

Abstract
Lung cancer is the leading cause of cancer death in the world. Schizandrin B (Sch B) is one of the main dibenzocyclooctadiene lignans present in the fruit of Schisandra chinensis (Schisandraceae). Sch B has multiple functions against cancer. The aim of this study was to determine the effect of Sch B on the proliferation, cell cycling, apoptosis and invasion of lung adenocarcinoma A549 cells by MTT, flow cytometry, wound healing and transwell invasion assays. Treatment with Sch B inhibited the proliferation of A549 cells in a dose-dependent manner. Sch B induced cell cycle arrest at G0/G1 phase by down-regulating the expression of cyclin D1, cyclin-dependent kinase (CDK)4, and CDK6, but up-regulating p53 and p21 expression in A549 cells. Furthermore, Sch B triggered A549 cell apoptosis by increasing Bax, cleaved caspase-3, 9, Cyto C, but decreasing Bcl-2 and PCNA expression. In addition, Sch B inhibited the invasion and migration of A549 cells by down-regulating the expressions of HIF-1, VEGF, MMP-9 and MMP-2. Therefore, Sch B has potent anti-tumor activity and may be a promising traditional Chinese medicine for human lung carcinoma.
Keywords: Schizandrin B, proliferation, cell cycling, invasion, migration, A549 cells
Introduction
Lung cancer is the leading cause of cancer mortality worldwide, and affects nearly 1.2 million deaths annually [1]. Currently, lung adenocarcinoma, one subtype of the non-small cell lung cancer (NSCLC), has become the most common histologic type of the lung cancers diagnosed [2]. Besides surgical resection of the tumor, platinum based therapeutic protocol is the standard chemotherapy for NSCLC, although target therapies, such as the epidermal growth factor receptor tyrosine kinase inhibitors (EGFR-TKIs), have been used for NSCLC patients. However, the therapeutic efficacy is limited due to diverse responses to these therapies. Furthermore, chemotherapy related severe side effects and resistance remains challenge, and results in a low rate of five-year survival in NSCLC patients [3]. Therefore, it is urgent for searching effective and safe drugs to improve the outcome of patients with NSCLC.
Schisandrin B (Sch B) is the most abundant dibenzo cyclooctadiene lignans present in the traditional Chinese medicinal herb Schisandra chinensis (Turcz.) Baill. A previous study has shown that Sch B can protect against oxidative stress-induced injury, including carbon tetrachloride-induced hepatotoxicity [4], myocardial ischemia/reperfusion injury [5], and brain oxidative damage [6]. Sch B is also capable of preventing and protecting from doxorubicin-induced acute and chronic cardiotoxicity [7,8]. In addition, Sch B has been revealed to possess multiple functions against cancers. Sch B enhances doxorubicin-induced apoptosis in cancer cells, but does not enhance its toxicities against healthy cells [7,9]. Sch B can also attenuate the invasion and metastasis of cancer [10]. However, little is known about the effects of Sch B on the proliferation, cell cycling, survival and invasion of NSCLC cells.
In this study, we investigated the anti-tumor activity of Sch B against human lung adenocarcinoma A549 cells and explored the potential mechanisms underlying the action of Sch B in regulating the proliferation, cell cycling, survival and invasion of A549 cells in vitro. Our findings indicated that Sch B had potent anti-tumor activity by inhibiting the proliferation, inducing cell cycle arrest at G0/G1 phrase, triggering cell apoptosis and attenuating cell invasion in A549 cells. Therefore, our findings may provide a new basis for the design of anti-NSCLC therapies.
Materials and methods
Materials
Sch B was purchased from Shanghai Yuanye Bio-Technology (Shanghai, China); Fetal calf serum (FCS) and 0.25% trypsin were obtained from Gibco BRL, (Grand Island, NY, USA); Trizol reagent, and real time polymerase chain reaction (RT-PCR) kit were purchased from Promega (Madison, WI, USA); 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT) was purchased from Sigma Chemical (St. Louis, MO, USA). The annexin-V/propidium iodide (PI) apoptosis detection kit was purchased from Tianjin Sungene Biotech (Tianjin, China). Antibodies against cleaved caspase-3, caspase-9, Bax, Bcl-2, cyclin D1, Cyclin-dependent kinase (CDK)4, CDK6, p53, p21, proliferating cell nuclear antigen (PCNA), matrix metalloproteinase (MMP)-2, MMP-9, hypoxia inducible factor (HIF)-1, vascular endothelial growth factor (VEGF) or β-actin were from Cell Signaling Technology (Beverly, MA, USA).
Cell line and culture
Human adenocarcinoma A549 cells were purchased from the Chinese Academy of Medical Sciences (Beijing, China). The cells were cultured in RPMI-1640 (Gibco BRL, Carlsbad, CA, USA) supplemented with 10% FCS and 100 units/mL penicillin and 0.1 mg/mL streptomycin (Gibco) at 37°C in a humidified atmosphere containing 5% CO2.
Cell proliferation assay
The effect of Sch B on the proliferation of A549 cells was examined by MTT assay. Briefly, A549 cells (5 × 103 cells/well) were cultured in 96-well plates overnight and treated in triplicate with Sch B at varying concentrations (0, 12.5, 25, or 50 µM) for 72 hours. During the last four-hour incubation, the cells were exposed to MTT (20 µl/well, 5 mg/ml). The resulting formazan was dissolved in 150 µL dimethyl sulfoxide (DMSO) and measured for the absorbance at 490 nm using a SpectraMax 190 Microplate Reader (Molecular Devices, Sunnyvale, CA, USA). The inhibitory rates of Sch B in cell proliferation were calculated by the formula of (optical density (OD) of controls-OD of experiments)/OD of controls. In addition, the morphology of individual groups of cells at 72 hours post treatment with Sch B was examined under a phase contrast microscope (CKX41, Olympus, Japan).
Colony formation assay
A549 cells (500 cells/well) were cultured in 6-well plates for six hours and treated in duplicate with Sch B (0, 12.5, 25, or 50 µM) for 72 h, followed by culturing for 14 days. The formed cell clones were stained with crystal violet in 50% methanol and 10% acetic acid, and the stained clones that had > 50 cells were counted in a blinded manner. The clone formation efficiency (CFE) was the ratio of the clone number to the planted cell number.
Flow cytometry
A549 cells (1 × 106 cells/well) were cultured in 6-well plates overnight and treated in triplicate with different concentrations (0, 12.5, 25, or 50 µM) of Sch B for 72 hours. The cells were harvested by trypsinization, and were stained with Annexin V-FITC and PI. The percentages of apoptotic cells were analyzed by flow cytometry analysis.
Furthermore, the harvested cells were fixed in 70% ethanol at 4°C overnight and treated with 10 mg/mL RNase and 1 mg/mL PI at 37°C in the dark for 30 min. The distribution of different phases of cells was analyzed by flow cytometry using ModFit LT software.
The cell migration and invasion assays
The impact of Sch B treatment on the invasion and migration of A549 cells was determined by transwell invasion and wound-healing assays, Briefly, A549 cells (5 × 103 cells/well) were cultured in FCS-free RPMI 1640 medium in the top chambers of 24-well transwell plates (0.4 µm, Corning Costar, Cambridge, MA) that had been coated with Matrigel (BD Biosciences) and the lower chambers were filled with RPMI-1640 supplemented with 10% FCS as a chemo-attractant for 72 hours. The different concentrations of Sch B (0, 12.5, 25 or 50 µM) were added in both the top and bottom chambers. The cells invaded on the bottom of the top chamber were stained crystal violet in 2% ethanol and counted at least 10 fields under the view of a phase-contrast microscope.
In addition, A549 cells (1 × 105 cells/well) were starved in 2% FCS medium in 24-well plates overnight and scratched with a small tip along the ruler. The scratched areas were washed with PBS to remove free cells and the cells were cultured in complete medium containing different concentrations of Sch B (0, 12.5, 25 or 50 µM) for 72 hours, followed by longitudinally photoimaging under a phase-contrast microscope. The distance between two edges of cultured cells was measured longitudinally.
Western blot analysis
A549 cells were treated with various concentrations of Sch B (0, 12.5, 25 or 50 µM) for 72 hours. The cells were harvested and lysed in a lysis buffer. After quantification of protein concentrations using a Bio-Rad detergent-compatible protein assay kit (Bio-Rad), the cell lysates (50-70 µg proteins/lane) were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) on 10% gels and transferred onto nitrocellulose membranes. Subsequently, the membranes were blocked with 5% (w/v) non-fat dry milk in Tris-buffered saline for 2 hours at room temperature and probed with primary antibodies (1:500 dilutions) for 2 hours at room temperature. Anti-β-actin served as the control. After being washed, the bound antibodies were detected with peroxidase-conjugated anti-rabbit or anti-mouse IgG and visualized using the enhanced chemiluminescent (ECL) detection reagent (Millipore, Billerica, MA). The relative levels of each target protein to the control were determined by densitometric scanning and quantitatively calculated using the ImageJ software.
Quantitative RT-PCR
A549 cells (5 × 105/well/well) were treated with, or without, Sch B for 72 hours and total RNA was extracted from individual groups of cells using TRIzol reagent (Invitrogen), followed by reversely transcribing into cDNA using an RT-PCR kit (Promega, Madison, WI, USA). The relative levels of target gene mRNA transcripts to the control GAPDH were determined by quantitative RT-PCR (qRT-PCR) on a Stratagene M x 3000P Real-Time PCR System (Stratagene, La Jolla, CA, USA) using the FastStart Universal SYBR Green Master reagent (Roche, Mannheim, Germany) and specific primers (Table 1). The relative levels of mRNA target gene mRNA transcripts to the control GAPDH were calculated by the 2-∆∆Ct.
Table 1.
Primer sequences used for qPCR analyses
| Gene | Forward primer | Reverse primer |
|---|---|---|
| GAPDH | 5’-AGAAGGCTGGGGCTCATTTG-3’ | 5’-AGGGGCCATCCACAGTCTTC-3’ |
| p53 | 5’-CCATCTACAAGCAGTCACAG-3’ | 5’-CAAATCTACAAGCAGTCACAG-3’ |
| p21 | 5’-ACTTCGACTTTGTCACCGAGA-3’ | 5’-GAGGCACAAGGGTACAAGACA-3’ |
| MMP-9 | 5’-CGAACTTCGACACTGACAAGAAGT-3’ | 5’-GCACGCTGGAATGATCTAAGC-3’ |
| MMP-2 | 5’-CTGGGTTTACCCCCTGATGTCC-3’ | 5’-AACCGGGGTCCATTTTCTTCTTT-3’ |
| Caspase-3 | 5’-GAAACCTCCGTGGATTCAAA-3’ | 5’-AGCCCATTTCAGGGTAATCC-3’ |
| Caspase-9 | 5’-TCCTGGTACATCGAGACCTTG-3’ | 5’-AAGTCCCTTTCGCAGAAACAG-3’ |
| Bax | 5’-ACCAAGAAGCTGAGCGAGTG-3’ | 5’-CCCAGTTGAAGTTGCCATCA-3’ |
| Bcl-2 | 5’-ACGACTTCTCCCGCCGCTAC -3’ | 5’-CCCAGCCTCCGTTATCCTG -3’ |
| PNCA | 5’-TCAAGAAGGTGTTGGAGGCA -3’ | 5’-TCGCAGCGGTAGGTGTCG -3 |
| Cyclin D1 | 5’-AGTTGCTGCAAATGGAACTG-3’ | 5’-AAAGGTCTGTGCATGTTTGC -3’ |
| CDK4 | 5’-TTTGATCTCATTGGATTGCC-3’ | 5’-AGGTCAGCATTTCCAGCAG -3’ |
| CDK6 | 5’-TGGAGTGTTGGCTGCATATT -3’ | 5’-ACAGGGCACTGTAGGCAGAT -3’ |
| HIF-1α | 5’-CCACAGGACAGTACAGGATG -3’ | 5’-TCAAGTCGTGCTGAATAATACC -3’ |
| VEGF | 5’-AAACCCTGAGGGAGGCTCC -3’ | 5’-TACTTGCAGATGTGACAAGCCG -3’ |
| Cytochrome c | 5’-ACAGGAGCCCTATCAGCTCT -3’ | 5’-AAAATCCGGCAAAGAAGAAT -3’ |
Statistical analysis
Data were analyzed using the SPSS 17.0 software. Unless noted, data are given as the mean ± SD. Groups were compared by one-way analysis of variance (ANOVA) and post hoc Bonferroni multiple comparisons test and Student’s t-test. A P value of < 0.05 was considered statistically significant.
Results
Sch B inhibits the proliferation and changes the morphology of A549 cells
The impact of Sch B on the proliferation of A549 cells was determined by MTT assay (Figure 1A). Treatment with 12.5 or 50 µM of Sch B for 48 hours significantly inhibited the proliferation of A549 cells (P < 0.05) and treatment with different doses of Sch B for varying periods inhibited the proliferation of A549 cells in a dose- and time-dependent manner. Morphologically, treatment with Sch B for 72 hours dramatically changed the morphology of A549 cells and many cells lost their adhesion and spindle shapes (Figure 1B). Hence, treatment with Sch B significantly inhibited the proliferation of A549 cells and changed their morphology.
Figure 1.
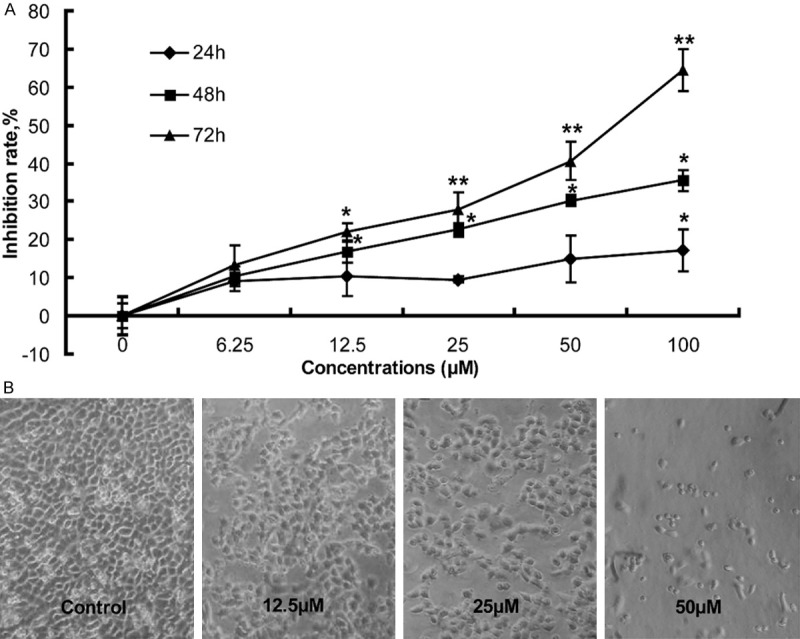
The effects of Sch B on the proliferation (A) and morphology of A549 cells (B). A549 cells were treated in triplicate with, or without, the indicated concentrations of Sch B for varying periods and their effects on the survival of A549 cells were determined by MTT, followed by calculating the inhibition rates (%). The inhibition rates of control cells without Sch B treatment were designated as 0%. Simultaneously, some cells were photo-imaged under a microscope. Data are representative images (magnification x 100) and expressed as the means ± SD of the inhibition of Sch B on the survival of A549 cells in individual groups of cells from three separate experiments. *P < 0.05, **P < 0.01 vs. the control, determined by ANOVA and post hoc Bonferroni multiple comparisons test.
Sch B inhibits the clonal formation of A549 cells
A549 cells were treated in triplicate with, or without, different concentrations of Sch B for three days and further cultured for 14 days. The cell clones were photo-imaged under a microscope and the clonal formation efficacy was calculated (Figure 2). First, treatment with Sch B obviously reduced the clonal size and numbers (Figure 2A, 2B). Quantitative analysis of individual clones with > 50 cells indicated that treatment with 25 µM Sch B significantly reduced the clonal formation efficacy of A549 cells (18.6 ± 3.6% vs. 45.5% ± 9.5%, P < 0.05) and treatment with 50 µM Sch B further reduced the clonal formation efficacy to 8.5 ± 7.1% (Figure 2C). These data indicated that treatment with Sch B inhibited the clonal formation of A549 cells in a dose-dependent manner.
Figure 2.
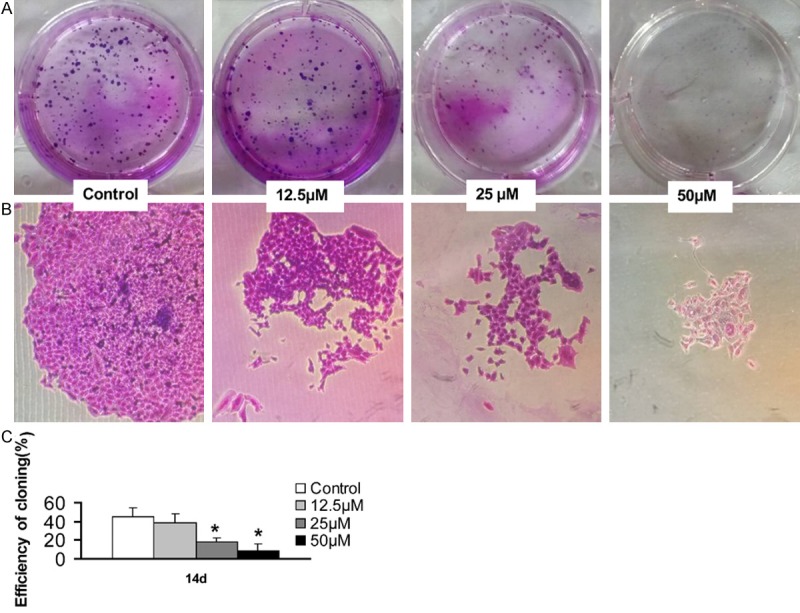
Sch B inhibits the clonal formation of A549 cells. A549 cells were treated in triplicate with, or without, the indicated concentrations of Sch B for three days and cultured for 14 days. The formed cell clones were photo-imaged and the clonal formation efficacy of A549 cells was quantitatively analyzed. Data are representative images (magnification x 100 or 400) and expressed as the means ± SD of each group of cells from three independent experiments. The clonal formation efficacy of cells without Sch B treatment was designated as 0%. *P < 0.05 vs. the control, determined by Student T test.
Sch B induces the apoptosis of A549 cells
To understand the potential mechanisms underlying the action of Sch B in inhibiting A549 cell proliferation, A549 cells were treated, or without, different concentrations of Sch B for 72 hours and the cells were stained with FITC-Annexin V and PI, followed by flow cytometry analysis. As shown in Figure 3A, the percentages of apoptotic cells in the Sch B-treated groups were significantly higher than that in the control group (P < 0.05). Furthermore, treatment with Sch B significantly increased the levels of cleaved caspase 3 and 9, Bax and cyto c expression, but decreased the levels of BcL-2 and PCNA expression in A549 cells. Collectively, treatment with Sch B triggered the apoptosis of A549 cells.
Figure 3.
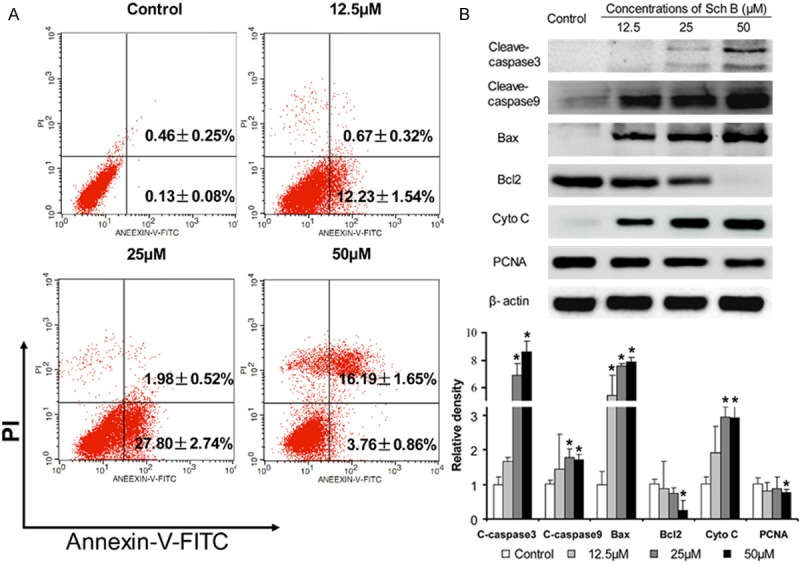
Treatment with Sch B induces the apoptosis of A549 cells. A549 cells were treated with, or without, different concentrations of Sch B for 72 h and stained with Annexin V-FITC/PI, followed by flow cytometry. The relative levels of target protein expression were determined by Western blot assay. Data are representative charts, images, and expressed as the mean ± SD of individual groups of cells from three independent experiments. A. Flow cytometry analysis of the percentages of apoptotic cells; B. Western blot analysis. *P < 0.05 vs. the control, determined by ANOVA and post hoc Bonferroni multiple comparisons test.
Sch B induces cell cycle arrest at G0/G1 phase in A549 cells
Next, the impact of Sch B treatment on the distribution of different phases of cell cycling was determined. A549 cells were treated with, or without, different concentrations of Sch B for 72 hours, fixed with 70% ethanol and treated with RNase I and PI. The distribution of different phases of cells was analyzed by flow cytometry. As shown in Figure 4A and 4B, treatment with Sch B significantly increased the percentages of G0/G1 phase of cells, indicating that treatment with Sch B induced cell cycle arrest at G0/G1 phase. Analysis of the relative levels of cell cycle regulator expression indicated that treatment with Sch B significantly reduced the relative levels of CDK4, CDK6, cyclin D1, but elevated the relative levels of p53 and p21 expression in A549 cells (Figure 4C, 4D). These data demonstrated that treatment with Sch B induced cell cycle arrest at G0/G1 phase.
Figure 4.
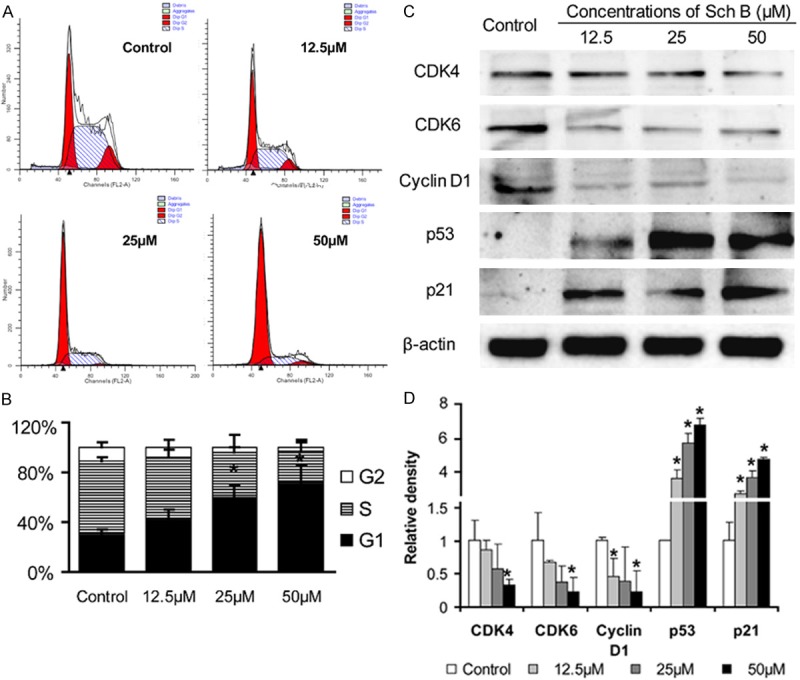
Treatment with SchB induces cell cycle arrest at G0/G1 phase. A549 cells were treated with, or without, different concentrations of Sch B for 72 hours, fixed, and treated with RNase I and PI. The distribution of different phases of cells was determined by flow cytometry. Data are representative histograms, images and expressed as the means ± SD of individual groups of cells from three independent experiments. A. Flow cytometry analysis. B. Quantitative analysis. C. Western blot analysis. D. Quantitative analysis of the relative levels of target protein to the GAPDH. *P < 0.05 vs. the control, determined by ANOVA and post hoc Bonferroni multiple comparisons test.
Sch B inhibits the invasion and migration of A549 cells
The effects of Sch B treatment on the migration and invasion of A549 cells were determined by the wound healing and transwell invasion assays. As shown in Figure 5A, treatment with Sch B inhibited the migration of A549 cells and the effects of Sch B treatment on inhibiting the migration of A549 cells were dose-dependent. Similarly, treatment with Sch B inhibited the invasion of A549 cells in a dose-dependent manner (P < 0.01). Furthermore analyses indicated that treatment with Sch B significantly reduced the relative levels of HIF-1α, VEGF, MMP2 and MMP9 expression, particularly with high concentration of Sch B in A549 cells.
Figure 5.
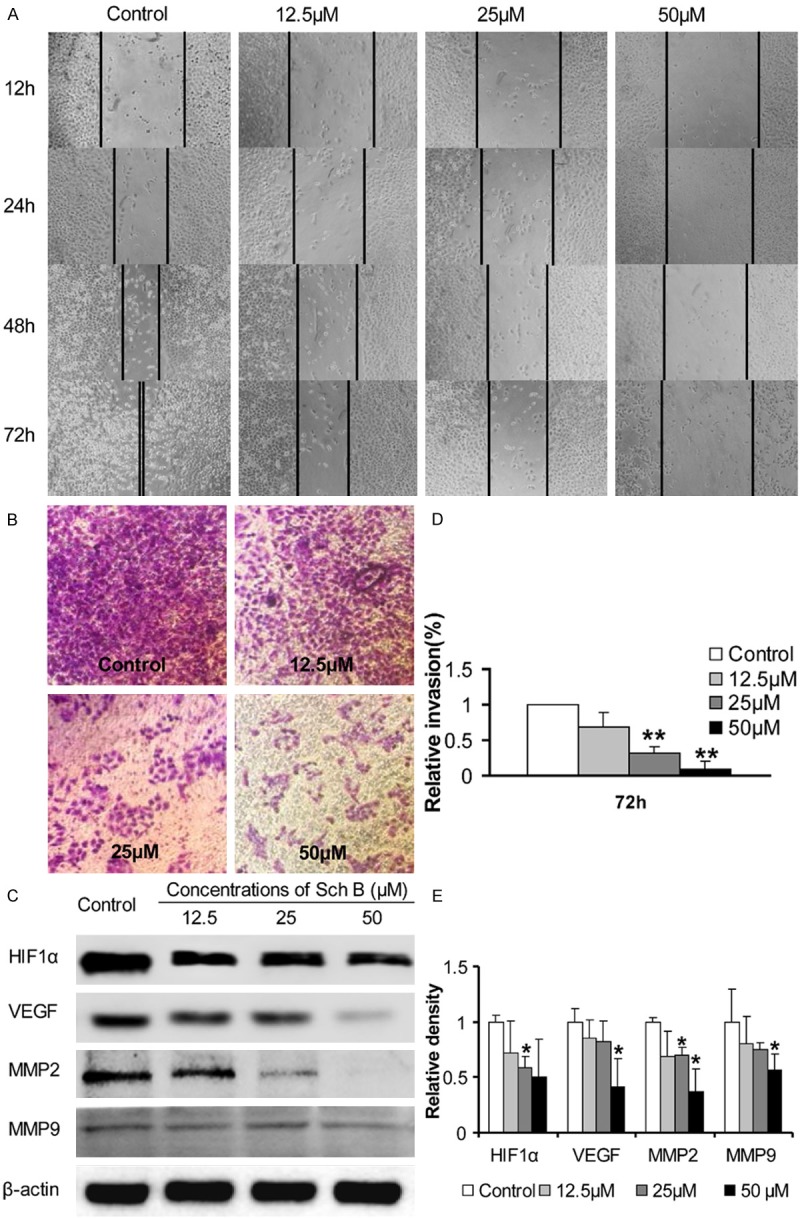
The effects of Sch B on the invasion and migration of A549 cells by inhibiting migration-related regulator expression. A549 cells were cultured, scratched, treated with different concentrations of Sch B. The distance between two edges was photo-imaged longitudinally. Furthermore, A549 cells were cultured in the top chambers that had been coated with matrigel and tested for their invasive responses to complete medium in the bottom chambers of 24-well transwell plates. Finally, the cells were harvested at 72 hours post treatment and the relative levels of HIF-1α, VEGF, MMP2 and MMP9 expression in individual groups of cells were determined by Western blot assays. Data are representative images and expressed as the means ± SD of individual groups of cells from three independent experiments. A. Photo-images of the wound healing areas. B. Photo-images of the invaded cells. C. Quantitative analysis of the numbers of invaded cells. D. Western blot analysis. E. Quantitative analysis of the relative levels of target proteins. *P < 0.05, **P < 0.01 vs. the control, determined by ANOVA and post hoc Bonferroni multiple comparisons test.
Sch B modulates the relative levels of regulator gene mRNA transcripts in A549 cells
Finally, the relative levels of different regulator gene mRNA transcripts to GAPDH were determined quantitative RT-PCR. Treatment with Sch B up-regulated the relative levels of caspase 3, caspase 9, p53, cyto C and Bax mRNA transcripts in A549 cells (P < 0.05, Figure 6). In contrast, treatment with Sch B significantly reduced the relative levels of Bcl-2 and PCNA mRNA transcripts in A549 cells (P < 0.05). These data further indicated that treatment with Sch B induced the apoptosis of A549 cells. Furthermore, treatment with Sch B significantly reduced the relative levels of CDK4, CDK6, cyclin D1, but elevated the level of p27 mRNA transcripts (P < 0.05), indicating that treatment with Sch B induced cell cycle arrest in A549 cells. In addition, treatment with Sch B significantly decreased the relative levels of HIF-1α, VEGF, MMP2 and MMP9 expression (P < 0.05), supporting that treatment with Sch B inhibited the migration and invasion of A549 cells.
Figure 6.
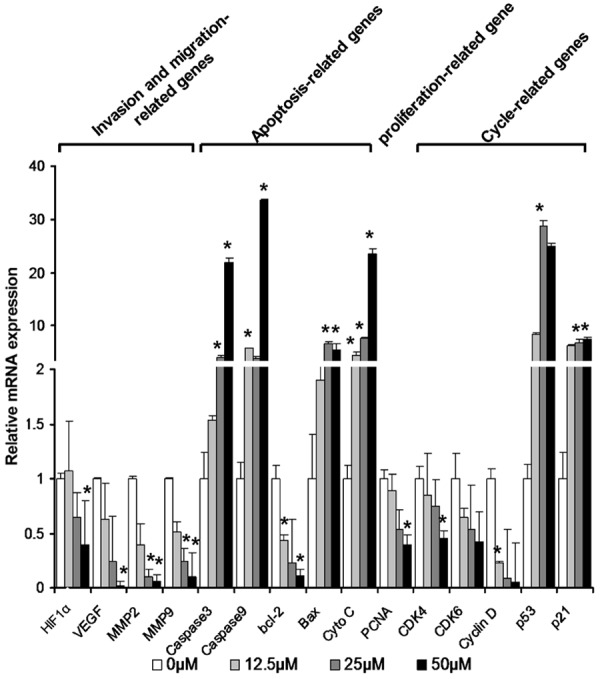
Sch B modulates the relative levels of regulator gene mRNA transcripts in A549 cells. A549 cells were treated with, or without, different concentrations of Sch B for 72 hours and total RNA was extracted from individual groups of cells, followed by reversely transcribing into cDNA. The relative levels of each target gene mRNA transcripts to GAPDH were determined by quantitative RT-PCR. Data are expressed as the means ± SD of individual groups of cells from three independent experiments. The relative levels of gene transcription in the cells without Sch B treatment were designated as 1. *P < 0.05, **P < 0.01 vs. the control, determined by ANOVA and post hoc Bonferroni multiple comparisons test.
Discussion
In this study, we examined the effects of Sch B on the proliferation, cell cycling, survival and invasion of A549 cells in vitro. We found that treatment with Sch B inhibited the proliferation of A549 cells in a dose-dependent manner and changed the morphology of A549 cells in vitro. Furthermore, Sch B treatment significantly reduced the levels of PCNA expression in A549 cells. In addition, Sch B induced cell cycle arrest at G0/G1 phase and triggered cell apoptosis in A549 cells. Cell cycle arrest is associated with inhibition of tumor cell proliferation [11,12]. Cell cycle progression is positively regulated by a series of cyclins and CDKs, but negatively regulated by CDK inhibitors of p21, p16, p27 and others. We found that treatment with Sch B significantly reduced the levels of CDK4, CDK6 and cyclin D1 expression, but elevated the levels of p21 expression in A549 cells. Cyclin D1 is the most outstanding G1-phase cyclin and has been reported as an oncogenic driver in cancer cells [13]. Cyclin-CDKs complexes regulate the cell cycling at various levels, such as the assembly of cyclin and CDK subunits. The G1/S-phase transition is negatively regulated by p21 and p27 through the degradation of Cyclin/CDK complexes [14,15]. The significant inhibition of Sch B on the levels of cyclin D1, CDK4 and CDK6, together with increased levels of p21 expression, should restrict the G1-S transition [16-18]. In addition, we detected significantly higher levels of p53 in the Sch B-treated A549 cells, which is associated with cell cycle arrest and apoptosis [19,20]. Given that p53 is an important regulator of p21 expression [20], it is possible that Sch B treatment may up-regulate p53 expression and in turn elevate the expression of p21, which inhibits the cyclin1/CDK complex, leading to cell cycle arrest at G0/G1 phase in A549 cells. To the best of our knowledge, this was the first study to reveal potent antitumor activity of Sch B in A549 cells. Our novel findings may aid in the design of new therapies for intervention of human NSCLC.
Induction of cell cycle arrest is associated with triggering cell apoptosis, and the cell cycling is usually disrupted in cancer cells [21-23]. To understand the molecular mechanisms underlying the action of Sch B in inhibiting the proliferation of A549 cells, we examined the effect of Sch B on the survival of A549 cells. We found that treatment with Sch B triggered the apoptosis of A549 cells. Cell apoptosis can be regulated positively by the Bcl-2 family members, such as Bax, Bad and others, and negatively regulated by anti-apoptotic Bcl-2, Bcl-xL and others. Apoptotic triggers can activate the mitochondrial pathway by inducing the formation of cytochrome c complex, which activates caspase 9 and in turn caspase 3, leading to cell apoptosis [24,25]. The balance of pro-apoptotic and anti-apoptotic factors is crucial for the development of apoptosis [26]. In this study, we found that Sch B treatment significantly increased the levels of Bax, cleaved caspase 3 and 9, but decreased the levels of Bcl-2 expression in A549 cells. Therefore, Sch B induced A549 cell apoptosis through the activating the mitochondrial pathway.
Cancer cells can migrate and invade the adjacent and distant tissues, which increases the risk for poor outcome of patients with NSCLC [27]. The migration and invasion of cancer cells are regulated by many factors, such as chemokines, their receptors, the EMT process, matrix degrading enzymes and antioxidant enzymes, such as MMPs, which mediate the degradation of extracellular matrix (ECM) proteins [28,29]. Furthermore, the migration and invasion can be modulated by hypoxic condition and angiogenesis regulators, including HIF-1α and VEGF [30,31]. We found that Sch B treatment significantly inhibited the migration and invasion of A549 cells, accompanied by significantly attenuating the expression of HIF-1α, VEGF, MMP2 and MMP9. Given that most tumor cells are highly responding to a hypoxic condition by up-regulating HIF-1α and VEGF to promote angiogenesis the significantly reduced levels of HIF-1α and VEGF, together with significantly decreased levels of MMP2 and MMP9 expression in the Sch B-treated A549 cells, may contribute to its inhibition on the proliferation, migration and invasion of A549 cells.
In conclusion, our data indicated that Sch B treatment significantly inhibited the proliferation of A549 cells by inducing their cell cycle arrest at G0/G1 phase through down-regulating cyclin D1, CDK4 and CDK6, but up-regulating p21 and p53 expression. Sch B treatment triggered the apoptosis of A549 cells by activating the mitochondrial pathway. Furthermore, Sch B treatment significantly inhibited the migration and invasion of A549 cells by reducing the levels of MMP2, MMP9, HIF-1α and VEGF expression. Therefore, our data suggest that Sch B may be a promising candidate for intervention of NSCLC and our findings may provide new insights into understanding the pharmacological mechanisms underlying anti-tumor activity of Sch B.
Acknowledgements
This study was supported by grants from the National Natural Science Foundation of China (number: 81472169, 81241069) and the Project 2014030 supported by Graduate Innovation Fund of Jilin University.
Disclosure of conflict of interest
None.
References
- 1.Sun S, Schiller JH, Gazdar AF. Lung cancer in never smokers--a different disease. Nat Rev Cancer. 2007;7:778–790. doi: 10.1038/nrc2190. [DOI] [PubMed] [Google Scholar]
- 2.Fan Z, Schraeder R. The changing pathology of lung cancer. Surg Oncol Clin N Am. 2011;20:637–653. doi: 10.1016/j.soc.2011.07.004. [DOI] [PubMed] [Google Scholar]
- 3.DeSantis CE, Lin CC, Mariotto AB, Siegel RL, Stein KD, Kramer JL, Alteri R, Robbins AS, Jemal A. Cancer treatment and survivorship statistics, 2014. CA Cancer J Clin. 2014;64:252–271. doi: 10.3322/caac.21235. [DOI] [PubMed] [Google Scholar]
- 4.Chiu PY, Leung HY, Siu AH, Poon MK, Ko KM. Schisandrin B decreases the sensitivity of mitochondria to calcium ion-induced permeability transition and protects against carbon tetrachloride toxicity in mouse livers. Biol Pharm Bull. 2007;30:1108–1112. doi: 10.1248/bpb.30.1108. [DOI] [PubMed] [Google Scholar]
- 5.Chen N, Chiu PY, Ko KM. Schisandrin B enhances cerebral mitochondrial antioxidant status and structural integrity, and protects against cerebral ischemia/reperfusion injury in rats. Biol Pharm Bull. 2008;31:1387–1391. doi: 10.1248/bpb.31.1387. [DOI] [PubMed] [Google Scholar]
- 6.Ko KM, Lam BY. Schisandrin B protects against tert-butylhydroperoxide induced cerebral toxicity by enhancing glutathione antioxidant status in mouse brain. Mol Cell Biochem. 2002;238:181–186. doi: 10.1023/a:1019907316129. [DOI] [PubMed] [Google Scholar]
- 7.Xu Y, Liu Z, Sun J, Pan Q, Sun F, Yan Z, Hu X. Schisandrin B prevents doxorubicin-induced chronic cardiotoxicity and enhances its anticancer activity in vivo. PLoS One. 2011;6:e28335. doi: 10.1371/journal.pone.0028335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li L, Pan Q, Han W, Liu Z, Li L, Hu X. Schisandrin B prevents doxorubicin-induced cardiotoxicity via enhancing glutathione redox cycling. Clin Cancer Res. 2007;13:6753–6760. doi: 10.1158/1078-0432.CCR-07-1579. [DOI] [PubMed] [Google Scholar]
- 9.Li L, Lu Q, Shen Y, Hu X. Schisandrin B enhances doxorubicin-induced apoptosis of cancer cells but not normal cells. Biochem Pharmacol. 2006;71:584–595. doi: 10.1016/j.bcp.2005.11.026. [DOI] [PubMed] [Google Scholar]
- 10.Liu Z, Zhang B, Liu K, Ding Z, Hu X. Schisandrin B attenuates cancer invasion and metastasis via inhibiting epithelial-mesenchymal transition. PLoS One. 2012;7:e40480. doi: 10.1371/journal.pone.0040480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pasetto LM, D’Andrea MR, Brandes AA, Rossi E, Monfardini S. The development of platinum compounds and their possible combination. Crit Rev Oncol Hematol. 2006;60:59–75. doi: 10.1016/j.critrevonc.2006.02.003. [DOI] [PubMed] [Google Scholar]
- 12.Voland C, Bord A, Peleraux A, Penarier G, Carriere D, Galiegue S, Cvitkovic E, Jbilo O, Casellas P. Repression of cell cycle-related proteins by oxaliplatin but not cisplatin in human colon cancer cells. Mol Cancer Ther. 2006;5:2149–2157. doi: 10.1158/1535-7163.MCT-05-0212. [DOI] [PubMed] [Google Scholar]
- 13.Kim JK, Diehl JA. Nuclear cyclin D1: an oncogenic driver in human cancer. J Cell Physiol. 2009;220:292–296. doi: 10.1002/jcp.21791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hedberg Y, Ljungberg B, Roos G, Landberg G. Retinoblastoma protein in human renal cell carcinoma in relation to alterations in G1/S regulatory proteins. Int J Cancer. 2004;109:189–193. doi: 10.1002/ijc.11665. [DOI] [PubMed] [Google Scholar]
- 15.Vermeulen K, Van Bockstaele DR, Berneman ZN. The cell cycle: a review of regulation, deregulation and therapeutic targets in cancer. Cell Prolif. 2003;36:131–149. doi: 10.1046/j.1365-2184.2003.00266.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sancar A, Lindsey-Boltz LA, Unsal-Kacmaz K, Linn S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu Rev Biochem. 2004;73:39–85. doi: 10.1146/annurev.biochem.73.011303.073723. [DOI] [PubMed] [Google Scholar]
- 17.Murray AW. Recycling the cell cycle: cyclins revisited. Cell. 2004;116:221–234. doi: 10.1016/s0092-8674(03)01080-8. [DOI] [PubMed] [Google Scholar]
- 18.Abukhdeir AM, Park BH. P21 and p27: roles in carcinogenesis and drug resistance. Expert Rev Mol Med. 2008;10:e19. doi: 10.1017/S1462399408000744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li L, Dai HJ, Ye M, Wang SL, Xiao XJ, Zheng J, Chen HY, Luo YH, Liu J. Lycorine induces cell-cycle arrest in the G0/G1 phase in K562 cells via HDAC inhibition. Cancer Cell Int. 2012;12:49. doi: 10.1186/1475-2867-12-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chipuk JE, Green DR. Cytoplasmic p53: bax and forward. Cell Cycle. 2004;3:429–431. [PubMed] [Google Scholar]
- 21.Dai HY, Liu L, Qin SK, He XM, Li SY. Lobaplatin suppresses proliferation and induces apoptosis in the human colorectal carcinoma cell Line LOVO in vitro. Biomed Pharmacother. 2011;65:137–141. doi: 10.1016/j.biopha.2010.12.001. [DOI] [PubMed] [Google Scholar]
- 22.Tsuruo T, Naito M, Tomida A, Fujita N, Mashima T, Sakamoto H, Haga N. Molecular targeting therapy of cancer: drug resistance, apoptosis and survival signal. Cancer Sci. 2003;94:15–21. doi: 10.1111/j.1349-7006.2003.tb01345.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Takeda K, Stagg J, Yagita H, Okumura K, Smyth MJ. Targeting death-inducing receptors in cancer therapy. Oncogene. 2007;26:3745–3757. doi: 10.1038/sj.onc.1210374. [DOI] [PubMed] [Google Scholar]
- 24.Park C, Choi YW, Hyun SK, Kwon HJ, Hwang HJ, Kim GY, Choi BT, Kim BW, Choi IW, Moon SK, Kim WJ, Choi YH. Induction of G1 arrest and apoptosis by schisandrin C isolated from Schizandra chinensis Baill in human leukemia U937 cells. Int J Mol Med. 2009;24:495–502. doi: 10.3892/ijmm_00000258. [DOI] [PubMed] [Google Scholar]
- 25.Eichhorn JM, Alford SE, Sakurikar N, Chambers TC. Molecular analysis of functional redundancy among anti-apoptotic Bcl-2 proteins and its role in cancer cell survival. Exp Cell Res. 2014;322:415–424. doi: 10.1016/j.yexcr.2014.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Burlacu A. Regulation of apoptosis by Bcl-2 family proteins. J Cell Mol Med. 2003;7:249–257. doi: 10.1111/j.1582-4934.2003.tb00225.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sleeman J, Steeg PS. Cancer metastasis as a therapeutic target. Eur J Cancer. 2010;46:1177–1180. doi: 10.1016/j.ejca.2010.02.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Verma S, Kesh K, Ganguly N, Jana S, Swarnakar S. Matrix metalloproteinases and gastrointestinal cancers: Impacts of dietary antioxidants. World J Biol Chem. 2014;5:355–376. doi: 10.4331/wjbc.v5.i3.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gencer S, Cebeci A, Irmak-Yazicioglu MB. Matrix metalloproteinase gene expressions might be oxidative stress targets in gastric cancer cell lines. Chin J Cancer Res. 2013;25:322–333. doi: 10.3978/j.issn.1000-9604.2013.06.05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ramakrishnan S, Subramanian IV, Yokoyama Y, Geller M. Angiogenesis in normal and neoplastic ovaries. Angiogenesis. 2005;8:169–182. doi: 10.1007/s10456-005-9001-1. [DOI] [PubMed] [Google Scholar]
- 31.Yamakuchi M, Lotterman CD, Bao C, Hruban RH, Karim B, Mendell JT, Huso D, Lowenstein CJ. P53-induced microRNA-107 inhibits HIF-1 and tumor angiogenesis. Proc Natl Acad Sci U S A. 2010;107:6334–6339. doi: 10.1073/pnas.0911082107. [DOI] [PMC free article] [PubMed] [Google Scholar]