Oncogene. 2003;22:4690–4701. doi: 10.1038/sj.onc.1206692. [DOI] [PubMed] [Google Scholar]