

Abstract
Purpose: Beta 2 glycoprotein I (β2GPI) has been shown the positive effect on diabetic atherosclerosis and retinal neovascularization. β2GPI can be reduced by thioredoxin-1, resulting in the reduced state of β2GPI. The possible protective effects of β2GPI and reduced β2GPI on diabetic nephropathy (DN) are not fully elucidated. The purpose of this study was to test a hypothesis that β2GPI and reduced β2GPI would improve DN in streptozotocin (STZ) induced diabetic mice and high-glucose (HG) exposed rat mesangial cell (RMC). Methods: The STZ-induced Balb/c mice and HG exposed RMCs were administrated with β2-GPI and reduced β2-GPI at different time and concentrations gradient respectively. The changes of glomerular structure and expression of collagen IV, TGF-β1, p38 MAPK and phospho-p38 MAPK in renal cortical and mesangial cells were observed by immunohistochemical techniques, quantitative real-time PCR and western blot with or without the treatment of β2-GPI and reduced β2-GPI. Results: β2GPI and reduced β2GPI improved early clinical and pathological changes of DN in STZ-diabetic mice. Treatment with β2GPI and reduced β2GPI in the STZ-diabetic mice and HG exposed RMCs resulted in decrease expression levels of TGF-β1 and collagen IV, with concomitant decrease in phospho-p38 MAPK expression. Conclusions: β2GPI and reduced β2GPI improved renal structural damage and kidney function. The renoprotective and antifibrosis effects of β2GPI and reduced β2GPI on DN were closely associated with suppressing the activation of the TGF-β1-p38 MAPK pathway.
Keywords: Beta 2 glycoprotein I, diabetic nephropathy, TGF-β1
Introduction
Diabetic nephropathy (DN) is the major microvascular complication of diabetes mellitus (DM) and is the most common cause of end-stage renal disease worldwide [1]. Clinically, DN is characterized by a reduction in the glomerular filtration rate and increasing proteinuria that eventually leads to progressive renal failure. Pathologically, DN is characterized by the glomerular mesangium hypertrophy caused by a proliferation of mesangial cells and excessive accumulation of extracellular matrix (ECM), which ultimately progresses to kidney fibrosis and glomerulosclerosis [2].
Transforming growth factor-β1 (TGF-β1) is a fibrogenic and inflammatory cytokine that plays a vital role in glomerulosclerosis and interstitial fibrosis in various renal diseases, including DN. The abnormal production of TGF-β1 induced by hyperglycemia causes an excessive accumulation of ECM proteins, such as collagen and fibronectin, which ultimately leads to mesangial expansion and glomerular basement membrane thickening. The mitogen-activated protein kinases (MAPK), a family of serine/threonine kinases, regulate intracellular signal transduction in response to various extracellular stimuli. MAPK subfamilies contain extracellular signal-regulated kinase (ERK) 1/2, c-Jun NH2-terminal kinase (JNK), and p38 mitogen-activated protein kinase (p38 MAPK) [3]. Recently, some evidence suggests that the activation of the p38 MAPK pathway, but not the canonical Smad signaling pathway, mediates TGF-β1-induced collagen and fibronectin expression during the development of DN [4].
Beta 2 glycoprotein I (β2GPI) is a 50-kDa protein that was first described in 1961[5], and since 1990 β2GPI has been identified as the most prominent antigen in antiphospholipid syndrome [6]. β2GPI is predominantly synthesized in hepatocytes, and its circulating plasma concentration is variable (50-500 µg/mL, 1-10 µM) [7]. β2GPI consists of five repeating amino acid domains. Domains I-IV include four cysteines each and have the conserved sequences, domain V has an extra 20 amino acid tail with a unique cysteine termination and is aberrant [8]. Recently, it has been shown that the disulfide bond between Cys288 to Cys326 in the domain V can be reduced by thioredoxin-1 (TRX-1) or protein disulfide isomerase (PDI) [9], resulting in the reduced state of β2GPI, referred to as reduced β2GPI.
Although β2GPI has been shown to be a participant in the autoimmune system [10,11], vascular thrombosis [12,13], infectious diseases [14] and others system, the function of β2GPI remains unclear. It has also reported that β2GPI is associated with accelerated atherosclerosis and enhanced oxidative stress [15,16]. High levels of β2GPI have been reported to decrease the risk of myocardial infarction in elderly men [17]. In addition, reduced β2GPI was found to protect endothelial cells from oxidative stress-induced injury [18]. We have previously found a positive effect of β2GPI and reduced β2GPI in diabetic atherosclerosis and retinal neovascularization [19]. Till now, no studies have examined the relationship between β2GPI and reduced β2GPI and DN. In this study, we hypothesize that β2GPI or reduced β2GPI has beneficial effects in DN. In addition, we investigated the renoprotective effects of β2GPI or reduced β2GPI in the TGF-β1-p38 MAPK pathway by examining the expression of total and phosphorylated p38 MAPK (p-p38 MAPK), TGF-β1, and collagen IV in diabetic mice and high glucose exposed rat mesangial cells.
Materials and methods
Materials and reagents
The rat mesangial cell (RMC) line, HBZY-1, was obtained from the American Type Culture Collection (ATCC number: CRL-2573). Fetal bovine serum (FBS), human serum albumin (HSA) and mannitol were purchased from Beijing Solarbio Science & Technology Co., Ltd. (Beijing, China). Streptozotocin (STZ) was purchased from Sigma (Aldrich, USA). Trizol Reagent was purchased from Invitrogen (Carlsbad, USA). A Reverse Transcription Kit was purchased from Thermo Scientific. SYBR® Premix Ex Taq TM reagent was purchased from TaKaRa Biotechnology, Inc. (Otsu, Japan). Antibodies against TGFβ1 and collagen IV were purchased from ABCAM (Cambridge, UK). Antibodies against p38 MAPK and p-p38 MAPK were purchased from Cell Signaling Technology, Inc. (Boston, USA). Antibodies against β-tubulin were purchased from Tianjin Sungene Biotech Co., Ltd. (Tianjin, China). Horseradish peroxidase labelled goat anti-rabbit IgG was purchased from Beijing ComWin Biotech Co., Ltd. (Beijing, China). The biotinylated goat anti-rabbit antibody and the Vectastain Elite ABC Staining Kit were purchased from Vector Laboratories (Burlingame, CA, USA). All other materials were of reagent grade.
Purification of β2GPI and preparation of reduced β2GPI
β2GPI was purified from normal human plasma using previously methods [20]. Plasma β2GPI was precipitated by 3% perchloric acid and isolated by heparin-sepharose affinity chromatography (HiTrap Heparin, GE Healthcare). LC-MS analysis was used to confirm this protein. The purity of β2GPI was confirmed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) on a 10% mini-gel. SDS-PAGE analysis of the protein sample showed the same band as the standard sample (Figure 1). The bicinchoninic acid (BCA) method was used to determine the concentration of β2GPI. Reduced β2GPI was prepared by previously methods [18]. Purified β2GPI (1 μM) was reduced by TRX-1 (3.5 μM) activated with dithiothreitol (DTT, 70 μM). The thiols of reduced β2GPI were protected by reduced glutathione. The reduced β2GPI was verified using a western blot and LC-MS analysis. Western blot analysis showed the new protein maintained the same immunologic activity as β2GPI. LC-MS analysis verified that domain V of the new protein had free thiols.
Figure 1.
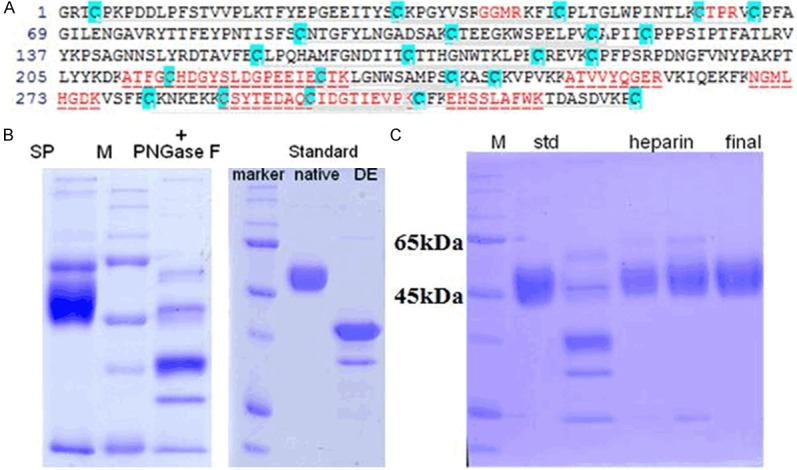
Verification of β2GPI. After purification, the sample fractions were digested by trypsin PNGase F followed by LC-MS analysis and SDS-PAGE analysis. A. The red coloured sequence is the sequence we found. B. SDS-PAGE result revealed that the digested fraction was consistent with standard protein. Thus, we verified that the fractions dly contaed β2GPI. C. After further purification with heparin-sepharose affinity chromatography, the sample fractions were analyzed with SDS-PAGE. The molecule weight of the sample was calculated according to the relationship between molecular weight of standard protein and relative mobility ratio, and the result was 50 kDa. SP, std: β2GPI standard protein; M: marker; DE: digested by trypsin PNGase F.
Experimental animals for diabetic nephropathy
Female Balb/c mice (8-weeks old, 18-20 g) were obtained from Peking University Laboratory Animal Center (Beijing, China). All animal procedures were performed according to the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. All animals were housed under standard conditions with unrestricted food and water. Mice were randomly divided into a diabetes mellitus group (n=40) and a normal control group (n=8) and were fed a high fat and standard chow diet respectively. After 8 weeks, the DM group was induced by intraperitoneal injection of STZ at 80 mg/kg three times for three days as previously described [21]. The normal control group was injected with an equal amount of sodium citrate buffer. Diabetic mice were confirmed by a plasma glucose concentration ≥16.7 mM after 72 hours injection. The STZ-induced diabetic mice were randomly divided into 5 groups (n=8) as follows: (a) and (b) β2GPI or reduced β2GPI for 3 weeks in which mice were induced by an intravenous injection of 20 μg of β2GPI or reduced β2GPI one time for 3 weeks; (c) and (d) β2GPI or reduced β2GPI for 6 weeks in which mice were injected with 20 μg of β2GPI or reduced β2GPI two times for 6 weeks (one time per 3 weeks); (e) diabetic control mice in which mice were injected with equal volumes of PBS two times for 6 weeks. The normal control group was simultaneously injected with equal volumes of PBS two times for 6 weeks also. After STZ or control injections, the blood glucose and body weight were monitored weekly. The mice were housed individually in metabolic cages. The 24 hours urine samples were collected on the day before the end of the experiment and were stored at -80°C. After 3 or 6 weeks of treatment, blood samples were collected from the retro-orbital venous plexus then mice were sacrificed and both kidneys were removed. Serum was collected by centrifugation at 3000 g for 15 min and stored at -80°C. Urine albumin concentration was determined using the mouse albumin ELISA kit (Assaypro, USA) according to the manufacturer’s instructions. The serum levels of glucose, urea nitrogen and creatinine were determined using an Automatic Biochemical Analyzer (Hitachi Co., Ltd., Japan). Kidney samples were weighed and frozen in liquid nitrogen and stored at -80°C or fixed in 10% neutral-buffered formalin.
Cell culture and treatment
RMCs between passages 3 and 8 were used in experiments. RMCs were cultured in Dulbecco’s modified Eagle Medium (DMEM) containing 5.5 mM glucose supplemented with 10% FBS and antibiotics, maintained at 37°C in a 95% air, 5% CO2 atmosphere. RMCs were seeded at a density of 1.0×106 cells/well in six-well plates. When the cells had grown to 70-80% confluence, RMCs were placed in serum-free DMEM for 24 hours and were then divided into eight groups for the next 24 hours as follows: (a) normal glucose in which RMCs were cultured in 5.5 mM glucose DMEM (NG group); (b) mannitol in which RMCs were cultured in medium composed of 19.5 mM mannitol and 5.5 mM glucose DMEM that was used as an osmotic control group (Mannitol group); (c) and (d) low or high-dose HSA in which RMCs were cultured in 25 mM glucose DMEM supplemented with 40 or 80 μg/ml HSA (HG plus HSA group) that was used as the high glucose control group; (e) and (f) low or high-dose β2GPI in which RMCs were cultured in 25 mM glucose DMEM supplemented with 40 or 80 μg/ml β2GPI (HG plus β2GPI group); and (g) and (h) low or high-dose reduced β2GPI in which RMCs were cultured in 25 mM glucose DMEM supplemented with 40 or 80 μg/ml reduced β2GPI (HG plus reduced β2GPI group). Cell lysates and RNA were collected and used for quantitative real-time PCR and western blot analysis.
Morphological and immunohistochemical analysis
The kidneys were embedded in paraffin and were sliced into 4 μm sections. The tissue slices were stained with periodic acid-Schiff (PAS) for general histological studies. For immunohistochemical analysis, the paraffin sections were deparaffinized and rehydrated. After blockage of endogenous peroxidase and antigen retrieval, the tissue slices were incubated with primary antibodies (TGF-β1 or collagen IV, diluted 1:200) at 4°C overnight. The slices were then washed with PBS and incubated with a biotinylated second antibody (diluted 1:200) (Vector, Burlingame, CA) at room temperature for 30 min. The slices were washed again with PBS and incubated with a Vectastain ABC kit (Vector, UK) according to the manufacturer’s instructions. Immunoreactivity was visualized with the chromogen 3,3’-diaminobenzidine (DAB, Sigma). The slices were counterstained with hematoxylin, dehydrated and mounted. Brown areas were judged as positive staining. Glomerular cross-sectional areas were assessed with light microscopy (Olympus IX-52, Olympus Optical, Tokyo, Japan). Semi-quantitative analysis of the ratio of positive staining areas to the glomerular areas was evaluated with the image analysis software Image Pro. Plus 6.0 (Media Cybernetics, Inc., Bethesda, MD, USA).
Quantitative real-time RT-PCR
Total RNA was extracted from kidneys or cultured cells with Trizol Regent and was reverse transcribed into cDNA with a Reverse Transcription Kit. RNA was quantitated by OD values at 260 nm and the integrity was verified by ethidium bromide staining of ribosomal 18S and 28S bands on agarose gels. Real-time PCR was performed with the SYBR® Premix Ex Taq TM reagent according to the manufacturer’s instructions. The following primers were used for TGF-β1 of STZ-diabetic mice: forward: 5’-AGG GCT ACC ATG CCA ACT TC-3’, reverse: 5’-CCA CGT AGT AGA CGA TGG GC-3’; for TGF-β1 of RMCs: forward: 5’-AGG GCT ACC ATG CCA ACT TC-3’, reverse: 5’-CCA CGT AGT AGA CGA TGG GC-3’; for collagen IV of STZ-diabetic mice: forward 5’-TCC AGG CCC CCC TGG AAC TGT-3’, reverse 5’-GAG GGC CTG GTT GGC CTG-3’; for collagen IV of RMCs: forward 5’-CCA TCT GTG GAC CAT GGC TT-3’, reverse 5’-GCG AAG TTG CAG ACG TTG TT-3’; for GAPDH: forward: 5’-CAA GGT CAT CCA TGA CAA CTT TG-3’, reverse: 5’-GTC CAC CAC CCT GTT GCT GTA G-3’. Water, as a replacement for cDNA, was used as negative control. GAPDH was used as an internal control. The mRNA expression levels were quantified with normalization to GAPDH. The results were calculated using the 2-△△CT method.
Western blot
The renal cortex tissue homogenates and RMCs after being washed with ice-cold PBS were lysed with RIPA lysis buffer. After incubation for 30 min on ice, the lysates were centrifuged at 12000 rpm for 15 min at 4°C and the supernatant was collected. The concentration of protein in the supernatant was determined with the Micro BCATM Protein Assay Reagent Kit according to the manufacturer’s instructions. The samples (30 μg protein per lane) were separated on a 10% SDS-PAGE and blotted onto a nitrocellulose transfer membrane. The membranes were blocked with 5% nonfat milk in TBST (20 mM Tris-HCl, pH 7.6, 137 mM NaCl and 0.01% Tween-20) for 60 min at room temperature and were then incubated with primary antibodies (against TGF-β1, collagen IV, p38 MAPK, p-p38 MAPK and β-tubulin) overnight at 4°C. The membranes were subsequently washed with TBST and incubated with horseradish peroxidase-conjugated secondary antibodies for 60 min at room temperature. After the incubation, the membranes were washed with TBST. Immunoreactive bands were detected with ECL reagents (Amersham Biosciences, UK) and exposure to Hyperfilm (GE Healthcare, UK). Band densities were measured using BandScan software and quantified by normalization to β-tubulin.
Statistical analysis
All of the results are expressed as the means ± SEM. Statistical analyses were performed using IBM SPSS Statistics 19.0 software. Statistical significance among multi-groups was determined using one way analysis of variance and Tukey’s test post hoc analysis. Statistical significance between two groups was determined using the unpaired Student’s t-test. P<0.05 was considered statistically significant.
Results
β2GPI and reduced β2GPI improve biochemical parameters and kidney hypertrophy in STZ-induced diabetic mice
After STZ injection, all mice became diabetic with significantly higher urine albumin, urea nitrogen and creatinine compared with the normal control group (P<0.05) (Table 1). However, the levels of urea nitrogen, creatinine and urine albumin were significantly lower in diabetic mice treated with β2GPI and reduced β2GPI compared with untreated diabetic mice (P<0.05), except in mice treated with β2GPI for 3 weeks.
Table 1.
The effects of β2GPI and reduced β2GPI on the biochemical parameters and renal function of STZ-induced diabetic mice at the end of the experimental period. Data are expressed as the mean ± S.E.M. (n = 8 for each group)
| Groups | NC | DM | DM + β2-GPI | DM + reduced β2GPI | ||
|---|---|---|---|---|---|---|
|
| ||||||
| 3 wk | 6 wk | 3 wk | 6 wk | |||
| Body weight (g) | 24.58±2.53 | 22.48±3.11 | 23.39±1.65 | 24.04±2.83 | 24.33±2.76 | 23.72±2.42 |
| Kidney weight (g) | 134.69±19.40 | 263.09±15.81* | 212.02±22.41* | 205.30±22.69*,# | 210.45±30.49* | 186.34±15.04# |
| Kidney/body weight ratio (mg/g) | 5.57±1.40 | 11.88±1.93* | 9.04±0.33* | 8.56±0.64*,# | 8.64±0.59*,# | 7.88±0.63# |
| Blood glucose (mmol/L) | 6.05±0.86 | 25.66±3.21* | 23.89±6.43* | 23.78±6.21* | 24.49±5.55* | 22.40±4.28* |
| Serum creatinine (μmmol/L) | 7.29±1.23 | 23.64±5.07* | 17.17±3.06*,# | 15.98±5.89*,# | 16.14±3.93*,# | 13.20±1.71*,# |
| Blood urea nitrogen (mmol/L) | 5.23±0.65 | 12.57±1.24* | 9.75±1.22* | 8.10±0.84*,# | 8.60±1.43*,# | 8.33±1.34*,# |
| Urine albumin (μg/24 h) | 27.26±8.74 | 230.99±40.01* | 161.41±31.03* | 132.88±28.61*,# | 140.58±25.72*,# | 116.88±18.52*,# |
C: normal control; DM: diabetic model control; β2GPI: 20 μg β2GPI; reduced β2GPI: 20 μg reduced β2GPI. NC and DM were treated with equal volumes of PBS. All the mice were administered via intravenous injection.
P<0.05 vs. NC group.
P<0.05 vs. DM group.
The kidney hypertrophy index (kidney/body weight ratio) increased significantly (P<0.05), while the body weight slightly decreased in diabetic mice compared with the normal control group (P>0.05) (Table 1). However, the degree of kidney hypertrophy index decreased after treatment with β2GPI and reduced β2GPI, particularly in mice treated with reduced β2GPI for 6 weeks.
β2GPI and reduced β2GPI improved glomerular morphological changes in STZ-induced diabetic mice
The mesangial expansion and capillary basement membrane thickening were observed in the diabetic control group after PAS staining (Figure 2). After treatment with β2GPI and reduced β2GPI, there was decreased mesangial expansion and capillary basement membrane thickening. As treatment time progressed, the improvements were evident. Particularly, after 6 weeks treatment with reduced β2GPI, the PAS staining showed nearly normal glomerular structure.
Figure 2.

Treatment with β2GPI and reduced β2GPI prevented mesangial expansion and capillary basement membrane thickening in STZ-induced diabetic mice. Glomerular histopathology analysis by PAS staiin. A. The pictures display representative glomeruli of PAS-stained sections with original magnification of ×400. Scale bar = 50 μm for all microrahs. B. The ratio of PAS-positive staining areas to the glomerular areas was evaluated by a semi-quantitative method. Data are expressed as the mean ± S.E.M. (n=8 for each group). *P<0.05 vs. NC roup, #P<0.05 vs. DM group. NC: normal control; DM: diabetic model control; β2GPI: 20 μg β2GPI; reduced β2GPI: 20 μg reduced β2GPI.
β2GPI and reduced β2GPI decreased the excessive accumulation of ECM proteins in STZ-induced diabetic mice
The STZ-induced diabetic mice exhibited an increase in collagen IV-positive areas accumulated in the mesangial matrix and capillary basement membranes compared with the normal control group (Figure 3A). After 3 weeks treatment with β2GPI and reduced β2GPI, collagen IV-positive areas decreased slightly compared with the diabetic control group. However, 6 weeks treatment with β2GPI and reduced β2GPI exhibited significantly less collagen IV-positive areas. In addition, collagen IV mRNA and protein expression from the diabetic control group were significantly higher than those from the normal control group (Figure 3B and 3C). After β2GPI and reduced β2GPI treatment, collagen IV mRNA and protein expression decreased significantly compared with the diabetic control group, particularly after 6 weeks treatment with reduced β2GPI.
Figure 3.

Treatment with β2GPI and reduced β2GPI decreased the excessive accumulation of ECM proteins in STZ-induced diabeti mce. A. Immunohistochemical staining of collagen IV in the above groups. Increased expression of collagen IV-positive areas (dark brown) was observed after STZ induction in the DM group and was reduced with β2GPI and reduced β2GPI as the treatment increasing. Original magnification: ×400. Scale bar = 50 μm for all micrographs. NC: normal control; DM: diabetic model control; β2GPI: 20 μg β2GPI; reduced β2GPI: 20 μg reducedβ2GPI. B and C. Quantification of collagen IV expression in the kidneys was performed using quantitative real-time RT-PCR and western blot. The mRNA and protein expression of collagen IV in kidneys were significantly increased after STZ induction in the DM group and were reduced with β2GPI, evidently with reduced β2GPI for 6 weeks. Data are expressed as the mean ± S.E.M. (n=8 for each group). *P<0.05 vs. NC roup, #P<0.05 vs. DM group. N: normal control; DM: diabetic model control; β2GPI: 20 μg β2GPI; reduced β2GPI: 20 μg reduced β2GPI.
β2GPI and reduced β2GPI inhibited the activation of TGF-β1 and p38 MAPK in STZ-induced diabetic mice
TGF-β1 mRNA and protein expression were significantly higher in the diabetic control group compared with the normal control group (Figure 4A and 4B). Although TGF-β1 mRNA expression was higher than the normal control group after treatment, it was lower than the diabetic control group. Meanwhile, the β2GPI and reduced β2GPI treated diabetic mice had decreased TGF-β1 protein expression compared with the diabetic and normal control groups. The reduction of TGF-β1 expression was most evident in the diabetic mice treated with reduced β2GPI for 6 weeks. In addition, the phosphorylation level of p38 MAPK significantly increased in the diabetic control group, whereas β2GPI and reduced β2GPI treatment inhibited p38 MAPK phosphorylation (Figure 4C). It was noted that the phosphorylation level of p38 MAPK in treated diabetic mice was lower than the normal control group.
Figure 4.
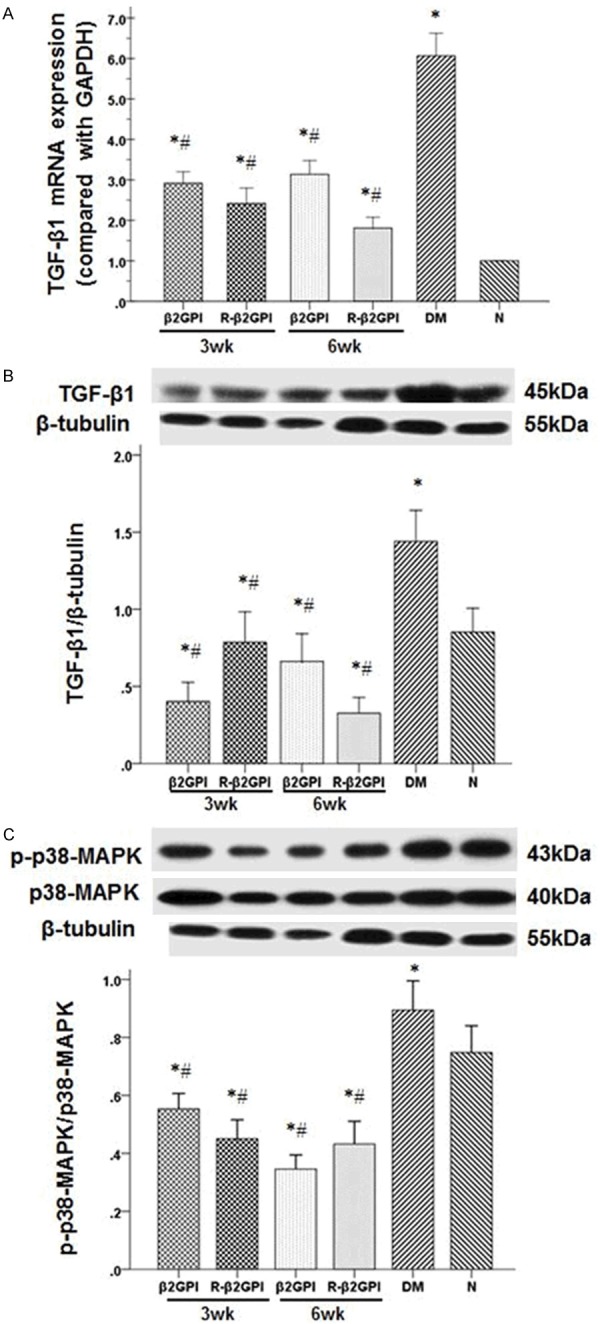
Treatment with β2GPI and reduced β2GPI inhibited the activation of TGF-β1 and p38 MAPK in STZ-induced diabeti mice. A and B. Quantification of TGF-β1 expression in the kidneys was performed using quantitative real-time RT-PCR and western blot. The mRNA and protein expression of TGF-β1 in kidneys were significantly increased after STZ induction in the DM group and were reduced with β2GPI, evidently with reduced β2GPI fo 6eeks. C. Quantification of p38MAPK and phospho-p38 MAPK expression in the kidneys was performed using western blot. The phosphorylation level of p38 MAPK significantly increased in the diabetic control group, whereas β2GPI and reduced β2GPI treatment inhibited p38 MAPK phosphorylation. Data are expressed as the mean ± S.E.M. (n=8 for each group). *P<0.05 vs. group, #P<0.05 vs. DM group. N: normal control; DM: diabetic model control; β2GPI: 20 μg β2GPI; reduced β2GPI: 20 μg reduced β2GI.
β2GPI and reduced β2GPI decreased the expression of collagen IV in high glucose-induced RMCs
Collagen IV mRNA and protein expression were significantly increased after high glucose stimulation for 24 hours (Figure 5A and 5B). These elevated expression levels decreased after β2GPI and reduced β2GPI treatment (P<0.05). As a hyperosmotic control, the mannitol group demonstrated no difference compared with the normal glucose group. Incredibly, the protein expression of collagen IV in β2GPI and reduced β2GPI groups were lower than the normal glucose group. Furthermore, collagen IV mRNA expression was slightly lower in the high-dose of β2GPI and reduced β2GPI compared with the low-dose groups. Collagen IV protein expression was lower in the high-dose reduced β2GPI group compared with the low-dose group.
Figure 5.
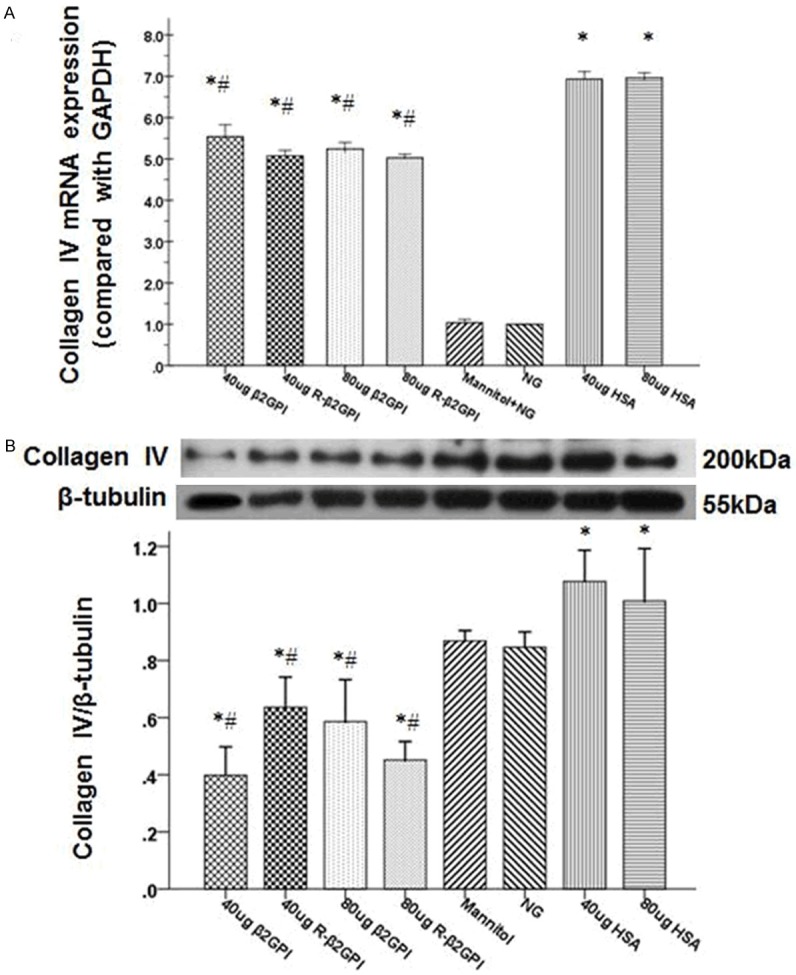
Treatment with β2GPI and reduced β2GPI decreased the expression of collagen IV in high glucose-indued RMCs. A and B. Quantification of collagen IV expression in high glucose-induced RMCs was performed using quantitative real-time RT-PCR and western blot. The mRNA and protein expression of collagen IV were significantly increased after high glucose stimulation. However, the elevated expression levels decreased after β2GPI and reduced β2GPI treatment with high glucose stimulation. Data are expressed as the mean ± S.E.M.. *P<0.05 vs. normal gluose group, #P<0.05 vs. high glucose congtrol group. NG: normal glucose group; Mannitol: osmotic control group; HSA: high glucose with HSA as high glucose control group; β2GPI: high glucose with β2GPI; reduced β2GPI: high glucose with reduced 2GPI.
β2GPI and reduced β2GPI inhibited the activation of TGFβ1 and p38 MAPK in high glucose-induced RMCs
High glucose stimulation increased TGF-β1 mRNA and protein expression compared with the normal glucose and mannitol groups (Figure 6A and 6B). Although TGF-β1 mRNA expression was higher than the normal glucose and mannitol group after β2GPI and reduced β2GPI treatment, it was lower than the high glucose group. TGF-β1 mRNA expression levels were significantly decreased with a high-dose of β2GPI and reduced β2GPI compared with a low-dose groups. Furthermore, TGF-β1 protein expression was significantly lower after β2GPI and reduced β2GPI treatment compared with the normal glucose group. In addition, the phosphorylation level of p38 MAPK significantly increased after high glucose stimulation. It was noted that p38 MAPK phosphorylation levels in the high glucose-induced RMCs treated with reduced β2GPI were lower than the normal glucose group, whereas treatment with β2GPI slightly inhibited p38 MAPK phosphorylation compared with the normal glucose group (Figure 6C).
Figure 6.
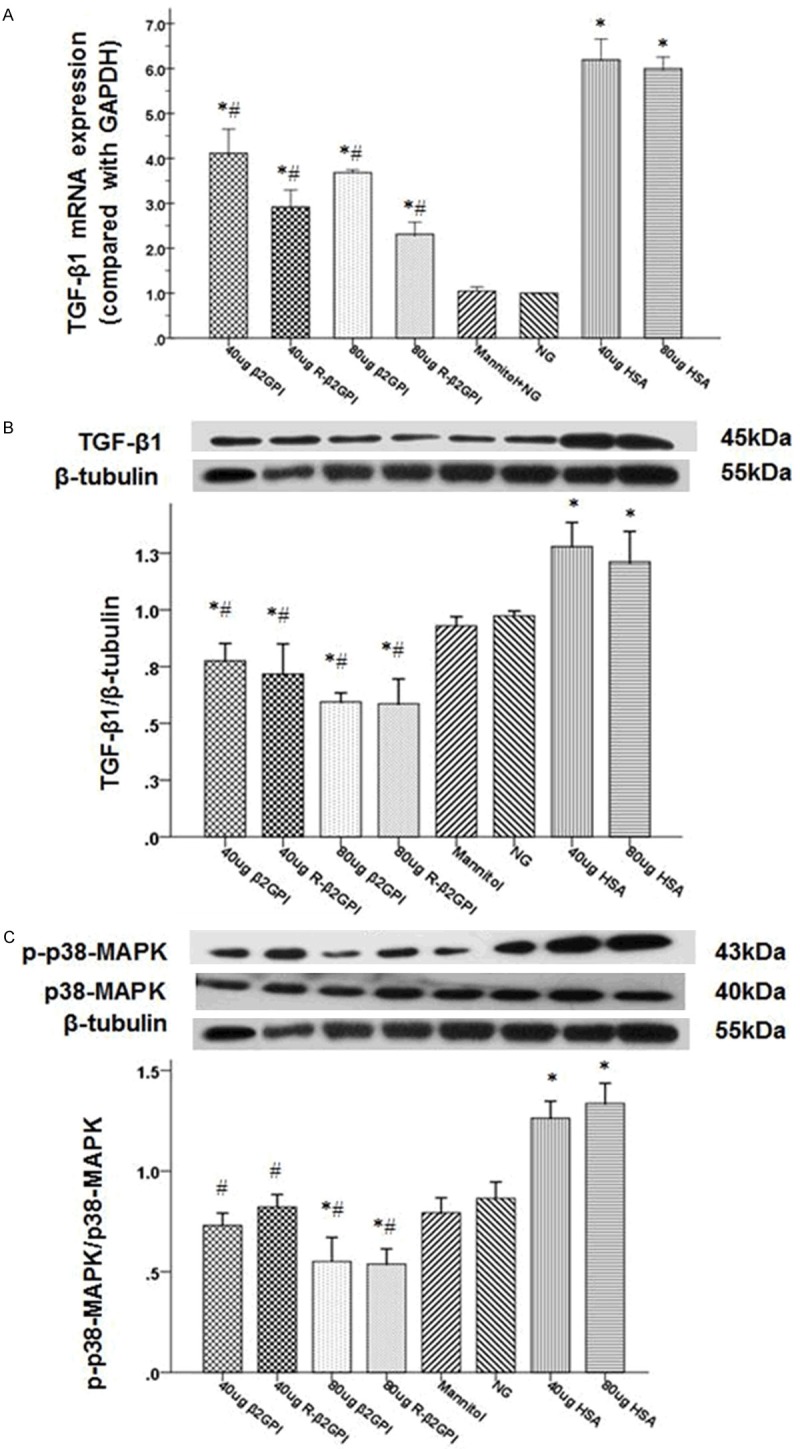
Treatment with β2GPI and reduced β2GPI inhibited the activation of TGF-β1 and p38 MAPK in high glucose-iduced RMs. A and B. Quantification of TGF-β1 expression in high glucose-induced RMCs was performed using quantitative real-time RT-PCR and western blot. The mRNA and protein expression of TGF-β1 were significantly increased after high glucose stimulation and were significantly decreased with a high-dose of β2GPI and reduced β2GPI compared witha ow-dose. C. Quantification of p38MAPK and phospho-p38 MAPK expression in high glucose-induced RMCs was performed using western blot. The phosphorylation level of p38 MAPK significantly increased after high glucose stimulation, whereas β2GPI and reduced β2GPI treatment inhibited p38 MAPK phosphorylation. Data are expressed as the mean ± S.E.M. (n=8 for each group). *P<0.05 v. N group, #P<0.05 vs. DM group. N: normal control; DM: diabetic model control; β2GPI: 20 μg β2GPI; reduced β2GPI: 20 μg reduced β2GPI.
Discussion
In the present study, we demonstrated that diabetic mice induced by a high-fat diet and STZ exhibited a number of early clinical and pathological characteristics of DN. Treatment with β2GPI and reduced β2GPI improved the above changes with time dependently, indicating their renoprotective effects during DN development. In vivo and in vitro studies have demonstrated that hyperglycemia can induce enhanced synthesis of ECM proteins such as collagen and fibronectin [22-24]. In this study, we determined that collagen IV expression was significantly increased in STZ-induced diabetic mice and high glucose-induced RMCs, which is consistent with previous reports. However, treatment with β2GPI and specifically reduced β2GPI effectively decreased collagen IV expression, indicating that β2GPI and reduced β2GPI could improve kidney fibrosis and glomerulosclerosis through the suppression of ECM proteins synthesis.
TGF-β1 has been recognized as an important inflammatory cytokine in DN as previously mentioned. The inhibition of TGF-β1 expression enhances DN treatment by suppressing matrix accumulation. Therefore, TGF-β1 has been proposed as an intervention target for DN treatment [25]. In the present study, the STZ-induced diabetic mice and the high glucose-induced RMCs had increased TGF-β1 mRNA and protein expression. However, treatment with β2GPI and reduced β2GPI could inhibit TGF-β1 mRNA and protein expression.
The activation of p38 MAPK, one of the downstream effectors of the TGF-β1 signaling cascades, has been reported to be involved in the progression of DN [26]. The effects of β2GPI and reduced β2GPI on TGF-β1-p38 MAPK activation in DN have not been elucidated. The phosphorylation levels of p38 MAPK significantly increased in the present study, which were positively correlated with TGF-β1 protein. The β2GPI and reduced β2GPI treatment decreased p38 MAPK phosphorylation levels as well as TGF-β1 expression in STZ-induced diabetic mice, and similar results were observed in high glucose-induced RMCs.
The proportion of reduced β2GPI is significantly lower in the antiphospholipid syndrome group than that in healthy individuals [27], which suggests that reduced β2GPI may play a protective role in our bodies. Reduced β2GPI was recently found to protect EAhy926 (fusion of HUVECs and the A549 carcinoma cell line) from oxidative stress induced endothelial cell damage and to display increased binding to von Willebrand factor (VWF) than non-reduced β2GPI in vitro [28]. At present, no studies have reported the correlation between β2GPI and reduced β2GPI and DN. Our study first indicated that both β2GPI and reduced β2GPI improved kidney fibrosis and decreased mesangial cells produce collagen IV by inhibiting TGF-β1 and p38 MAPK phosphorylation expression. Although reduced β2GPI have the better effect on kidney fibrosis, β2GPI have the same renoprotective effect. It is possible that β2GPI can switch between an oxidized and reduced state under harsh conditions. As described previously, domain V of β2GPI can be reduced by TRX-1 resulting the functional disulfide bond (Cys288-Cys326) opened, resulting in some functional changes. The opened functional disulfide bond could be anchored to the cells membrane, providing the appropriate interface to react with cell surface proteins such as TGF-β1, resulting inhibiting the activation of TGF-β1-p38 MAPK signal pathway. TRX-1 is ubiquitously expressed and is also present on mesangial cells in DN [29]. Given β2GPI’s high concentration in plasma makes it easily available for reactions with TRX-1, resulting in the reduced formation of β2GPI. Thus, in this study β2GPI and reduced β2GPI have been showed the similar renoprotective and antifibrosis effects.
In summary, the present study confirmed that β2GPI and reduced β2GPI improved renal dysfunction and kidney fibrosis as well as decreased collagen IV and TGF-β1 mRNA and protein expression in STZ-induced diabetic mice and high glucose-induced RMCs. Moreover, the present studies demonstrated that the renoprotective and antifibrosis effects of β2GPI and reduced β2GPI in DN were closely associated with suppressing the activation of the TGF-β1-p38 MAPK pathway. Currently, the function of β2GPI and reduced β2GPI has been poorly understood, the definitive molecular mechanism of β2GPI and reduced β2GPI inhibition of the TGF-β1-p38 MAPK pathway in DN needs to be investigated in future.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 30971393 and No. 81070645), the Tianjin natural science fund (No. 10JCYBJC12000).
Disclosure of conflict of interest
None.
References
- 1.Rossing P. Diabetic nephropathy: worldwide epidemic and effects of current treatment on natural history. Curr Diab Rep. 2006;6:479–483. doi: 10.1007/s11892-006-0083-y. [DOI] [PubMed] [Google Scholar]
- 2.Raptis AE, Viberti G. Pathogenesis of diabetic nephropathy. Exp Clin Endocrinol Diabetes. 2001;109(Suppl 2):S424–S437. doi: 10.1055/s-2001-18600. [DOI] [PubMed] [Google Scholar]
- 3.Seger R, Krebs EG. The MAPK signaling cascade. FASEB J. 1995;9:726–735. [PubMed] [Google Scholar]
- 4.Choi ME, Ding Y, Kim SI. TGF-beta signaling via TAK1 pathway: role in kidney fibrosis. Semin Nephrol. 2012;32:244–252. doi: 10.1016/j.semnephrol.2012.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schultze HE, Heide K, Haupt H. Über ein bisher unbekanntes niedermolekulares Beta2-Globulin des Humanserums. Naturwissenschaften. 1961;48:719. [Google Scholar]
- 6.McNeil HP, Simpson RJ, Chesterman CN, Krilis SA. Anti-phospholipid antibodies are directed against a complex antigen that includes a lipid-binding inhibitor of coagulation: beta 2-glycoprotein I (apolipoprotein H) Proc Natl Acad Sci U S A. 1990;87:4120–4124. doi: 10.1073/pnas.87.11.4120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rioche M, Masseyeff R. Synthesis of plasma beta 2 glycoprotein I by human hepatoma cells in tissue culture. Biomedicine. 1974;21:420–423. [PubMed] [Google Scholar]
- 8.Schwarzenbacher R, Zeth K, Diederichs K, Gries A, Kostner GM, Laggner P, Prassl R. Crystal structure of human beta2-glycoprotein I: implications for phospholipid binding and the antiphospholipid syndrome. EMBO J. 1999;18:6228–6239. doi: 10.1093/emboj/18.22.6228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.de Groot PG, Meijers JC. beta(2)-Glycoprotein I: evolution, structure and function. J Thromb Haemost. 2011;9:1275–1284. doi: 10.1111/j.1538-7836.2011.04327.x. [DOI] [PubMed] [Google Scholar]
- 10.Petri M. Update on anti-phospholipid antibodies in SLE: the Hopkins’ Lupus Cohort. Lupus. 2010;19:419–423. doi: 10.1177/0961203309360541. [DOI] [PubMed] [Google Scholar]
- 11.Agar C, de Groot PG, Marquart JA, Meijers JC. Evolutionary conservation of the lipopolysaccharide binding site of beta(2)-glycoprotein I. Thromb Haemost. 2011;106:1069–1075. doi: 10.1160/TH11-05-0333. [DOI] [PubMed] [Google Scholar]
- 12.Jankowski M, Vreys I, Wittevrongel C, Boon D, Vermylen J, Hoylaerts MF, Arnout J. Thrombogenicity of beta 2-glycoprotein I-dependent antiphospholipid antibodies in a photochemically induced thrombosis model in the hamster. Blood. 2003;101:157–162. doi: 10.1182/blood-2002-05-1310. [DOI] [PubMed] [Google Scholar]
- 13.Arad A, Proulle V, Furie RA, Furie BC, Furie B. beta(2)-Glycoprotein-1 autoantibodies from patients with antiphospholipid syndrome are sufficient to potentiate arterial thrombus formation in a mouse model. Blood. 2011;117:3453–3459. doi: 10.1182/blood-2010-08-300715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nilsson M, Wasylik S, Morgelin M, Olin AI, Meijers JC, Derksen RH, de Groot PG, Herwald H. The antibacterial activity of peptides derived from human beta-2 glycoprotein I is inhibited by protein H and M1 protein from Streptococcus pyogenes. Mol Microbiol. 2008;67:482–492. doi: 10.1111/j.1365-2958.2007.05974.x. [DOI] [PubMed] [Google Scholar]
- 15.Vaarala O. Antiphospholipid antibodies and atherosclerosis. Lupus. 1996;5:442–447. doi: 10.1177/096120339600500522. [DOI] [PubMed] [Google Scholar]
- 16.Staub HL, Franck M, Ranzolin A, Norman GL, Iverson GM, von Muhlen CA. IgA antibodies to beta2-glycoprotein I and atherosclerosis. Autoimmun Rev. 2006;6:104–106. doi: 10.1016/j.autrev.2006.06.014. [DOI] [PubMed] [Google Scholar]
- 17.de Laat B, de Groot PG, Derksen RH, Urbanus RT, Mertens K, Rosendaal FR, Doggen CJ. Association between beta2-glycoprotein I plasma levels and the risk of myocardial infarction in older men. Blood. 2009;114:3656–3661. doi: 10.1182/blood-2009-03-212910. [DOI] [PubMed] [Google Scholar]
- 18.Ioannou Y, Zhang JY, Passam FH, Rahgozar S, Qi JC, Giannakopoulos B, Qi M, Yu P, Yu DM, Hogg PJ, Krilis SA. Naturally occurring free thiols within beta 2-glycoprotein I in vivo: nitrosylation, redox modification by endothelial cells, and regulation of oxidative stress-induced cell injury. Blood. 2010;116:1961–1970. doi: 10.1182/blood-2009-04-215335. [DOI] [PubMed] [Google Scholar]
- 19.Yu P, Passam FH, Yu DM, Denyer G, Krilis SA. Beta2-glycoprotein I inhibits vascular endothelial growth factor and basic fibroblast growth factor induced angiogenesis through its amino terminal domain. J Thromb Haemost. 2008;6:1215–1223. doi: 10.1111/j.1538-7836.2008.03000.x. [DOI] [PubMed] [Google Scholar]
- 20.Lin KY, Wang HH, Lai ST, Pan JP, Chiang AN. beta(2)-glycoprotein I protects J774A. 1 macrophages and human coronary artery smooth muscle cells against apoptosis. J Cell Biochem. 2005;94:485–496. doi: 10.1002/jcb.20314. [DOI] [PubMed] [Google Scholar]
- 21.Zhang R, Zhou SJ, Li CJ, Wang XN, Tang YZ, Chen R, Lv L, Zhao Q, Xing QL, Yu DM, Yu P. C-reactive protein/oxidised low-density lipoprotein/beta2-glycoprotein I complex promotes atherosclerosis in diabetic BALB/c mice via p38mitogen-activated protein kinase signal pathway. Lipids Health Dis. 2013;12:42. doi: 10.1186/1476-511X-12-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Noh H, Ha H, Yu MR, Kang SW, Choi KH, Han DS, Lee HY. High glucose increases inducible NO production in cultured rat mesangial cells. Possible role in fibronectin production. Nephron. 2002;90:78–85. doi: 10.1159/000046318. [DOI] [PubMed] [Google Scholar]
- 23.Yu Y, Lyons TJ. A lethal tetrad in diabetes: hyperglycemia, dyslipidemia, oxidative stress, and endothelial dysfunction. Am J Med Sci. 2005;330:227–232. doi: 10.1097/00000441-200511000-00005. [DOI] [PubMed] [Google Scholar]
- 24.Mahimainathan L, Das F, Venkatesan B, Choudhury GG. Mesangial cell hypertrophy by high glucose is mediated by downregulation of the tumor suppressor PTEN. Diabetes. 2006;55:2115–2125. doi: 10.2337/db05-1326. [DOI] [PubMed] [Google Scholar]
- 25.McGowan TA, Zhu Y, Sharma K. Transforming growth factor-beta: a clinical target for the treatment of diabetic nephropathy. Curr Diab Rep. 2004;4:447–454. doi: 10.1007/s11892-004-0055-z. [DOI] [PubMed] [Google Scholar]
- 26.Hayashida T, Poncelet AC, Hubchak SC, Schnaper HW. TGF-beta1 activates MAP kinase in human mesangial cells: a possible role in collagen expression. Kidney Int. 1999;56:1710–1720. doi: 10.1046/j.1523-1755.1999.00733.x. [DOI] [PubMed] [Google Scholar]
- 27.Ioannou Y, Zhang JY, Qi M, Gao L, Qi JC, Yu DM, Lau H, Sturgess AD, Vlachoyiannopoulos PG, Moutsopoulos HM, Rahman A, Pericleous C, Atsumi T, Koike T, Heritier S, Giannakopoulos B, Krilis SA. Novel assays of thrombogenic pathogenicity in the antiphospholipid syndrome based on the detection of molecular oxidative modification of the major autoantigen beta2-glycoprotein I. Arthritis Rheum. 2011;63:2774–2782. doi: 10.1002/art.30383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Passam FH, Rahgozar S, Qi M, Raftery MJ, Wong JW, Tanaka K, Ioannou Y, Zhang JY, Gemmell R, Qi JC, Giannakopoulos B, Hughes WE, Hogg PJ, Krilis SA. Redox control of beta2-glycoprotein I-von Willebrand factor interaction by thioredoxin-1. J Thromb Haemost. 2010;8:1754–1762. doi: 10.1111/j.1538-7836.2010.03944.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Advani A, Gilbert RE, Thai K, Gow RM, Langham RG, Cox AJ, Connelly KA, Zhang Y, Herzenberg AM, Christensen PK, Pollock CA, Qi W, Tan SM, Parving HH, Kelly DJ. Expression, localization, and function of the thioredoxin system in diabetic nephropathy. J Am Soc Nephrol. 2009;20:730–741. doi: 10.1681/ASN.2008020142. [DOI] [PMC free article] [PubMed] [Google Scholar]