

Abstract
Variant microRNA (miRNA) expression is a character of many cancer types. The combined analysis of miRNA and messenger RNA (mRNA) expression profiles is crucial to identifying links between deregulated miRNAs and oncogenic pathways. The aim of this study was to screen several novel genes associated with renal cell carcinoma (RCC), and analyze the gene functions and signal pathways which were critical to RCCs with DNA microarray. The gene expression profile of GSE6344 was downloaded from Gene Expression Omnibus database, including 10 RCC samples and 10 healthy controls. Compared with the control samples, differentially expressed genes (DEGs) of RCC was identified. The selected DEGs were further analyzed using bioinformatics methods. Gene ontology (GO) enrichment analysis was performed using Gene Set Analysis Toolkit and protein-protein interaction (PPI) network was constructed with prePPI. Then, pathway enrichment analysis to PPI network was performed using WebGestalt software. We found that a total of 521 DEGs were down-regulated and 473 DEGs were up-regulated in RCC samples compared to healthy controls. A total of 15 remarkable enhanced functions and 17 suppressed functions were identified. PPI nodes of high degrees, such as RHCG, RALYL, SLC4A1, UMOD and CA9, were obtained. The DEGs were classified and significantly enriched in cytokine and cytokine receptor pathway. The hub genes we find from RCC samples are not only biomarkers, but also may provide the groundwork for a combination therapy approach for RCCs.
Keywords: Microarray profiling, oncogenes, potential biomarkers, critical pathways, renal cell carcinoma
Introduction
Renal cell carcinoma (RCC), is a kidney cancer that originates in the lining of the proximal convoluted tubule, and known to be the most lethal of all the genitourinary tumors [1,2]. It is also the most common type of kidney cancer in adults, responsible for approximately 80% of cases [3]. RCC is relatively resistant to radiation therapy and chemotherapy, although some cases respond to immunotherapy [4]. So it is clear that early diagnosis and medical intervention seems vital in decreasing mortality and promoting total quality of life, novel molecular markers about kidney cancer that can help individually evaluate risk of patient outcome and predict the prognosis are urgently required, as well as the prediction of therapy effect and advocating personalized treatment [5,6]. The major difficulty in RCC is the constitution that this disease is not one entity but rather a collection of different types of tumors, each possessing distinct genetic characteristics, histological features, and, to some extent, clinical phenotypes [7].
MicroRNAs (miRNAs) are a class of small non-coding RNAs that can repress gene expression through translational repression or messenger RNA deadenylation and decay by base pairing to partially complementary sites [8,9]. miRNAs were shown to negatively regulate gene expression at the post-transcriptional level by binding to the 3’-untranslated region (3’-UTR) of target mRNAs. miRNAs have been shown to be involved in tumour progression and metastasis in kidney and other carcinomas [10]. Next-generation small RNA-Sequencing (sRNA-Seq) allows for unbiased quantitative and qualitative sncRNA profiling [11]. When compared to miRNA array platforms, sRNA-Seq additionally enables the discovery of novel miRNAs as well as the detection of other differentially expressed sncRNAs like small nucleolar RNAs (snoRNAs) and transfer RNA (tRNA)-derived fragments that can mimic miRNA function [12].
Several microarray based studies have demonstrated 21 to 34 differentially expressed miRNAs between ccRCC and normal kidney tissue [13]. SRNA-Seq studies reported more than 100 differentially regulated miRNAs, some of which might serve as diagnostic and prognostic markers [14]. Nevertheless, these studies lack detailed information about miRNA targets and bioinformatical analysis is often only focused on miRNAs currently known to miRbase [15].
Here we used omiRas to analyze a publicly available dataset (GEO: GSE6344) published by Gumz M et al., comprising twenty sRNA-Seq libraries of ten ccRCCs and ten adjacent control tissues from the same patient in order to identify sncRNAs with deregualted expression across all cases [16]. After outlier detection with principle component analysis samples of nine patients were used for downstream analysis.
We detected 104 sncRNAs as differentially expressed between the groups. Among these were several miRNAs without previous implication in kidney cancer development, like miR-147a. Additionally, we detected seven snoRNAs and two tRNA derived fragments as differentially expressed between ccRCC and control tissues. We connected the deregulated miRNAs to biological pathways composed of differentially expressed genes under potential post-transcriptional control of these miRNAs. To do so, we utilized another publicly available mRNA-Sequencing (RNA-Seq) dataset. The “interaction network tool” of omiRas allows for the construction of interaction networks of miRNAs and mRNAs, interrogating the information from several miRNA-mRNA interaction databases. Therefore, we in silico assigned functions to significantly deregulated miRNAs and defined miRNAs implicated in the carcinogenesis of ccRCC [17,18].
However, the paramount regulator and distinct molecular mechanism of RCC has yet to be evaluated as the complexity of its pathogenesis. In this present paper, we aimed to screen several novel tumor suppressor genes and explore the molecular mechanism of RCC with a computational analysis. The most significant DEGs which served as potential biomarkers may provide a new sight on RCC clinical therapy.
Materials and methods
Dataset collection and Affymetrix microarray
The gene profile of GSE6344 was downloaded from Gene Expression Omnibus (http://www.ncbi.nlm. nih.gov/geo), which was based on the platform of Affymetrix Human Genome U133B Array condensed [19]. This expression dataset was deposited by Gumz ML. A total of 20 samples were derived from 10 ccRCC patients and 10 age- and gender- matched healthy control subjects.
Data preprocessing and differential expression analysis
Then we converted the probe-level data in CEL files into expression measures. For each sample, the expression values of all probes for a given gene were reduced to a single value by taking the average expression value [20]. After that, we imputed missing data and performed quartile data normalization. The multtest package in R was used to identify differentially expressed genes (DEGs) in patients with RCC compared to healthy controls. To circumvent the multi-test problem which might induce too much false positive results, the Benjamini & Hochberg (BH) method was used to adjust the raw P-values into false discovery rate (FDR) [21]. The FDR less than 0.05 and the absolute logFC larger than 1 were chosen as cut-off criteria.
Gene ontology enrichment analysis
To produce a dynamic, controlled vocabulary that can be applied to all eukaryotes, Gene Ontology (GO) analysis has been used frequently in functional study large scale genomic and transcriptional data [22]. DEGs were separated into two sets based on different expression behavior, and then the GO analysis was performed using Gene Set Analysis Toolkit suit [23]. A P-value less than 0.05 was considered statistically significant.
Construction and analysis of the protein-protein interaction (PPI) network
Protein-protein interaction network is a database of predicted and experimentally determined protein-protein interactions (PPI) using a Bayesian framework that combines structural, functional, evolutionary and expression information for yeast and human [24]. In this analysis, the most significant up- or down-regulated DEGs were screened, and a PPI network was constructed by collecting interactions from prePPI database.
Pathway enrichment analysis
For functional annotation the DEGs, the over-represented KEGG categories in pathways was identified using KAAS [25]. The P-value less than 0.05 was chosen as cut-off criterion. Up and downregulated genes in ccRCC were mapped to functional Gene Ontology (GO) categories using DAVID Bioinformatics Resources 6.7. Genes within enriched (FDR < 0.05) categories were committed to the STRING database to determine protein-protein interactions of their gene products. Additionally, miRNAs that might be causative for the deregulation of genes within the category were detected as described above. PCA and hierarchical clustering of the differentially expressed miRNAs were performed and visualized with R 3.0.2. Networks of genes from the same GO category were visualized with Cytoscape. Visualizations of annotation statistics for each library were taken from omiRas.
Results
Differential gene expression in RCC samples
After normalization, the data was performed differential expression analysis. The genes with P-value less than 0.05 and logFC absolute value over than 1, were considered as DEGs. A total of 521 DEGs were down-regulated and 473 DEGs were up-regulated in RCC samples compared to healthy controls. Among these 984 DEGs, nuclear factor kappa (NF-κB) was the most significantly up-regulated gene and Mitogen-activated protein kinase (MAPK) was down-regulated the most (P=3.1E-04, logFC=7.56; P=3.2E-06, logFC=-9.47, respectively). In addition, cytokine receptors, such as interleukin 17 receptor alpha (IL17RA) and colony stimulating factor 1 receptor (CSF1R), were also up-regulated (P=3.2E-06, logFC=3.17; P=4.6E-04, logFC=2.18, respectively) (Figure 1).
Figure 1.

Normalization of gene expressions in the ccRCC sample and control samples. The midcourt line in the box was the median value. After normalization, the median values were approximated with an average value of 7, which represents that the gene expression values in different samples are at the same levels and comparable.
mRNA-protein interaction network and potential miRNA upstream regulators by IPA
Interaction networks of DEGs were constructed as shown in Figure 2. According to the results, interaction relationship of 250 DEGs was obtained. A high level of connectivity was shown in the network constructed based on Mitogen-activated protein kinase (Figure 2). PPI nodes extracellular signal-regulated kinase (ERK), interleukin 6 (IL6), bone morphogenetic protein receptor, type II (BMPR2), P38 MAPK and natural cytotoxicity triggering receptor 1 (NCR1) were of high degrees in the network, all of which were obtained from RCC samples. In addition, the PPI network based on MAPK was also constructed, with imperfect connections due to the absence of several key hubs, as is shown in Figure 2.
Figure 2.
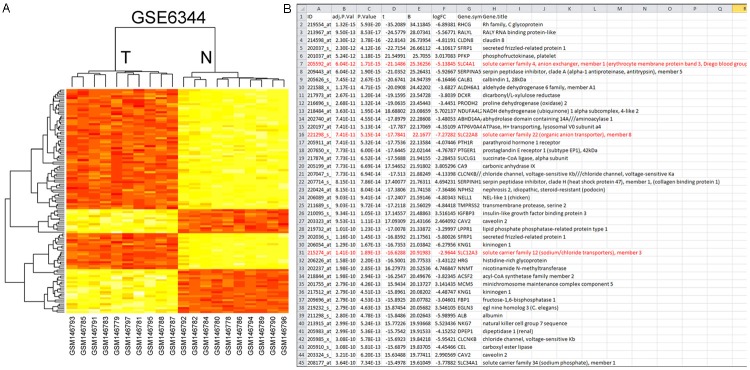
Interaction networks of DEGs constructed with prePPI. The line represents that the two genes may have an interaction based on their expression, evolution or structures. A. Hierarchical clustering of miRNA in kidney tissue samples. Kidney tissue samples were clustered according to the expression miRNAs between tumor tissue and normal tissue. Data from each miRNA were median centered. Samples are in columns and miRNAs in rows. N normal, T tumor. The P vales for these miRNAs were less than 0.05 in tumor tissue compared with normal tissue. B. PPI network based on SLC family.
Potential biomarkers and critical pathways in RCCs
We performed a functional enrichment analysis using Gene Set Analysis Toolkit and identified 11 remarkable enhanced functions and 13 suppressed functions (Figure 3, FDR < 0.05). The DEGs was classified with GO analysis according to their functions. Among these enhanced functions, the most remarkable up-regulation was immune response based on cytokine and cytokine receptor enrichment (GO: 0006657, FDR = 5.6E-27). The other significant functions included defense response (GO: 0006948, FDR = 6.1E-12), response to wounding (Go: 0009623, FDR = 1.7E-13), leukocyte activation (GO: 0057324, FDR = 1.4E-06), positive regulation of immune system process (GO: 0003214, FDR = 2.6E-12), inflammatory response (GO: 0005658, FDR = 1.4E-07) and so on. Genes, such as toll-like receptor (TLR) family, IL super-family, chemokine (C-X-C motif) ligand 12 (CXCL12), TAP binding protein (TAPBP), C-type lectin domain family 4 member A (CLEC4A), major histocompatibility complex class I (HLA), complement component 3a receptor 1 (C3AR1) and integrin alpha L (ITGAL), were involved in GO analysis.
Figure 3.

HGF network in renal cancer samples. Functional enrichment of DEGs between RCC sample and healthy samples (P < 0.05). Among these enhanced functions, the most remarkable up-regulation was immune response based on cytokine and cytokine receptor enrichment (GO: 0006955, FDR = 5.8E-29) and the most evidently downward modulation function in RCC was oxidation reduction which regulated by hormones (GO: 0055114, FDR = 3.9E-36), respectively.
The most evidently downward modulation function in RCC was oxidation reduction which regulated by hormones (GO: 0057216, FDR = 3.7E-24). The other significantly suppressed function included ion transport (GO: 0005421, FDR = 9.7E-05), response to organic substance (GO: 0013021, FDR = 3.0E-05) and generation of precursor metabolites and energy (GO: 0005986, FDR = 7.7E-11). The response to hormone stimulus was also suppressed (GO: 0008627, FDR = 5.4E-01), which in turn weaken the chemical homeostasis.
Cytokine and cytokine receptor pathway enrichment in RCCs
Pathway enrichment analysis of all genes involved in PPI was performed with NF-κB. Only cytokine and cytokine receptor pathway was enriched with a remarkable FDR less than 0.001 in PPI network of MAPK. Genes associated with cytokine and cytokine receptor pathway were chemokine, hematopoietin, WNT, TNF family and TGF-β family proteins genes. Five chemokines genes participated in this pathway, including IL6, NOB1, ERK1/2, JNK, P38 MAPK and Sclcl4. The primary hematopoietins were cytokine receptors, such as SOS, CRK and SLC4A1. TGF-β family proteins, such as PROC, ALB, VEGFRA, E-cadherin, β-catenin and their receptor TGF-β, were also involved in these pathways (Figure 4).
Figure 4.

PI3K/AKT network in renal cancer samples. Genes associated with cytokine and cytokine receptor pathway were chemokine, hematopoietin, VEGF family, TNF family and TGF-β family proteins genes.
Discussion
Our study links coding- and non-coding transcriptome data of normal and ccRCC tissue from two distinct studies. By the use of several miRNA-mRNA interaction databases available in omiRas we are able to provide new insights into the influence of aberrant miRNA expression on hundreds of deregulated genes [26]. Small non-coding RNA has drawn more attention in the recent years due to their role in the gene transcriptional and posttranscriptional regulation [27]. Nearly, 30% of gene expression in the human body is regulated by miRNAs. Recently, targeted therapies were developed to interfere with the transduction of key signaling pathways or to inhibit the function of tumour-specific molecules in malignant ccRCC [28].
In the present paper, we identified 984 DEGs in RCC patient samples compared to the control samples. DEGs in RCCs were highly associated with immune response, hormone response and defense response, which may play important roles in tumor initialization and migration. Pathway enrichment demonstrates that only cytokine and cytokine receptor pathway was enhanced in RCC patients compared to healthy controls. Genes obtained from the results of pathway enrichment analysis are the subject of our investigation. Among these DEGs, MAPK and NOB1 were the most significantly down-regulated and up-regulated genes, respectively. In addition, cytokine receptors, such as ERK1/2 and P38 MAPK, were also up-regulated.
MAPK signaling pathways can induce either cell proliferation or cell survival depending on the cell type and stimulus, the activation of the MAPK pathway has been associated with renal cancer proliferation [29]. The three main members that integrate the MAPK family in mammalian cells are ERK1/2, JNK and p38 MAPK, which are important in the control of cell differentiation, proliferation and apoptosis [30]. NOB1 protein is a key factor linking the proteasome and cellular growth, and therefore investigation of the NOB1 function will shed some light on the mechanism of growth control by the ubiquitin-proteasome pathway [31]. Perhaps the inhibition of the proteasome leads to stabilization of proteins that increase phosphorylation of the three key components in the MAPK pathway [27].
Others reported that TGF-β1 can also influence signal transduction pathways, such as nuclear factor kappa-B (NF-κB) and PI3K/Akt signal pathway [32,33]. Although the precise function of TGF-β1 in carcinogenesis is unknown, previous studies showed that TGF-β1 may regulate a set of genes in cell growth and proliferation, which may be important in cancer development and cancer cell proliferation Due to these functions of E-cadherin, it plays a key tumor suppressor role in suppressing the invasiveness of cancer cells [34,35]. Cadherin switch is a key change during EMT, during which the normal expression of E-cadherin is replaced by the abnormal expression of N- or P-cadherin [36]. This downregulation of Ecadherin is associated with the release of β-catenin, which then migrates to the nucleus and activates WNT signaling resulting in the EMT and metastasis [37,38]. Based on the analysis above, E-cadherin and β-catenin have often been used to monitor the progress of EMT during embryonic development and cancer progression [39,40].
In conclusion, though the DEGs and relevant genes may provide a new way into the therapy approach for ccRCCs, however, the expression of these genes was not confirmed by real-time PCR and the function in RCCs was not evaluated. The potential biomarkers and hub genes, such as ERK1/2, P38 MAPK, VEGFRA, SCLC4A1 and TGF-β, only provided potential targets for RCC therapy, and further effort to confirm the hypothesis was still in great needed.
Disclosure of conflict of interest
None.
References
- 1.Prince J, Bultman E, Hinshaw L, Drewry A, Blute M, Best S, Lee FT Jr, Ziemlewicz T, Lubner M, Shi F, Nakada SY, Abel EJ. Patient and Tumor Characteristics can Predict Non-Diagnostic Renal Mass Biopsy Findings. J Urol. 2015;193:1899–904. doi: 10.1016/j.juro.2014.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Leslie S, Gill IS, de Castro Abreu AL, Rahmanuddin S, Gill KS, Nguyen M, Berger AK, Goh AC, Cai J, Duddalwar VA, Aron M, Desai MM. Renal tumor contact surface area: a novel parameter for predicting complexity and outcomes of partial nephrectomy. Eur Urol. 2014;66:884–893. doi: 10.1016/j.eururo.2014.03.010. [DOI] [PubMed] [Google Scholar]
- 3.Forauer AR, Dewey BJ, Seigne JD. Cancer-free survival and local tumor control after impendence-based radiofrequency ablation of biopsy-proven renal cell carcinomas with a minimum of 1-year follow-up. Urol Oncol. 2014;32:869–876. doi: 10.1016/j.urolonc.2014.03.016. [DOI] [PubMed] [Google Scholar]
- 4.Crisan N, Ivan C, Gherman V, Neiculescu C, Coman I. Tumor enucleation with zero ischemia for renal cell carcinoma by robotic retroperitoneal approach. Urol J. 2014;11:1721–1723. [PubMed] [Google Scholar]
- 5.Yang FQ, Yang FP, Li W, Liu M, Wang GC, Che JP, Huang JH, Zheng JH. Foxl1 inhibits tumor invasion and predicts outcome in human renal cancer. Int J Clin Exp Pathol. 2014;7:110–122. [PMC free article] [PubMed] [Google Scholar]
- 6.Zeng L, Bai M, Mittal AK, El-Jouni W, Zhou J, Cohen DM, Zhou MI, Cohen HT. Candidate tumor suppressor and pVHL partner Jade-1 binds and inhibits AKT in renal cell carcinoma. Cancer Res. 2013;73:5371–5380. doi: 10.1158/0008-5472.CAN-12-4707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Khella HW, Bakhet M, Allo G, Jewett MA, Girgis AH, Latif A, Girgis H, Von Both I, Bjarnason GA, Yousef GM. miR-192, miR-194 and miR-215: a convergent microRNA network suppressing tumor progression in renal cell carcinoma. Carcinogenesis. 2013;34:2231–2239. doi: 10.1093/carcin/bgt184. [DOI] [PubMed] [Google Scholar]
- 8.Ishihara T, Seki N, Inoguchi S, Yoshino H, Tatarano S, Yamada Y, Itesako T, Goto Y, Nishikawa R, Nakagawa M, Enokida H. Expression of the tumor suppressive miRNA-23b/27b cluster is a good prognostic marker in clear cell renal cell carcinoma. J Urol. 2014;192:1822–1830. doi: 10.1016/j.juro.2014.07.001. [DOI] [PubMed] [Google Scholar]
- 9.Chen Z, Tang ZY, He Y, Liu LF, Li DJ, Chen X. miRNA-205 is a candidate tumor suppressor that targets ZEB2 in renal cell carcinoma. Oncol Res Treat. 2014;37:658–664. doi: 10.1159/000368792. [DOI] [PubMed] [Google Scholar]
- 10.Sanders I, Holdenrieder S, Walgenbach-Brunagel G, Von Ruecker A, Kristiansen G, Muller SC, Ellinger J. Evaluation of reference genes for the analysis of serum miRNA in patients with prostate cancer, bladder cancer and renal cell carcinoma. Int J Urol. 2012;19:1017–1025. doi: 10.1111/j.1442-2042.2012.03082.x. [DOI] [PubMed] [Google Scholar]
- 11.Hinton A, Hunter SE, Afrikanova I, Jones GA, Lopez AD, Fogel GB, Hayek A, King CC. sRNA-seq analysis of human embryonic stem cells and definitive endoderm reveals differentially expressed microRNAs and novel IsomiRs with distinct targets. Stem Cells. 2014;32:2360–2372. doi: 10.1002/stem.1739. [DOI] [PubMed] [Google Scholar]
- 12.Vidal EA, Moyano TC, Krouk G, Katari MS, Tanurdzic M, McCombie WR, Coruzzi GM, Gutierrez RA. Integrated RNA-seq and sRNA-seq analysis identifies novel nitrate-responsive genes in Arabidopsis thaliana roots. BMC Genomics. 2013;14:701. doi: 10.1186/1471-2164-14-701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wotschofsky Z, Busch J, Jung M, Kempkensteffen C, Weikert S, Schaser KD, Melcher I, Kilic E, Miller K, Kristiansen G, Erbersdobler A, Jung K. Diagnostic and prognostic potential of differentially expressed miRNAs between metastatic and non-metastatic renal cell carcinoma at the time of nephrectomy. Clin Chim Acta. 2013;416:5–10. doi: 10.1016/j.cca.2012.11.010. [DOI] [PubMed] [Google Scholar]
- 14.Ramankulov A, Lein M, Johannsen M, Schrader M, Miller K, Loening SA, Jung K. Serum amyloid A as indicator of distant metastases but not as early tumor marker in patients with renal cell carcinoma. Cancer Lett. 2008;269:85–92. doi: 10.1016/j.canlet.2008.04.022. [DOI] [PubMed] [Google Scholar]
- 15.Zigeuner R, Ratschek M, Rehak P, Schips L, Langner C. Value of p53 as a prognostic marker in histologic subtypes of renal cell carcinoma: a systematic analysis of primary and metastatic tumor tissue. Urology. 2004;63:651–655. doi: 10.1016/j.urology.2003.11.011. [DOI] [PubMed] [Google Scholar]
- 16.Gumz ML, Zou H, Kreinest PA, Childs AC, Belmonte LS, LeGrand SN, Wu KJ, Luxon BA, Sinha M, Parker AS, Sun LZ, Ahlquist DA, Wood CG, Copland JA. Secreted frizzled-related protein 1 loss contributes to tumor phenotype of clear cell renal cell carcinoma. Clin Cancer Res. 2007;13:4740–4749. doi: 10.1158/1078-0432.CCR-07-0143. [DOI] [PubMed] [Google Scholar]
- 17.Pal SK, He M, Tong T, Wu H, Liu X, Lau C, Wang JH, Warden C, Wu X, Signoretti S, Choueiri TK, Karam JA, Jones JO. RNA-seq reveals aurora kinase-driven mtor pathway activation in patients with sarcomatoid metastatic renal cell carcinoma. Mol Cancer Res. 2015;13:130–137. doi: 10.1158/1541-7786.MCR-14-0352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li P, Conley A, Zhang H, Kim HL. Whole-Transcriptome profiling of formalin-fixed, paraffin-embedded renal cell carcinoma by RNA-seq. BMC Genomics. 2014;15:1087. doi: 10.1186/1471-2164-15-1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Davis S, Meltzer PS. GEOquery: a bridge between the Gene Expression Omnibus (GEO) and BioConductor. Bioinformatics. 2007;23:1846–1847. doi: 10.1093/bioinformatics/btm254. [DOI] [PubMed] [Google Scholar]
- 20.Zhu Y, Davis S, Stephens R, Meltzer PS, Chen Y. GEOmetadb: powerful alternative search engine for the Gene Expression Omnibus. Bioinformatics. 2008;24:2798–2800. doi: 10.1093/bioinformatics/btn520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Boyle J. Gene-Expression Omnibus integration and clustering tools in SeqExpress. Bioinformatics. 2005;21:2550–1. doi: 10.1093/bioinformatics/bti355. [DOI] [PubMed] [Google Scholar]
- 22.Baker BA, Pine PS, Chatterjee K, Kumar G, Lin NJ, McDaniel JH, Salit ML, Simon CG Jr. Ontology analysis of global gene expression differences of human bone marrow stromal cells cultured on 3D scaffolds or 2D films. Biomaterials. 2014;35:6716–6726. doi: 10.1016/j.biomaterials.2014.04.075. [DOI] [PubMed] [Google Scholar]
- 23.Caniza H, Romero AE, Heron S, Yang H, Devoto A, Frasca M, Mesiti M, Valentini G, Paccanaro A. GOssTo: a stand-alone application and a web tool for calculating semantic similarities on the Gene Ontology. Bioinformatics. 2014;30:2235–2236. doi: 10.1093/bioinformatics/btu144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Veres DV, Gyurko DM, Thaler B, Szalay KZ, Fazekas D, Korcsmaros T, Csermely P. ComPPI: a cellular compartment-specific database for protein-protein interaction network analysis. Nucleic Acids Res. 2015;43:D485–93. doi: 10.1093/nar/gku1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Espinosa-Soto C, Immink RG, Angenent GC, Alvarez-Buylla ER, de Folter S. Tetramer formation in Arabidopsis MADS domain proteins: analysis of a protein-protein interaction network. BMC Syst Biol. 2014;8:9. doi: 10.1186/1752-0509-8-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ye S, Yang L, Zhao X, Song W, Wang W, Zheng S. Bioinformatics method to predict two regulation mechanism: TF-miRNA-mRNA and lncRNA-miRNA-mRNA in pancreatic cancer. Cell Biochem Biophys. 2014;70:1849–1858. doi: 10.1007/s12013-014-0142-y. [DOI] [PubMed] [Google Scholar]
- 27.Li W, Liu M, Feng Y, Xu YF, Huang YF, Che JP, Wang GC, Yao XD, Zheng JH. Downregulated miR-646 in clear cell renal carcinoma correlated with tumour metastasis by targeting the nin one binding protein (NOB1) Br J Cancer. 2014;111:1188–1200. doi: 10.1038/bjc.2014.382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li XY, Luo QF, Wei CK, Li DF, Li J, Fang L. MiRNA-107 inhibits proliferation and migration by targeting CDK8 in breast cancer. Int J Clin Exp Med. 2014;7:32–40. [PMC free article] [PubMed] [Google Scholar]
- 29.Mizuno R, Oya M, Hara S, Matsumoto M, Horiguchi A, Ohigashi T, Marumo K, Murai M. Modulation of bcl-2 family proteins in MAPK independent apoptosis induced by a cdc25 phosphatase inhibitor Cpd 5 in renal cancer cells. Oncol Rep. 2005;14:639–644. [PubMed] [Google Scholar]
- 30.Dygai AM, Zhdanov VV, Miroshnichenko LA, Udut EV, Zyuz’kov GN, Simanina EV, Sherstoboev EY, Chaikovskii AV, Stavrova LA, Burmina YV, Khrichkova TY, Reichart DV, Goldberg VE. Role of PI3K, MAPK/ERK1/2, and p38 in Implementation of the Proliferative and Differentiation Potential of Erythroid Progenitors after Blood Loss. Bull Exp Biol Med. 2015;158:417–20. doi: 10.1007/s10517-015-2775-2. [DOI] [PubMed] [Google Scholar]
- 31.Che JP, Li W, Yan Y, Liu M, Wang GC, Li QY, Yang B, Yao XD, Zheng JH. Expression and clinical significance of the nin one binding protein and p38 MAPK in prostate carcinoma. Int J Clin Exp Pathol. 2013;6:2300–2311. [PMC free article] [PubMed] [Google Scholar]
- 32.Baer C, Oakes CC, Ruppert AS, Claus R, Kim-Wanner SZ, Mertens D, Zenz T, Stilgenbauer S, Byrd JC, Plass C. Epigenetic silencing of miR-708 enhances NF-kappaB signaling in chronic lymphocytic leukemia. Int J Cancer. 2015 doi: 10.1002/ijc.29491. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 33.Suman S, Kallakury BV, Fornace AJ Jr, Datta K. Protracted Upregulation of Leptin and IGF1 is Associated with Activation of PI3K/Akt and JAK2 Pathway in Mouse Intestine after Ionizing Radiation Exposure. Int J Biol Sci. 2015;11:274–283. doi: 10.7150/ijbs.10684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kang H, Lee M, Jang SW. Celastrol inhibits TGF-beta1-induced epithelial-mesenchymal transition by inhibiting Snail and regulating E-cadherin expression. Biochem Biophys Res Commun. 2013;437:550–556. doi: 10.1016/j.bbrc.2013.06.113. [DOI] [PubMed] [Google Scholar]
- 35.Li W, Kidiyoor A, Hu Y, Guo C, Liu M, Yao X, Zhang Y, Peng B, Zheng J. Evaluation of transforming growth factor-beta1 suppress Pokemon/epithelial-mesenchymal transition expression in human bladder cancer cells. Tumour Biol. 2015;36:1155–1162. doi: 10.1007/s13277-014-2625-2. [DOI] [PubMed] [Google Scholar]
- 36.Tang O, Chen XM, Shen S, Hahn M, Pollock CA. MiRNA-200b represses transforming growth factor-beta1-induced EMT and fibronectin expression in kidney proximal tubular cells. Am J Physiol Renal Physiol. 2013;304:F1266–27. doi: 10.1152/ajprenal.00302.2012. [DOI] [PubMed] [Google Scholar]
- 37.Borthwick LA, Gardner A, De Soyza A, Mann DA, Fisher AJ. Transforming Growth Factor-beta1 (TGF-beta1) Driven Epithelial to Mesenchymal Transition (EMT) is Accentuated by Tumour Necrosis Factor alpha (TNFalpha) via Crosstalk Between the SMAD and NF-kappaB Pathways. Cancer Microenviron. 2012;5:45–57. doi: 10.1007/s12307-011-0080-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li R, Wang Y, Liu Y, Chen Q, Fu W, Wang H, Cai H, Peng W, Zhang X. Curcumin inhibits transforming growth factor-beta1-induced EMT via PPARgamma pathway, not Smad pathway in renal tubular epithelial cells. PLoS One. 2013;8:e58848. doi: 10.1371/journal.pone.0058848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Miao ZF, Li WY, Wang ZN, Zhao TT, Xu YY, Song YX, Huang JY, Xu HM. Lung cancer cells induce senescence and apoptosis of pleural mesothelial cells via transforming growth factor-beta1. Tumour Biol. 2015;36:2657–65. doi: 10.1007/s13277-014-2888-7. [DOI] [PubMed] [Google Scholar]
- 40.Jia Y, Wu D, Yun F, Shi L, Luo N, Liu Z, Shi Y, Sun Q, Jiang L, Wang S, Du M. Transforming growth factor-beta1 regulates epithelial-mesenchymal transition in association with cancer stem-like cells in a breast cancer cell line. Int J Clin Exp Med. 2014;7:865–872. [PMC free article] [PubMed] [Google Scholar]