

Abstract
Berberine (BBR) can potentially be used as a drug against non-alcoholic fatty liver disease (NAFLD) and diabetes. Our previous study found that BBR could change the pattern of DNA methylation. But the mechanisms underlying berberine are still far from completely understood. In this study, the function of L-PK in cell metabolism was explored, and high-fat-diet induced SD rats NAFLD models were created. The NAFLD rats were randomly grouped to be oral administration with BBR at a dosage of 200 mg/kg daily. Then DNA methylation and histone acetylation around the L-type Pyruvate Kinase (L-PK) gene were examined. In the results, we found that L-PK had a positive effect on cell proliferation, glucose utilization and triglyceride metabolism. However, the expression of L-PK was reduced in the livers of NAFLD rats, in accord with the decrease of DNA hypermethylation and histone deacetylation in the regulatory regions of L-PK. Notably, BBR treatment can restore the expression of L-PK by the demethylation of L-PK promoter and the increase in acetylation levels of histone H3 and H4 around L-PK, which indicated that BBR may be a potential drug for epigenetic-included diseases.
Keywords: Berberine, L-type pyruvate kinase, non-alcoholic fatty liver disease, DNA methylation, histone acetylation
Introduction
Non-alcoholic fatty liver disease (NAFLD) is a highly prevalent disease. Previous Studies have shown that epigenetics contributes to the progress of NAFLD. Many key genes, such as PPARGC1A [1] and MTTP [2], were abnormally expressed in NAFLD due to the aberrant epigenetic modifications.
Berberine (BBR) is the major pharmacological component of a Chinese herb named Coptis Chinensis. Reports showed that BBR had a strong effect on decreasing plasma glucose [3] and ameliorating dyslipidemia [4]. The proposed mechanisms of BBR included the inhibition of gluconeogenesis [5] and the stimulation of glycolysis [6]. However, the mechanisms underlying BBR are still elusive. BBR could change DNA methylation pattern and stimulate glycolysis [6,7]. We therefore hypothesized that BBR treatment could reduce blood glucose and ameliorate dyslipidemia by restoring the expression of abnormally expressed glycolysis genes in through epigenetic mechanisms. L-type pyruvate kinase (L-PK) is the third rate-limiting enzyme in glycolysis, which has close relationship with NAFLD and diabetes [8,9]. The current study was to determine the contribution of epigenetics to the decrease in L-PK expression and the epigenetic mechanism of BBR in the recovery of L-PK expression in NAFLD.
Materials and methods
Animal models and histological analysis
NAFLD rat model was created as we described previously [2]. Briefly, four-week-old Male Wistar rats were divided into two groups: the control group (ND) was fed a standard chow diet (12.5% fat) (SLACOM, Shanghai, CN), and the HFD group was fed a high-fat diet (51% fat) (SLACOM, Shanghai, CN) for 24 weeks. After 8 weeks of feeding, the HFD rats were randomly selected to receive either BBR (Sigma-Aldrich, Steinheim, UK) at a dosage of 200 mg/kg daily (HFD+BBR group) (n=5), or an equal volume of vehicle (0.5% methylcellulose, HFD group) (n=5) by gavage for 16 weeks. Rats fed normal diet received an equal volume of vehicle (0.5% methylcellulose, ND group) as control group (n=5).
Body weight and food intake were recorded weekly. Intraperitoneal glucose tolerance test (IPGTT) was conducted on 14-hours fasted rats before the rats were sacrificed. 1.5 g/kg D-glucose of body weight were injected. Blood samples were collected from saphenous vein immediately before glucose administration and 15, 30, 60 and 120 minutes thereafter. The plasma was separated by centrifugation, and aliquots were stored at -80°C for further analysis. Glucose values were obtained immediately after sampling using a glucose reagent strip and a glucometer (MediSense Precision PCx, Cherry Hill, NJ).
At the end of the experiment, the rats were sacrificed by decapitation after a 14-hour fast. Blood samples were collected and livers were quickly removed from both Wistar and SD rats. Serum level of glucose was assayed. Hepatic pathological changes (including H&E and steatosis) were analyzed. All experimental procedures involving the use of animals were reviewed and approved by the Animal Use and Care Committee of Fudan University. Each experiment was performed following the guidelines of the local committee for care and use of laboratory animals.
Histological analysis
The livers of rats were removed and subsequently fixed in phosphate-buffered 10% formalin. The right lateral lobule of the liver was then divided into 2 sections at the long middle line, one of which was embedded in paraffin blocks and the other in O.C.T. compound. A section from each paraffin block was stained with hematoxylin and eosin (HE) to examine the pathologic structures of the liver and serial cryosections were stained with Sudan III to evaluate lipid droplets.
L-PK expression analysis
RNA was isolated using Trizol (Invitrogen, Carlsbad, CA). Quantitative PCR (q-PCR) was performed using a 7900HT (ABI, Foster City, CA). Primers used for q-PCR were as follows: β-actin - 5’-GATTACTGCCCTGGCTCCTA-3’ (Forward) and β-actin - 5’-TCATCGTACTCCTGCTTGCT-3’ (Reverse); L-PK - 5’-CTCCCCACTCAGCTACAGACC-3’ (Forward) and L-PK - 5’-CCCTTCACAATTTCCACCTCC-3’ (Reverse).
Western blot was carried out using antibodies against L-PK (ab6191, Abcam, HK) and β-actin (ab8227, Abcam, HK) following standard procedures [2].
L-PK enzyme activity was measured by the formation of NAD+ in the presence of lactate dehydrogenase using a pyruvate kinase enzyme activity assay kit (Jiancheng, Nanjing, CN).
Plasmid construction
Human L-PK expression plasmid named pEGFP-L-PK was constructed, using primers: 5’-GGAAGATCTCG-ATGGAAGGGCCAGCGGGGTATCTGCG-3’ (Forward) and 5’-CGGAATTCGGATATGCTTAGCACCCGCATGAT-GTT-3’ (Reverse). An L-PK-RNAi plasmid with the target site GCCTCAAGGAGATGATCAAGG was purchased (GenePharma, Shanghai, CN).
Cell culture and analysis
QSG7701 and 293T cells were cultured in 1640 and DMEM-h media respectively, supplemented with 10% (v/v) fetal bovine serum in an atmosphere of 5% CO2 at 37°C. Cell proliferation was detected using the Cell Counting Kit (Dojindo, Rockville, US). Aliquots (100 µl) of cell in suspension were dispensed into the wells of a 96-well plate (5,000 cells/well). After 24 hours of pre-incubation, 10 µl of CCK-SK solution was added to each well. The plates were incubated for 1 hour at 37°C, and the absorbance at 450 nm was measured using a microplate reader.
The glucose concentration of the media was determined by the glucose oxidase method using a Glucose Reagent Kit (Kexin, Nanjing, CN). After converting the absorbance value to a concentration, the glucose consumption was calculated by the glucose concentrations of blank wells subtracting the remaining gulcose in cell plates wells [10].
Intracellular Triglyceride content was measured. Cells plated in 24-well plates were treated with 4% paraformaldehyde for 1 hour. Cells were washed 3 times with PBS and then exposed to 0.1 mg/ml Oil Red O (Sigma-Aldrich, Steinheim, UK) for 1 hour, followed by 3 more washes with PBS. Next, 100 µl isopropanol was added to each well. The triglyceride absorbance value was measured 30 min later using a spectrophotometer at 490 nm.
Quantification of DNA methylation
Genomic DNA was prepared from rat livers using the SDS/proteinase K method and then subjected to sodium bisulfite treatment. Rat L-PK promoter (-280 bp to +190 bp) was amplified by PCR using the L-PK-BSP primers: -5’-AGTTTGTTTTAAGTGGGGTTTT-3’ (Forward) and -5’-CCAAATCTATCCCCAAACTACAA-3’ (Reverse). The methylation level was measured using sodium bisulfite treatment method according to our previously reporting [11].
L-PK promoter activity assay
Rat L-PK promoter was amplified by PCR using primers 5’-AACTCGAGCCTTGTGGGGTAGGATGC-3’ (Forward) and 5’-GACAAGCTTTTCCAGCTCCAGGCAATT-3’ (Reverse). The fragment was inserted into pGL3-promoter plasmid named pGL3-L-PK.
PGL3-L-PK plasmid was methylated in vitro using the bacterial methylase SssI (New England BioLabs, Beverly, MA), which methylates all cytosine bases (at C5) within the double-stranded dinucleotide sequence 5’..CG..3’. A mock treatment of pDNA used the same procedure except that the SssI methylase was omitted. Methylated and mock-treated pGL3-L-PK were transfect into 293T cells using Lipofectamine 2000 (Invitrogen, Carlsbad, CA). After 24 h, the cells were measured using a Dual-Luciferase Reporter Assay System (Promega, Madison, WI) as previously described [1].
Chromatin immunoprecipitation assays (ChIP)
Rat liver tissues and the EZ ChIP Kit (Millipore, Lake Placid, NY) were used for ChIP, following the manufacturer’s instructions. ChIP was conducted with antibodies against ChREBP (ab92809, Abcam, HK), histone Ace-H3 (06-599, Millipore, CA), Ace-H4 (06-598, Millipore, CA), H3K9ace (ab10812, Abcam, HK), H3K18ace (ab1191, Abcam, HK), H4K8ace (ab15823, Abcam, HK), H4K12ace (ab46983, Abcam, HK) and H3K9me3 (ab8898, Abcam, HK). The purified DNA was amplified using q-PCR and determined by the comparative ΔΔCT method. The following primers were used for ChIP-PCR: ChREBP-L-PK - 5’-CACTCCCTGGTTCCTGGACT-3’ (Forward) and - 5’-TGAGTCCTGGTTAAAGTATAACC-3’ (Reverse); L-PK (-1200 bp) - 5’-TCAGGAGGATCCCCAGAGTA-3’ (Forward) and - 5’-ATGACCTTGCCTTGTTGACC-3’ (Reverse); L-PK (-350 bp) - 5’-CTCCCGGAGGTAAGAAGAGG-3’ (Forward) and - 5’-TTTAGCCGAGGTGAGGCTAT-3’ (Reverse); L-PK (0 bp) - 5’-CTTAACCAGGACTCATCTCATCTG-3’ (Forward) and - 5’-TAGAGCACCCGGTTTAGAAGC-3’ (Reverse); L-PK (+450 bp) - 5’-ACAAAGTAGCGGGTGTGTGA-3’ (Forward) and - 5’-GGAAACTCAGGCTCTTGCAC-3’ (Reverse); and L-PK (+2000 bp) - 5’-TATGAGGCCCTGTTTCAAGG -3’ (Forward) and - 5’-CTGTGCTGGGTTAAGGGTGT-3’ (Reverse).
Statistical analysis
All results are presented as the means ± SD. The differences between groups were evaluated for statistical significance using Student’s t test. Significance was defined as P < 0.05.
Results
Berberine reduces the liver fat and restores the downregulated expression of L-PK in the livers of NAFLD rats
Rat NAFLD model were created using high-fat diet. As shown in Table 1, the body weight, visceral fat and liver of NAFLD rats were much higher than rats of the control group (P < 0.05). A significant reduction in the three weight gain was observed following berberine treatment, whereas the vehicle treated mice showed normal body weight gain during the experimental period (P < 0.05). With HFD treatment, fasting blood glucose increased slightly in HFD rats (P > 0.05). While berberine administration resulted in a small reduction in fasting glucose level combined with the HFD group (P > 0.05). A higher lipid content was detected in the livers of NAFLD rats compared with controls, as visualized using H&E and Sudan III staining (Figure 1A-C). To assess glucose homeostasis in HFD and berberine treatment groups, we performed glucose tolerance tests. Plasma glucose level of the HFD group in the fasting state were higher than those of controls at all time points examined, although the difference was statistically significant only at 30 and 120 minutes, indicative of significantly impaired glucose tolerance in HFD SD rats. In contrast, berberine administration resulted in a significant reduction combined with a significant improvement in glucose level in HFD rats (Figure 1D). The mRNA level, protein levels and enzyme activity of L-PK were all significantly reduced in the livers of rats with NAFLD compared with control rats (P < 0.01) (Figure 1E-G). Berberine treatment markedly recovered the decreased expression of L-PK induced by NAFLD (P < 0.01) (Figure 1E-G).
Table 1.
General parameters of BBR treatment for 16 weeks
| Parameter | ND | HFD | HFD + BBR |
|---|---|---|---|
| Serum Glucose (mmol/L) | 5.16±1.53 | 5.22±1.33 | 4.92±1.17 |
| Body Weight (g) | 719.8±68.0 | 846.2±87.8* | 723.1±92.3# |
| Visceral Fat (g) | 45.1±18.9 | 74.7±23.1* | 42.7±14.3# |
| Liver Weight (g) | 19.0±3.3 | 38.9±5.8* | 30.6±6.1# |
n=6.
P<0.05 vs. ND;
**P<0.01 vs. ND.
P<0.05 vs. HFD;
##P<0.01 vs. HFD. ND, control group; HFD, high-fat diet-induced NAFLD group; HFD+BBR, Berberine treatment group.
Figure 1.
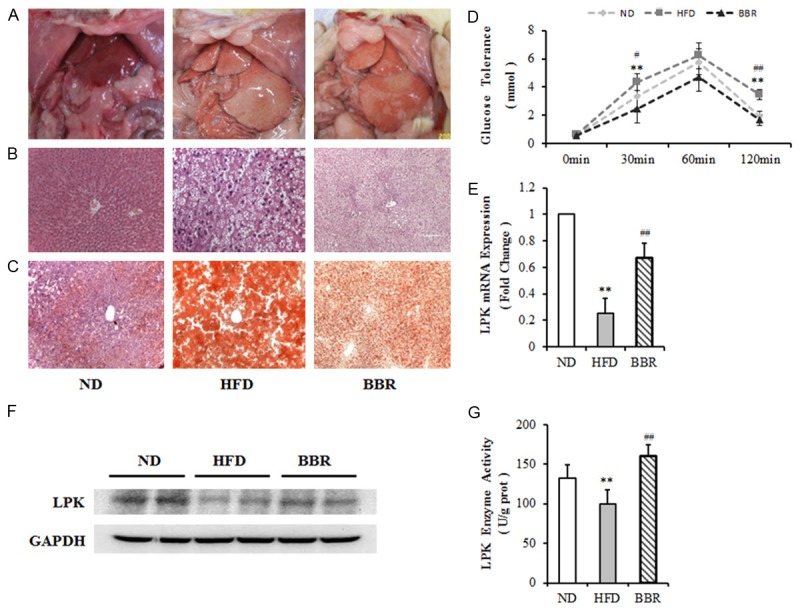
SD rat NAFLD models were created using a high-fat diet. A. Livers from normal and NAFLD rats stained with H&E (Hematoxylin and Eosin). C. Livers stained with Sudan III. D. Plasma glucose were measured before and 15, 30, 60 and 120 min after an oral glucose load. E. L-PK mRNA expression in the livers of rat NAFLD models. F. L-PK protein level in the livers of rat NAFLD models. G. L-PK enzyme activity in the livers of NAFLD models. n≥6. *P<0.05; **P<0.01 vs. NC. #P<0.05; ##P<0.01 vs. HFD. NC, control group; HFD, high-fat diet-induced NAFLD group; BBR, Berberine treatment group.
L-PK had a positive effect on cell proliferation, glucose utilization and triglyceride metabolism
To determine the correlation between L-PK and fat metabolism, the L-PK overexpression plasmid or the siRNA-based silencing of L-PK (siL-PK) plasmid was transfected into QSG7701 cells. At 48 hours following transfection of the L-PK expression plasmid into QSG7701 cells, L-PK mRNA level had increased several thousand-fold compared with the control group (Figure 2A), but the enzyme activity was only 24% higher (P < 0.01) (Figure 2B). In QSG7701 cells, cell proliferation notably increased (P < 0.05) (Figure 2C), the rate of glucose utilization was significantly accelerated (P < 0.01) (Figure 2D), and less intracellular triglyceride was detected (P < 0.01) (Figure 2E).
Figure 2.
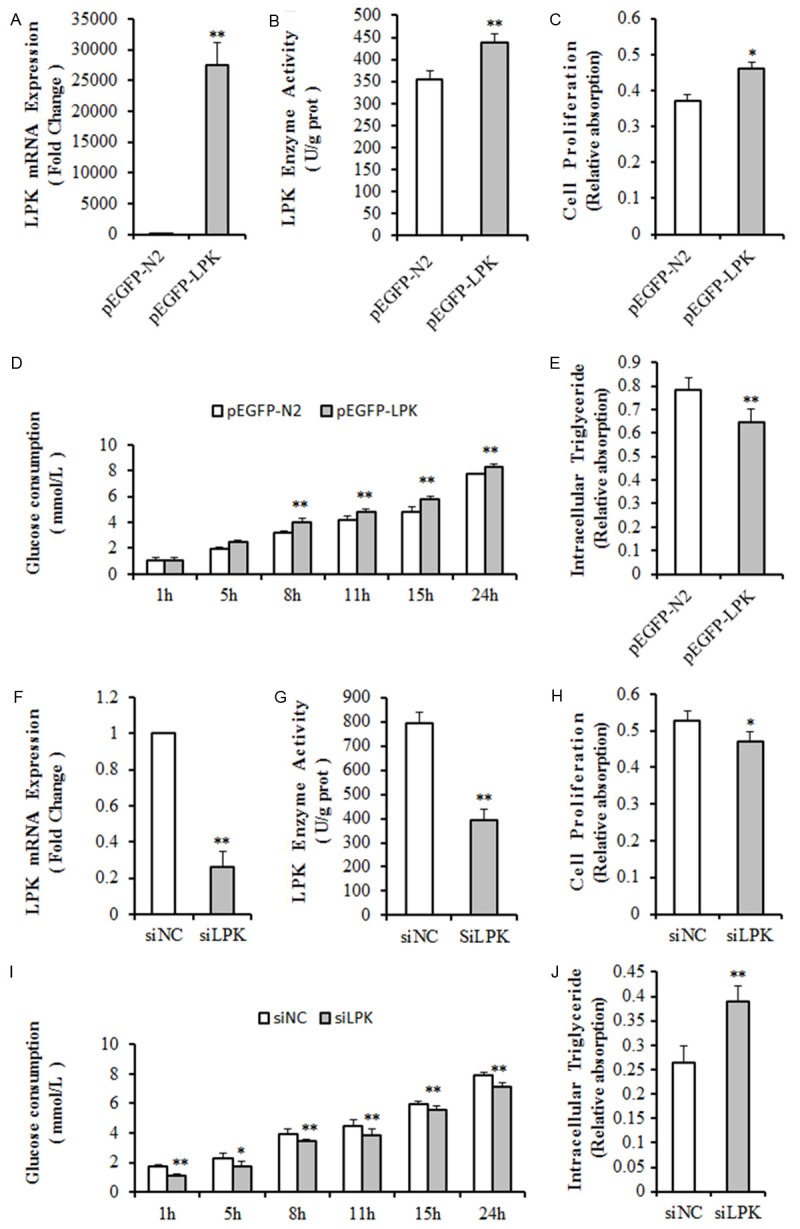
QSG7701 cells transfected with an L-PK overexpression plasmid or a L-PK-RNAi plasmid. QSG7701 cells 48 hours after transfection with an L-PK expression plasmid were examined for (A) L-PK mRNA expression, (B) L-PK enzyme activity, (C) cell proliferation, (D) glucose consumption, and (E) intracellular triglyceride. QSG7701 cells 48 hours after transfection with a siL-PK plasmid were examined for (F) L-PK mRNA expression, (G) L-PK enzyme activity, (H) cell proliferation, (I) glucose consumption, and (J) intracellular triglyceride. n≥3. *P<0.05; **P<0.01. pEGFP-N2, transfected with plasmid pEGFP-N2; pEGFP-L-PK, transfected with an L-PK expression plasmid; siNC, transfected with a control plasmid; siL-PK, transfected with L-PK-RNAi plasmid.
Meanwhile, the L-PK mRNA level and enzyme activity decreased by 84.2% (P < 0.01) (Figure 2F) and 50.5% (P < 0.01) (Figure 2G), in QSG7701 cells after 48 hours with siL-PK transfection. In addition, these cells had slowed cell proliferation (P < 0.05) (Figure 2H), decelerated glucose utilization (P < 0.01) (Figure 2I) and increased storage of intracellular triglyceride (P < 0.01) (Figure 2J).
Berberine reduced the hypermethylation of the L-PK promoter in the livers of NAFLD rats
To determine whether epigenetic modifications were involved in the decrease in L-PK mRNA levels, the methylation level of L-PK promoter was examined. 14 CpG sites were measured (Figure 3A). The mean methylation level of L-PK promoter was 45 ± 9% in control group, 67 ± 6% in NAFLD group, and 46 ± 7% in BBR group, which suggested that the L-PK promoter in NAFLD group exhibited higher methylation levels at all CpG sites than the promoters in the other two groups, while the control and berberine-treated rats exhibited a similar reduced level of methylation.
Figure 3.
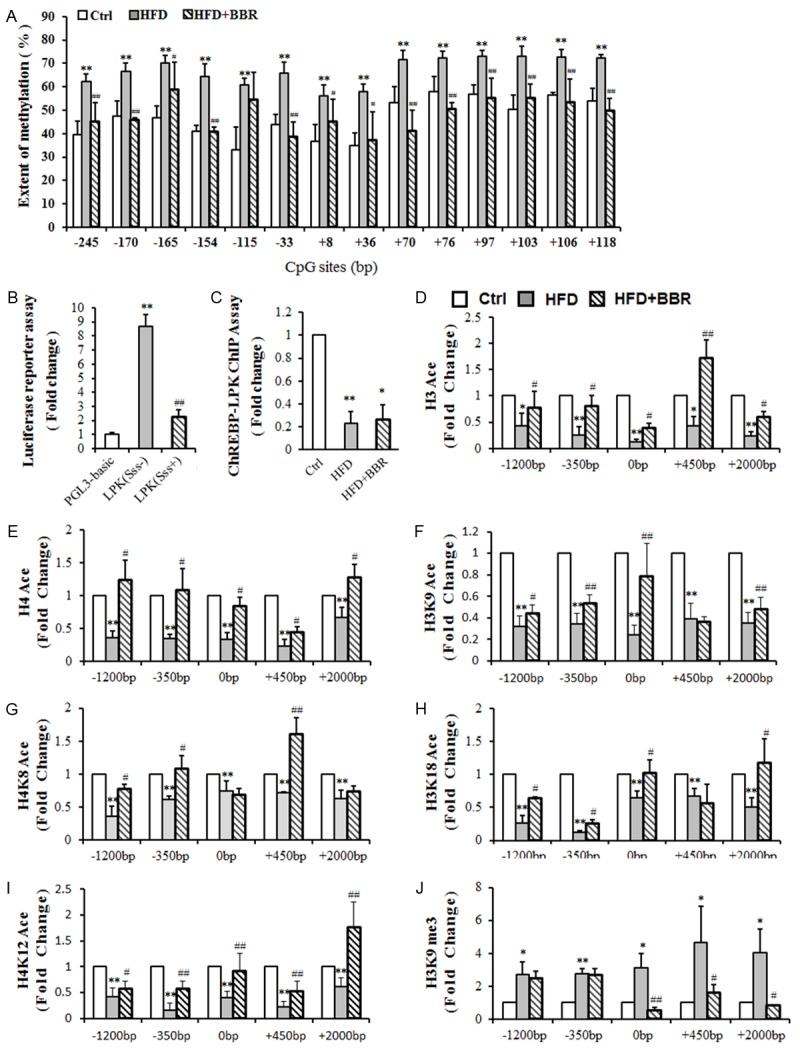
Epigenetic modifications in L-PK. A. Methylation level of the L-PK promoter. B. L-PK promoter activity assay in vitro. C. ChREBP-L-PK binding in vivo using a ChIP assay. D. Histone H3 acetylation status around the L-PK gene. E. Histone H3K9 acetylation status around the L-PK gene. F. Histone H3K18 acetylation status around the L-PK gene. G. Histone H4 acetylation status around the L-PK gene. H. Histone H4K8 acetylation status around the L-PK gene. I. Histone H4K12 acetylation status around the L-PK gene. J. Histone H3K9me3 methylation status around the L-PK gene. n≥4. *P<0.05; **P<0.01 vs. Ctrl or PGL3-basic. #P<0.05; ##P<0.01 vs. HFD or L-PK (Sss-). Ctrl, control group; HFD, high-fat diet-induced NAFLD group; HFD+BBR, Berberine treatment group; pGL3-basic, transfected with plasmid pGL3-Basic; L-PK (Sss-), transfected with the pGL3-L-PK promoter (unmethylated); L-PK (Sss+), transfected with the pGL3-L-PK promoter (methylated).
Luciferase reporter assay was carried out to certify the effect of L-PK promoter hypermethylation on L-PK transcription. The results revealed that the methylated reporter plasmid reduced luciferase activity by 74.3% relative to the unmethylated plasmid (Figure 3B), indicating that hypermethylation of the L-PK promoter region has a great effect on the inhibition of L-PK gene transcription activities.
ChREBP is an important transcription factor that has a positive effect on L-PK transcription. The amount of ChREBP binding to L-PK promoter was significantly reduced by 76.8% in the livers of rats with NAFLD compared with control rats. BBR treatment slightly increased the amount of ChREBP binding to L-PK, but the increase was not significant (Figure 3C).
BBR treatment increased the down-regulation of histone acetylation around L-PK in the livers of NAFLD rats
ChIP assay was further determined to reveal whether histone deacetylation was involved in reducing L-PK mRNA expression. Antibodies against methylated histone H3K9me3 and acetylated histone H3, H3K9, H3K18, H4, H4K8, and H4K12 were used in this experiment. DNA from the immunoprecipitation was isolated, and q-PCR was performed using primers spanning the -1200 bp, -350 bp, 0 bp, +450 bp and +2000 bp regions around L-PK.
As shown in Figure 3D and 3E, the levels of acetylated histone H3 and H4 in the -1200 bp, -350 bp, 0 bp, +450 bp and +2000 bp regions of L-PK were significantly reduced in the NAFLD group compared with those of the control group (P < 0.01). BBR treatment elevated the acetylation level of both histones H3 and H4 at all 5 positions in genomic DNA around L-PK. The trend of acetylated histone residues H3K9, H3K18, H4K8 and H4K12 was similar to that of histones H3 and H4, which were significantly reduced in the L-PK gene regions of -1200 bp, -350 bp, 0 bp, +450 bp and +2000 bp (P < 0.01) (Figure 3F-I). BBR treatment significantly increased the acetylation level of histone residues H3K9, H3K18, H4K8, H4K12 in most of the regions examined (P < 0.05).
Histone H3K9me3 methylation was also examined and was significantly higher in all 5 regions examined in NAFLD rats (P < 0.05). BBR treatment markedly reduced the histone H3K9me3 methylation level in the 0 bp, +450 bp and +2000 bp regions of L-PK (P < 0.05) (Figure 3J).
Discussion
L-PK has close relationships with NAFLD and diabetes [8,9]. Here, we demonstrate that L-PK had a positive effect on cell proliferation, glucose metabolism and triglyceride metabolism. BBR exhibited a strong effect on weight loss induced by NAFLD. More importantly, BBR achieved its function by restoring the expression of L-PK, which is down-regulated in NAFLD. Furthermore, we found that BBR treatment could partially counteract the HFD-elicited dysregulation of L-PK through epigenetic mechanisms.
DNA methylation is the most intensely studied epigenetic modification. Many genes, such as GcK [7] and PPARGC1A [1], have been found having abnormal expression caused by altered DNA methylation. The hypermethylation of L-PK promoter could lead to decreased L-PK transcription activity and reduced binding of ChREBP to L-PK. BBR could decrease the NAFLD-induced hypermethylation of L-PK. The same result was found in our previous research on MTTP [2], a key gene in lipid metabolism, indicating that demethylation maybe universal for BBR.
Histone acetylation is also important for gene expression. It is generally accepted that acetylation of the lysines on the tails of histones H3 (K9, K14 and K18) and H4 (K5, K8, K12 and K16) correlates strongly with gene transcription activity [12-14]. The acetylation levels of histones H3, H3K9, H3K18, H4, H4K8 and H4K12 were all significantly reduced around L-PK gene in the NAFLD group. Histone modifications regulate the accessibility of chromatin and gene activity [15-17]. The decreased acetylation of these lysines on the tails of histones H3 and H4 around the L-PK gene promoter may directly lead to reduced gene activity. BBR treatment significantly increased the acetylation levels of histones H3, H3K9, H3K18, H4, H4K8 and H4K12, which are decreased in NAFLD, in almost all positions around the L-PK gene. The histone H3K9me3 methylation level was significantly increased in the NAFLD group, and BBR treatment decreased the histone H3K9me3 methylation level in several of the regions around L-PK.
In summary, DNA methylation and histone deacetylation contribute to the decreased expression of L-PK, which has a positive effect on cell proliferation, glucose utilization and triglyceride metabolism, in the livers of rats with NAFLD. BBR treatment restored the expression of L-PK by the demethylation of L-PK promoter and the increase in acetylation levels of histone H3 and H4 around L-PK, indicating that BBR may be a good drug for epigenetic-included diseases.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (81170786, 81001114 and 81071739), general grants (20124354) from the Shanghai Municipal and Family Planing, and the Ph.D. Programs Foundation of the Ministry of Edu-cation of China (20110071110028).
Disclosure of conflict of interest
None.
References
- 1.Ribel-Madsen R, Fraga MF, Jacobsen S, Bork-Jensen J, Lara E, Calvanese V, Fernandez AF, Friedrichsen M, Vind BF, Højlund K, Beck-Nielsen H, Esteller M, Vaag A, Poulsen P. Genome-wide analysis of dna methylation differences in muscle and fat from monozygotic twins discordant for type 2 diabetes. PLoS One. 2012;7:e51302. doi: 10.1371/journal.pone.0051302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chang X, Yan H, Fei J, Jiang M, Zhu H, Lu D, Gao X. Berberine reduces methylation of the MTTP promoter and alleviates fatty liver induced by a high-fat diet in rats. J Lipid Res. 2010;51:2504–15. doi: 10.1194/jlr.M001958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang H, Wei J, Xue R, Wu J, Zhao W, Wang ZZ, Wang SK, Zhou ZX, Song DQ, Wang YM, Pan HN, Kong WJ, Jiang JD. Berberine lowers blood glucose in type 2 diabetes mellitus patients through increasing insulin receptor expression. Metabolism. 2010;59:285–92. doi: 10.1016/j.metabol.2009.07.029. [DOI] [PubMed] [Google Scholar]
- 4.Zhang Q, Xiao X, Feng K, Wang T, Li W, Yuan T, Sun X, Sun Q, Xiang H, Wang H. Berberine moderates glucose and lipid metabolism through multipathway mechanism. Evid Based Complement Alternat Med. 2011:2011. doi: 10.1155/2011/924851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xia X, Yan J, Shen Y, Tang K, Yin J, Zhang Y, Yang D, Liang H, Ye J, Weng J. Berberine improves glucose metabolism in diabetic rats by inhibition of hepatic gluconeogenesis. PLoS One. 2011;6:e16556. doi: 10.1371/journal.pone.0016556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yin J, Gao Z, Liu D, Liu Z, Ye J. Berberine improves glucose metabolism through induction of glycolysis. Am J Physiol Endocrinol Metab. 2008;294:E148–56. doi: 10.1152/ajpendo.00211.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jiang M, Zhang Y, Liu M, Lan MS, Fei J, Fan W, Gao X, Lu D. Hypermethylation of hepatic glucokinase and l-type pyruvate kinase promoters in high-fat diet-induced obese rats. Endocrinology. 2011;152:1284–9. doi: 10.1210/en.2010-1162. [DOI] [PubMed] [Google Scholar]
- 8.Wang H, Hays NP, Das SK, Craig RL, Chu WS, Sharma N, Elbein SC. Phenotypic and molecular evaluation of a chromosome 1q region with linkage and association to type 2 diabetes in humans. J Clin Endocrinol Metab. 2009;94:1401–8. doi: 10.1210/jc.2008-2132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hasstedt SJ, Chu WS, Das SK, Wang H, Elbein SC. Type 2 diabetes susceptibility genes on chromosome 1q21-24. Ann Hum Genet. 2008;72:163–9. doi: 10.1111/j.1469-1809.2007.00416.x. [DOI] [PubMed] [Google Scholar]
- 10.Yin J, Hu RM, Chen MD, Tang JF, Li FY, Yang Y, Chen J. Effects of berberine on glucose metabolism in vitro. Metabolism. 2002;51:1439–43. doi: 10.1053/meta.2002.34715. [DOI] [PubMed] [Google Scholar]
- 11.Jiang M, Zhang Y, Fei J, Chang X, Fan W, Qian X, Chen J. Rapid quantification of DNA methylation by measuring relative peak heights in direct bisulfite-PCR sequencing traces. Lab Invest. 2010;90:282–90. doi: 10.1038/labinvest.2009.132. [DOI] [PubMed] [Google Scholar]
- 12.Lusser A. Acetylated, methylated, remodeled: chromatin states for gene regulation. Curr Opin Plant Biol. 2002;5:437–43. doi: 10.1016/s1369-5266(02)00287-x. [DOI] [PubMed] [Google Scholar]
- 13.DoeneckE D, Gallwitz D. Acetylation of histones in nucleosomes. Mol Cell Biochem. 1982;44:113–28. doi: 10.1007/BF00226895. [DOI] [PubMed] [Google Scholar]
- 14.Li B, Carey M, Workman JL. The role of chromatin during transcription. Cell. 2007;128:707–19. doi: 10.1016/j.cell.2007.01.015. [DOI] [PubMed] [Google Scholar]
- 15.Kurdistani SK, Grunstein M. Histone acetylation and deacetylation in yeast. Nat Rev Mol Cell Bio. 2003;4:276–84. doi: 10.1038/nrm1075. [DOI] [PubMed] [Google Scholar]
- 16.Berger SL. Histone modifications in transcriptional regulation. Curr Opin Genet Dev. 2002;12:142–8. doi: 10.1016/s0959-437x(02)00279-4. [DOI] [PubMed] [Google Scholar]
- 17.Kornberg RD, Lorch YL. Twenty-five years of the nucleosome, fundamental particle of the eukaryote chromosome. Cell. 1999;98:285–94. doi: 10.1016/s0092-8674(00)81958-3. [DOI] [PubMed] [Google Scholar]