

Abstract

Exogenously and endogenously produced reactive oxygen species attack the base and sugar moieties of DNA showing a preference for reaction at 2′-deoxyguanosine (dG) sites. In the present work, dG was oxidized by HO• via the Fe(II)-Fenton reaction or by X-ray radiolysis of water. The oxidized lesions observed include the 2′-deoxynucleosides of 8-oxo-7,8-dihydroguanine (dOG), spiroiminodihydantoin (dSp), 5-guanidinohydantoin (dGh), oxazolone (dZ), 5-carboxamido-5-formamido-2-iminohydantoin (d2Ih), 5′,8-cyclo-2′-deoxyguanosine (cyclo-dG), and the free base guanine (Gua). Reactions conducted with ascorbate or N-acetylcysteine as a reductant under aerobic conditions identified d2Ih as the major lesion formed. Studies were conducted to identify the role of O2 and the reductant in product formation. From these studies, mechanisms are proposed to support d2Ih as a major oxidation product detected under aerobic conditions in the presence of the reductant. These nucleoside observations were then validated in oxidations of oligodeoxynucleotide and λ-DNA contexts that demonstrated high yields of d2Ih in tandem with dOG, dSp, and dGh. These results identify dG oxidation to d2Ih to occur in high yields leading to a hypothesis that d2Ih could be found from in cells stressed with HO•. Further, the distorted ring structure of d2Ih likely causes this lesion to be highly mutagenic.
Introduction
Cellular redox status is a dynamic state that balances reducing and oxidizing species, and if the balance is thrown off in the cell, dysfunction arises.1 The hydroxyl radical (HO•) is a powerful oxidizing species found in the cell and causes redox imbalance due to its particularly high redox potential (2.31 V vs NHE, pH 7; HO•(+H+)/H2O).2 The Fenton reaction can generate HO• when a redox-active metal effects cleavage of H2O2, a metabolic product that can easily diffuse within and between cells.3 Hydrogen peroxide is weakly reactive, but Fe(II) species have the capability to generate HO• plus Fe(III) via the Fenton reaction (Reaction 1). Furthermore, reducing agents in the cell such as ascorbate (Asc) allow this reaction to be catalytic in iron by reducing Fe(III) back to Fe(II) (Reaction 2).4,5 The unfortunate events that occurred at Fukushima, Japan in 2011 remind us of the dangers of radiation. The high-energy photons released upon radioactive decay are capable of yielding HO• via radiolysis of water (Reactions 3 and 4). The iron-Fenton reaction and X-ray radiolysis of water represent two methods for generating hydroxyl radicals that can oxidize biomolecules6,7 such as DNA.4,5,8 DNA damage plays a significant role in aging, and the development of diseases such as cancer.8
| 1 |
| 2 |
| 3 |
| 4 |
DNA is vulnerable to oxidative insults from HO•, and the damage is particularly deleterious because it can cause mutations that are heritable to daughter cells. Notable progress has been made in the discovery of DNA oxidation pathways resulting from radical oxygen species, such as HO•, during the past three decades.4,9−12 In DNA, the most susceptible site is 2′-deoxyguanosine (dG) because the guanine heterocycle has the lowest redox potential (1.29 V vs NHE, pH 7; dG•(−H+)/dG) among the four DNA bases.13 The nucleoside dG is a prime target for direct oxidation or electron transfer-mediated oxidation that can result from HO•.10,13,14 Oxidative damage to dG yields products that result from the initial product-forming reaction occurring at one of three sites that include the C5 or C8 positions of the heterocyclic ring, or the 2-deoxyribose unit (Figure 1). These products are identified by their characteristic mass changes from the parent nucleoside dG (M).
Figure 1.

Structures of dG and its oxidation products. Box A: products observed from initial HO• attack at C8. Box B: products observed from initial HO• attack at C5. Box C: guanine-derived products observed when oxidation occurs on 2-deoxyribose.
Sugar oxidation is a commonly observed reaction pathway of dG damage resulting from HO•.12 Products observed along this pathway include 5′,8-cyclo-2′-deoxyguanosine (cyclo-dG) diastereomers derived from H• atom abstraction at C5′, and release of the free base guanine (Gua) after hydroxylation of C1′. Direct base release in DNA leads to strand breaks due to the lability of the abasic site so formed.15−17 The yields of these products are highly dependent on the reaction conditions.12 Base oxidation of dG leads to two lesions resulting from reaction at C8 that include 8-oxo-7,8-dihydro-2′-deoxyguanosine (dOG), observed in high yield under aerobic oxidizing conditions, or the ring-opened product 2,6-diamino-4-hydroxy-5-formamidopyrimidine-2′-deoxyribonucleoside (Fapy-dG) observed under anaerobic reducing conditions.18−20 Both dOG and Fapy-dG are mutagenic compounds found in vivo,9 while dOG is a biomarker monitored to assay the extent of cellular oxidative stress.21 Because dOG has a lower redox potential (0.74 V vs NHE, pH 7; dOG•(−H+)/dOG) than dG,13 it is readily oxidized to an electrophilic intermediate that reacts with water at C5 to ultimately yield the hydantoin lesions spiroiminodihydantoin-2′-deoxyribonucleoside (dSp) and 5-guanidinohydantoin-2′-deoxyribonucleoside (dGh).8,22−25 These lesions have been observed in vivo and are also highly mutagentic.26−29
Additionally, oxidation of dG leads to products resulting from initial chemistry occurring at the C5 position of the purine. 2,5-Diaminoimidazolone-2′-deoxyribonucleoside (dIz) and its hydrolysis product 2,2,4-triamino-2H-oxazol-5-one-2′-deoxyribonucleoside (dZ) are four-electron oxidation products resulting from exposure to HO• or one-electron oxidants under aerobic conditions.8,18,30−34 Recent studies concerning dG oxidation have identified 5-carboxamido-5-formamido-2-iminohydantion-2′-deoxyribonucleoside (d2Ih) as a product from Fenton chemistry with copper or lead, activation of KHSO5 with nickel or manganese complexes, CO3•- oxidations, and the epoxidizing agent dimethyldioxirane.8,10,35−40 These observations have led us to the question of whether d2Ih is in fact a major product from dG oxidation by the hydroxyl radical generated either from the Fe(II)-mediated Fenton reaction or by X-ray radiolysis of water.
Toward this goal, we investigated the formation and yield of dG-oxidation products from the Fe(II)-mediated Fenton reaction or X-ray radiolysis of water while monitoring the effect of reductant concentration (ascorbate or N-acetylcysteine) on their yields in the nucleoside, short oligodeoxynucleotide, and λ-DNA contexts. These studies identify d2Ih to be a major guanine oxidation product observed when reactions were conducted with low millimolar amounts of reductant, conditions that mimic the presence of reducing agents such as glutathione, urate, or ascorbate in cells.41 The results of these studies led to proposed mechanisms for the formation of d2Ih and to discussions of its possible biological significance.
Results and Discussion
Identification of Oxidation Products
The nucleoside dG (3 mM) was initially chosen for oxidation by the Fe(II)-mediated Fenton reaction or X-ray irradiation in the absence or presence of ascorbate (Asc) or N-acetylcysteine (NAC). All reactions were conducted with 75 mM NaPi buffer (pH 7.4) at 22 °C. The reaction mixtures were initially analyzed by reversed-phase UPLC-ESI+-MS. This allowed identification of guanine (Gua), 8-oxo-7,8-dihydroguanine (OG), dOG, and the diastereomers of cyclo-dG after comparison to known standards. The void volume from this analysis was collected and reanalyzed with a Hypercarb column equipped with an ESI+-MS detector. This method allowed detection of Gh, dZ, and the diastereomers of d2Ih, dGh, and dSp. Because dIz hydrolyzes to dZ during preparation of the void volume, only dZ was quantified. The Hypercarb column allowed the separation of the diastereomer products of d2Ih and dSp for which the absolute configurations for the peaks had been previously determined.8,42,62 The two diastereomers of dGh are interconvertible and were not quantified individually.44 Further support for the identification of d2Ih, dSp, and dZ was obtained by ESI+-MS/MS fragmentation of the free bases from in-source fragmentation of the N-glycosidic bond of the parent nucleosides. The product dGh failed to be fragmented. HRMS was conducted to further support the identification of each product (Supporting Information Figures S3–S13).
Product Quantification from the Fe(II)-Mediated Fenton Reaction
Product quantification was conducted with reversed-phase HPLC while monitoring peak elution at 240 nm; product peak areas were integrated and normalized based on the product’s extinction coefficient at 240 nm (ε240 nm).8 The extinction coefficient for d2Ih has not been experimentally determined, and due to its instability toward N-glycosidic bond hydrolysis it was not determined in these studies.43 However, our previous time-dependent density functional theory studies on the UV–vis and ECD properties for dSp and d2Ih allow us to use these previous results to determine the relative difference in extinction coefficient between these two compounds at the wavelength monitored.42,43 These calculations determined the ε240 nm for d2Ih to be ∼30% less than that of dSp; therefore, the ε240 nm value used to quantify d2Ih was calculated to be 2290 L·mol–1·cm–1.45 The Fenton reaction was initially conducted with the Fe(II)/EDTA catalyst and 10 mM H2O2 under aerobic conditions to give 5% conversion to product. We elected to conduct reactions to a low overall yield so that primary oxidation products could be identified and compared. In this reaction, the highest absolute product yields observed were dZ (1.5%) and Gua (1.5%) in equal amounts, with the mass balance completed by low yields of cyclo-dG (0.2%), dOG (0.4%), dSp (0.9%), and d2Ih (0.4%, Table 1). In the next reaction, the same conditions were applied with the addition of 2 mM Asc that led to a 7-fold increase in product formation, as well as a significant change in product distribution. In the presence of Asc, the major dG product detected was d2Ih (13.4%) with lower yields of the 2-deoxyribose oxidation products cyclo-dG (0.9%) and Gua (7.1%), as well as OG (0.6%) and Gh (<0.1%); also, lower yields of dZ (2.7%), dOG (4.9%), dSp (5.6%), and dGh (0.4%) were observed (Table 1). These Asc-dependent studies revealed two key observations: (1) the reaction yields dramatically increased (7-fold) with Asc because the Fe(II)/EDTA complex can redox cycle in the presence of a reductant (Reaction 2), and (2) d2Ih was the major product observed when Asc was present. Because dSp and dGh are further oxidation products of the precursor dOG,22 the yield of d2Ih (13.4%) was compared to the combined yields of dOG, dSp, and dGh (10.9%), and it was found that the yield of d2Ih was higher than that of dOG and its oxidation products.
Table 1. Absolute Yields for Oxidation of dG by the Fe(II)-Mediated Fenton Reactiona.
| absolute
yield (%) |
||||
|---|---|---|---|---|
| product | Fe(II)/H2O2/+Asc/+O2 | Fe(II)/H2O2/–Asc/+O2 | Fe(II)/H2O2/+Asc/low O2b | Fe(II)/H2O2/–Asc/low O2b |
| dOG | 4.9 | 0.4 | 6.6 | 1.9 |
| dSp | 5.6 | 0.9 | 6.0 | 3.9 |
| dGh | 0.4 | <0.1 | 2.0 | 1.9 |
| Gh | <0.1 | <0.1 | N.D. | N.D. |
| dZ | 2.7 | 1.5 | N.D. | N.D. |
| d2Ih | 13.4 | 0.4 | 3.3 | 1.0 |
| cyclo-dG | 0.9 | 0.2 | 1.9 | 1.2 |
| Gua | 7.1 | 1.5 | N.D. | N.D. |
| OG | 0.6 | <0.1 | N.D. | N.D. |
| dG conversion % | 35.8 | 5.0 | 20.0 | 9.9 |
Reported values represent the average of three trials that have errors of ∼8% of the value. N.D. = not detected.
Low O2 represents a partial reduction in the O2 concentration by bubbling argon into the sample for 10 min that was later found to be insufficient to completely remove O2.
In the next set of reactions, the role of O2 was studied. First, a reaction was conducted with dG in the presence of Fe(II)/EDTA and H2O2 under low O2 conditions achieved by bubbling argon into the sample for 10 min that was insufficient to completely remove O2. In this reaction, the overall conversion of dG to product was 10%, and the major product observed was dSp (3.9%), with low yields of dOG (1.9%), dGh (1.9%), d2Ih (1.0%), and the sugar oxidation product cyclo-dG (1.2%, Table 1). The addition of 2 mM Asc to the low O2 reaction increased the dG conversion by 2-fold (20%) and changed the product distribution, giving dOG (6.6%) and dSp (6.0%) in high yield and low yields of d2Ih (3.3%), dGh (2.0%), and cyclo-dG (1.9%, Table 1). These low O2 studies indicated the yield of d2Ih was at a minimum under these conditions. Additionally, the major products observed under low O2 conditions were dOG and its oxidation product dSp.
Where is Fapy-dG? Many literature sources point to the presence of Fapy-dG as a guanine oxidation product formed under anaerobic, reducing conditions.46−48 In our present low O2 studies, this elusive compound was not observed, even after exhaustive searching of the LC-MS data. This led us to probe deeper into the formation of Fapy-dG, using the following conditions to make this compound from dG by the iron-Fenton reaction. First, the reaction had to be rigorously purged with argon for >30 min prior to addition of H2O2 with 2 mM Asc, and the HPLC mobile phase was changed from running buffered [20 mM NH4OAc (pH 7)] water and MeCN to unbuffered ddH2O and MeCN. These changes allowed observation of a broad hump after the void volume in the reversed-phase HPLC chromatogram with a UV–vis signature consistent with Fapy-dG (λmax = 270 and 218 nm),46 and a defined peak that eluted after dG with a similar UV–vis profile.46 The broad hump was consistent with literature reports because Fapy-dG exists as anomers of two different sugar configurations.46,47 LC-ESI+-MS of all these new peaks only found masses consistent with the Fapy free base, as well as a peak with mass Fapy + 18 (Figure S24). These observations identify that Fapy-dG is not stable to laboratory manipulation as are d2Ih, dOG, dSp, or dGh, and it appears to be sensitive to nucleophilic buffers in the HPLC mobile phase, e.g., NH4OAc. Further test reactions that had argon bubbled through them for a few seconds up to 1 min before H2O2 addition did not furnish the Fapy peaks. The Fapy peaks only appeared after purging the system with argon for >30 min. To reiterate, in cases with low O2 and Asc present, dOG was the major product observed, and when O2 was at ambient concentrations with Asc present, the major product was d2Ih. These observations identify Fapy-dG to be very challenging to obtain, as it is only observed under extreme anoxic conditions (∼100% argon and ∼0% O2), conditions under which cells cannot survive. If Fapy-dG was formed in a cell, detection of the nucleoside would be problematic due to the hydrolytic instability of the N-glycosidic bond. This later feature is in contrast to d2Ih, dOG, dSp, and dGh that are all at least stable enough to allow their characterization under very mild conditions (22 °C in water buffered at pH 7).
Finally, Fapy-dG has a low redox potential (1.1 V vs NHE)47 rendering it subject to further oxidation; however, this is also the case for dOG (0.7 V vs NHE)13 that has an even lower redox potential, and we detect and quantify this intermediate species and its further oxidation products dSp and dGh. Thus, the conditions outlined do not simply over-oxidize Fapy-dG causing its loss. We conclude that Fapy-dG is not a major contributor to the product distribution of guanosine oxidation by a hydroxyl radical under cellularly relevant conditions.
Reducing-Agent-Dependent Studies for the Iron-Mediated Fenton Reaction
In cells, two main small-molecule reductants are found in high concentrations, ascorbate and glutathione, found at concentrations of 0.8–3 mM and 1–10 mM, respectively.41 Knowing that d2Ih yields increased when Asc was present during the Fe(II)-mediated Fenton oxidation of dG (Table 1), we then studied the role of increasing reductant concentrations on the relative yields of the products. In addition, the nature of the reductant was studied, in which we chose N-acetylcysteine (NAC) as a model for glutathione. Note that protection of the amine group of cysteine with an acetyl group prevents amine adduct formation with dG oxidation intermediates.49,50
Titration of Asc from 0–5 mM into the Fe(II)-mediated Fenton oxidation of dG provided the following trends in the relative product distributions (Figure 2A): (1) As the Asc concentration increased, the reaction yield increased, supporting the role of Asc in allowing the iron catalyst to redox cycle between Fe(II) and Fe(III) and thereby increasing the production of HO• (Reactions 1 and 2). (2) The amount of 2-deoxyribose oxidation decreased (∼10%) as the concentration of Asc increased. This observation negates the involvement of Asc in sugar oxidation, and it is likely that Asc inhibits this pathway. (3) The relative yields of dOG, dSp, and dGh slightly increased (∼6%) with more Asc. (4) The Asc concentration was shown to be critical in the relative yields of the C5 products, dZ and d2Ih. When Asc was not present, the major C5-oxidation product was dZ (also a major product overall); however, as Asc was titrated into the reaction, the relative yield of dZ decreased and d2Ih increased until it was the major product of the reaction at Asc >2 mM (Figure 2A). Within the range of physiological Asc concentrations (1–3 mM),41d2Ih was always the major oxidation product detected (Figure 2A). Moreover, when the study was conducted with variable NAC concentrations, the same trends were observed (Figure 2B). Again, the most notable observation is that d2Ih was the major oxidation product observed in reactions conducted with >2 mM thiol concentrations (Figure 2B), even when considering a comparison of d2Ih (∼37%) with the combined yields of dOG, dSp, and dGh (∼30%, Figure 2A and 2B with 2 mM reductant).
Figure 2.
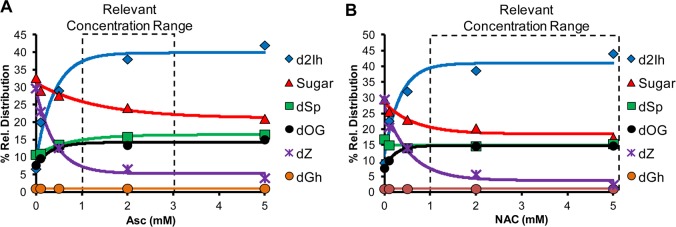
Effect of reductant concentration on relative product distributions observed from the Fe(II)-mediated Fenton reaction. The reactions were conducted with 3 mM dG, 10 μM Fe(II)/EDTA, 10 mM H2O2 in 75 mM NaPi buffer (pH 7.4) at 22 °C while varying the concentration of Asc (A) or NAC (B) from 0–5 mM. Data points represent the average of three trials with an error in each value of 3–10%.
H2O2 Concentration-Dependent Studies for the Iron-Mediated Fenton Reaction
In the next set of studies, dG oxidation product distribution versus the H2O2 concentration (0–10 mM) was monitored with a static amount of reductant (0.8 mM Asc or 3 mM NAC, Figure 3A and 3B, respectively). Key observations from these studies include the following: (1) As expected, the reaction yield increased as more H2O2 was added to the reaction (Supporting Information, Figures S16 and S17). (2) With increased H2O2 concentrations, the flux of HO• increased, and the relative yield of dSp increased at the expense of dOG. This observation supports dOG as the intermediate leading to dSp.10,11 (3) Both dZ and d2Ih increased as a function of H2O2 concentration (0–10 mM); moreover, the amount of d2Ih increased 4-fold when Asc was the reductant while dZ increased 3-fold (Figure 3A). In the H2O2-dependent studies with NAC, d2Ih increased 13-fold while dZ increased 3-fold when H2O2 increased from 0 to 10 mM (Figure 3B). These studies demonstrate that the yield of d2Ih was maximal under conditions that boost formation of HO•.
Figure 3.
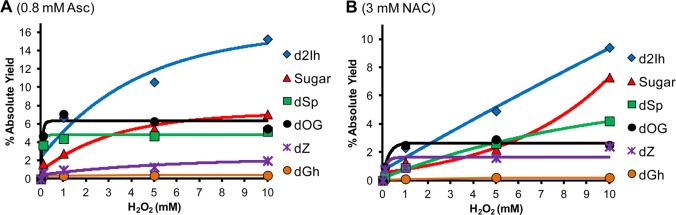
Effect of H2O2 concentration on the absolute product distributions from the Fe(II)-Fenton reaction. The reactions were conducted with 3 mM dG, 10 μM Fe(II)/EDTA, and with either 0.8 mM Asc (panel A) or 3 mM NAC (panel B) in 75 mM NaPi (pH 7.4) at 22 °C, while varying the concentration of H2O2 from 0–10 mM. Data points represent the average of three trials with an error in each value of 5–11%.
Product Quantification from X-ray-Mediated Oxidation of dG
Next, oxidation of dG was conducted with an X-ray source at a dose rate of 25 Gy/min for 30 min (total dose = 750 Gy). Initial studies without a reductant gave a 45% conversion of dG to product. The major oxidation products observed were dZ (14.7%), dSp (10.8%), and Gua (12.4%) and in low absolute yields were d2Ih (2.9%), dOG (1.4%), and dGh (1.4%). Next, Asc (2 mM) was added to the reaction to give the following changes in the yields. (1) The absolute conversion of dG to product decreased with Asc (2.5-fold), as expected, because Asc quenches radical reactions.51 (2) The major products were now d2Ih (6.3%), dSp (3.9%), and Gua (4.1%), with the mass balance completed by low yields of dZ (1.1%), dOG (2.5%), cyclo-dG (0.5%), and dGh (<0.1%). The most striking observation was that d2Ih was the major oxidation product when Asc was present during X-ray mediated oxidations; however, this is in contrast to studies without Asc where dZ was the major oxidation product quantified (Table 2). When considering the yield of d2Ih (6.3%) in comparison to the combined yields of dOG, dSp, and dGh (6.4%), the initial split between d2Ih and the C8 products was nearly equal with X-ray irradiation when reductant was present. Because the X-ray reactions required the reaction vessel to be open to the atmosphere, studies were not conducted under anaerobic conditions.
Table 2. Absolute Yields for Oxidation of dG by X-ray Radiolysis of Watera.
| Absolute
Yield (%) |
||
|---|---|---|
| product | X-ray +Asc/+O2 | X-ray –Asc/+O2 |
| dOG | 2.5 | 1.4 |
| dSp | 3.9 | 10.8 |
| dGh | 0.2 | 1.4 |
| Gh | <0.1 | 0.5 |
| dZ | 1.1 | 14.7 |
| d2Ih | 6.3 | 2.9 |
| cyclo-dG | 0.5 | 0.1 |
| Gua | 4.1 | 12.4 |
| OG | 0.2 | N.D. |
| dG conversion % | 18.8 | 45.3 |
Reported values represent the average of three trials, and the error in each value was 4–10%. N.D. = not detected.
Reducing-Agent-Dependent Studies for the X-ray Mediated Oxidations
Reactions with and without Asc suggest a major role for the reductant in the product distributions for X-ray mediated oxidations (Table 2). Therefore, studies were conducted with the reductant Asc or NAC titrated (0–5 mM) into the X-ray-mediated reaction mixture while monitoring the product yields (Figure 4A and 4B). From these studies, the following observations were made in X-ray-mediated oxidations: (1) As the reducing agent concentration increased, the reaction yield significantly decreased (Supporting Information, Figures S19 and S21). (2) The relative yield of dOG increased (6-fold) as a function of reductant concentration (Figure 4A and 4B), while the yield of dSp decreased (1.5-fold) and dGh decreased (3-fold). This result supports the conclusion that the reductant quenched the further oxidation of dOG to the hydantoins. (3) The yields of the sugar oxidation products were not significantly changed by the increase in reductant concentration (Figure 4A and 4B). (4) The reductant had the most dramatic effect on the C5-pathway products, dZ and d2Ih. When the reductant was not present, dZ was the major product and d2Ih was observed in very low yield; however, when Asc or NAC was increased to relevant concentrations, d2Ih was the major oxidation product observed with very little dZ detected.
Figure 4.
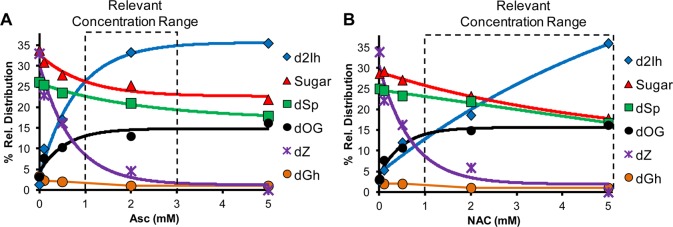
Effect of reductant concentration on the dG-oxidation product yield from the X-ray reaction. The reactions were conducted with 3 mM dG in 75 mM NaPi (pH 7.4) at 22 °C with a 30 min exposure to 25 Gy/min X-ray source (total dose = 750 Gy) while varying the concentration of Asc (panel A) or NAC (panel B) from 0–5 mM. Data points represent the average of three trials with an error in each value of 3–10%.
Total X-ray Dose Effect on dG Oxidation Product Distributions
In the next set of reactions, dG was irradiated with X-rays at a rate of 25 Gy/min while varying the time (i.e., total dose) and keeping the Asc concentration static (2 mM). As the total dose of X-rays increased from 25 to 750 Gy, the following trends were observed. (1) As expected, the overall reaction yield increased (Supporting Information Figure S22). (2) The yield of dSp and dGh increased at the expense of dOG with increasing X-ray dose. (3) The yield of sugar chemistry increased with increasing X-ray dose. (4) The yield of dZ and d2Ih both increased as a function of the X-ray dose, and d2Ih was always the major C5-oxidation pathway product, as well as being the major product of the reaction overall (Figure 5).
Figure 5.
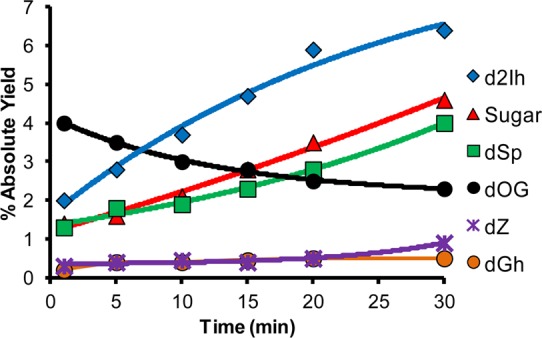
Effect of X-ray irradiation time on dG-oxidation product distributions. The reactions were conducted with 3 mM dG, 2 mM Asc in 75 mM NaPi (pH 7.4) at 22 °C while varying the exposure time (0–30 min) to an X-ray source that was delivered at a rate of 25 Gy/min. Data points represent the average of three trials with an error in each value of 3–10%.
Proposed Pathways Leading to dG Oxidation Products
Under all of the conditions studied, products derived from sugar oxidation were observed. Oxidation at the 5′-carbon yields the 5′ carbon-centered radical that attacks C8 of the heterocyclic ring of dG to furnish an intermediate radical that loses another electron and proton to furnish cyclo-dG (Scheme 1). Oxidation at the 1′, 2′, 3′, 4′, or 5′ carbons also yields a carbon-centered radical that reacts with O2, and many of these pathways ultimately cause cleavage of the N-glycosidic bond releasing Gua (Scheme 1).12,15 Evidence of these sugar oxidation pathways was determined by quantification of Gua release. The yield of cyclo-dG in the Fenton reaction was maximal under anaerobic reducing conditions and was observed to be at a minimum under aerobic nonreducing conditions (Table 1). In contrast, base release to give Gua was observed to be maximal under aerobic and reducing conditions and not observable under anaerobic conditions (Table 1). These results further support the role of O2 in effecting base release from 2-deoxyribose oxidation.12 Moreover, this reaction was not sensitive to the presence of the reductant. However, under anaerobic conditions the 5′-carbon radical was not trapped by O2, but rather added into the heterocyclic ring at C8 leading to cyclo-dG, and again this reaction was not sensitive to the presence of the reductant. Also observed in very low yield were OG and Gh, two products that are derived from further oxidation of Gua (Table 1). The observation of Gh and not Sp under these conditions (pH 7.4 and 22 °C) is consistent with a previous observation that oxidations of the free base OG only give Gh and not Sp.52 In the X-ray-mediated oxidation of dG, O2-dependent studies could not be conducted; therefore, only product dependence on the reductant was studied. In this study, the products derived from sugar oxidation were not greatly affected by the presence of the reductant (Table 2). Other products resulting from oxidation of the 2-deoxyribose sugar in dG were not observed by LC-ESI+-MS (Figure S3).
Scheme 1. Proposed Pathway for Products Derived from Sugar Oxidation.

For the sake of brevity, only oxidation of the 1′-carbon leading to Gua is shown.
Oxidation of dG at the C8 position yields dOG and Fapy-dG.4,10 Mechanistically, these products form when HO• adds at the C8 carbon to yield 8-HO-dG• (Scheme 2). This intermediate can be further oxidized to yield dOG, or be reduced and ring opened to yield Fapy-dG (Scheme 2).4,10 Under all conditions, dOG was observed; however, we did not detect Fapy-dG in these studies without exhaustive removal of O2 (Figure S24). In both the Fenton and X-ray oxidations, the yield of dOG was at a maximum when the reductant was present, which appears to be inconsistent with previous results;20 however, the current studies were conducted under aerobic conditions (Tables 1 and 2), while the previous studies were conducted under anaerobic conditions. Because dOG can undergo further oxidation to hydantoin products (see below),22 the presence of the reductant suppresses the further oxidation and addresses why dOG concentrations are highest under oxidations conducted with the reductant. In conclusion, HO•-mediated oxidation of dG via Fe(II)-Fenton chemistry and X-ray irradiation effects oxidation at C8 of dG to yield dOG as the dominant product along this pathway under low oxidant flux with O2 and the reductant present (Figures 3A, 3B, and 5). Furthermore, as the oxidant flux increases, the yield of dOG decreases due to further oxidation to dSp.
Scheme 2. HO•-Mediated Oxidation of dG at C8 Leading to dOG and Fapy-dG.

The low redox potential of dOG renders this nucleoside labile toward further oxidation to yield the hydantoins dSp and dGh (Scheme 3).22,54 Two-electron oxidation of dOG followed by water attack at C5 yields 5-HO-dOG that bifurcates along two pathways to either dSp or dGh.22,43,44,54 Acyl migration to dSp dominates under conditions of higher pH (pH > 5.8) and unencumbered contexts such as nucleosides, single-stranded DNA, and G-quadruplexes, while the yield of dGh is highest at low pH (pH < 5.8) or sterically encumbered contexts such as double-stranded DNA.22,43,44,54,55 These and previous observations explain why dSp was the major hydantoin observed in all unencumbered nucleoside studies. As previously stated, the yield of dOG was highest when the reductant was present at relevant concentrations (Figures 2 and 4). In contrast, the yield of dSp was highest in the absence of the reductant. This observation supports dOG being the stable intermediate that leads to dSp, as further oxidation of dOG to dSp was quenched with added reductant (Figures 2 and 4). These data, in which dOG was obtained in greater yield than the hydantoins under conditions with physiologically relevant reductant, further explain why concentrations of dOG are generally much greater than hydantoins in vivo.26,56
Scheme 3. Proposed Pathway for Products Derived from Further Oxidation of dOG.
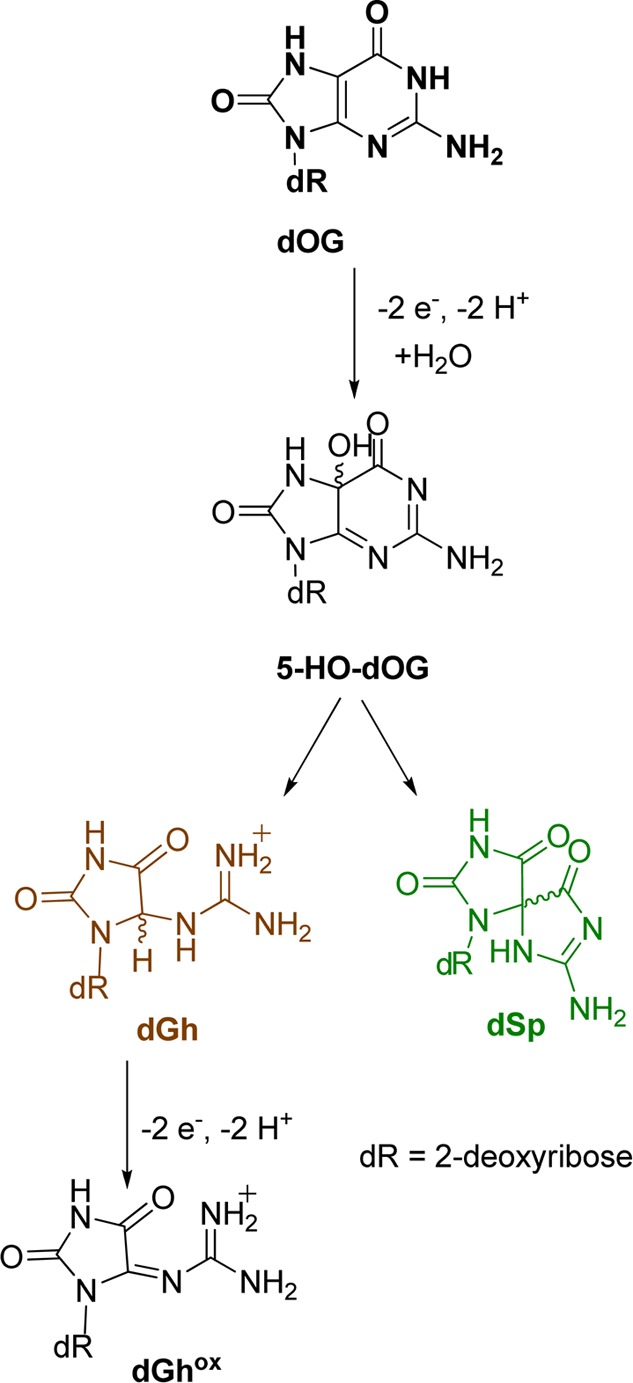
Oxidations of dOG have detected a four-electron oxidation product dehydroguanidinohydantoin-2′-deoxyribonucleoside (dGhox), or formally a six-electron oxidation product of dG (Scheme 3).57 This compound has a short half-life and decomposes to yield oxaluric acid that further hydrolyzes to liberate oxalate and ultimately 2′-deoxyribosyl-urea.58 These analytical conditions allow identification of oxaluric acid (unpublished result from our laboratory); however, under the current HO•-mediated oxidations, neither dGhox nor oxaluric acid were detected. In these studies, low product conversion was maintained, preventing hyperoxidation of dG to dGhox.
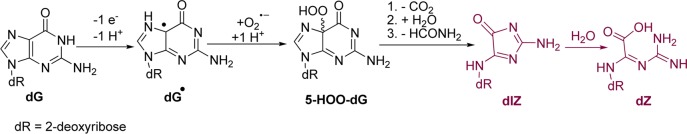
The products dZ and d2Ih result from oxidation of dG followed by initial product forming chemistry occurring at C5. Previous HO•-mediated oxidations of dG detected dZ as a major product under aerobic and nonreducing conditions, which we observed in these studies (Tables 1 and 2).59,60 Mechanistically, dZ was proposed to form from one-electron oxidation of dG (dG•+/dG•(−H+), pKa ∼3.9)55 to a neutral radical that couples with O2/O2•– to ultimately yield a hydroperoxy intermediate (5-HOO-dG). Next, 5-HOO-dG decomposes through a multistep process to dIz followed by hydration to dZ (Scheme 4).54,56,57 Additional support for the role of O2/O2•– in the product defining step leading to dZ was observed in the reactions under anaerobic conditions, for which dZ was not detected (Table 1). Further, as the reducing agent was titrated into the reactions, the yield of dZ dramatically decreased, an observation consistent with a previous report.59
Scheme 4. Proposed Pathway for Oxidation of dG Followed by Initial Reaction at C5 Leading to dZ.
When the oxidations were conducted under aerobic conditions with relevant concentrations of the reductant (Asc or NAC), d2Ih was the major product detected (Figures 3, 4, and 5). In Scheme 5 a mechanism is proposed to explain this observation. Formation of HO• via the Fe(II)-Fenton reaction or X-ray radiolysis of water (Reactions 1 – 4) effects the one-electron oxidation of dG followed by proton loss to yield dG•. Next, dG• couples with O2•– to ultimately yield 5-HOO-dG(24) that is the intermediate susceptible to reduction. When the reductant was not present, 5-HOO-dG decomposes to yield dIz; however, when the reductant was present, the hydroperoxyl group can be reduced to the alcohol 5-HO-dG. Next, 5-HO-dG undergoes acyl migration to reduced-dSp (dSpred) that hydrates at C8 and ring opens to yield d2Ih (Scheme 5A). This proposed mechanism provides a role for O2 and the reducing agent in the formation of d2Ih that was observed in the product distribution studies (Figures 2 and 4). The proposed mechanistic steps from 5-HO-dG to d2Ih were previously proposed from dG oxidations with KHSO5 catalyzed by a Mn–porphyrin complex.36,61
Scheme 5. Proposed Pathways for d2Ih Product Derived from C5 Pathway Oxidation.

Because d2Ih was also observed under anaerobic conditions (Table 1) there must be an alternative pathway for its formation that is not O2 dependent. In Scheme 5B, such a mechanism is proposed in which HO• adds to C5 yielding 5-HO-dG• that proceeds through a second one-electron oxidation and deprotonation to yield 5-HO-dG; acyl migration and hydration then yield d2Ih (Scheme 5B). This second pathway provides a route to d2Ih under anaerobic and nonreducing reaction conditions. The alternative pathway must be a minor reaction channel to d2Ih, because added reductant significantly increases d2Ih formation supporting the mechanism in Scheme 5A.
Product formation upon oxidation of dG initially occurs on the sugar leading to base release or cyclo-dG (Scheme 1), on C5 of the heterocyclic base leading to d2Ih and dZ (Schemes 4 and 5), or on C8 of the heterocyclic base leading to Fapy-dG and dOG (Scheme 2), in which dOG is further oxidized to dSp and dGh (Scheme 3). Inspection of the product distributions allows ranking the initial dG site of reactivity toward oxidation under each condition. The focus will be on reactions conducted under aerobic conditions with and without the addition of Asc (Tables 1 and 2). The iron-Fenton reaction without the addition of Asc focused on the sugar, C5, and C8 carbons in a ratio of 1.3:1.5:1.0, respectively, with the C5 products showing the highest yield. A comparison of the C5 and C8 product distributions show that C5 products are ∼150% of the C8 products. When Asc (2 mM) was added to the mixture, the ratio of reactivity was 1.0:1.9:1.3 for the sugar, C5, and C8 carbons, respectively (Table 1). The addition of Asc to the iron-Fenton reaction suppressed oxidation of the sugar, and initial reaction at the C5 was still greater than C8. More interestingly, C5 products were still ∼150% more than the C8 products. This observation is consistent with the proposed mechanistic role of Asc in product formation, in which it reduces intermediate species that have already reacted at C5 or C8 (Scheme 5).
The X-ray mediated oxidation of dG without Asc yielded sugar, C5, and C8 products in a 1.0:1.4:1.0 ratio, respectively (Table 2). Initial reactivity at C5 was the dominant pathway, and it was ∼140% greater than the C8 pathway, similar to that observed for the iron-Fenton mediated oxidation of dG. When Asc was added to the mixture the ratio of reactivity at the sugar, C5, and C8 sites on dG was 1.0:1.5:1.4, respectively. Overall, Asc decreased the sugar oxidation products (Table 2) and the C5 and C8 products were found in nearly the same yield. The reason C5 and C8 products were observed in similar yields has to do with dZ. The presence of Asc dramatically diminished the yield of dZ in the X-ray mediated oxidations, leading to less C5 products and similar yields of C5 and C8 products. These comparisons support a greater level of oxidation occurring at the heterocyclic ring of the dG nucleoside compared to the sugar particularly with Asc present.
Use of a Hypercarb column allowed separation of the d2Ih and dSp diastereomers for which the absolute configurations and elution order on this HPLC column are known.42,62 In the current studies, the R and S isomers of d2Ih were observed in a 1:2 ratio, respectively, for both oxidation reactions studied, while the R and S isomers of dSp were observed in a 1:1 ratio. Furthermore, these ratios did not change under any of the reaction conditions studied. The observation that the d2Ih isomers were not in a 1:1 ratio points to steric hindrance during the defining point of the reaction that determines product stereochemistry and was likely caused by the 2-deoxyribose sugar. In contrast to this result, the diastereomer ratio of d2Ih found from the copper-mediated Fenton oxidation was 2:1.8 In the other d2Ih studies, the diastereomer identity and ratios were not stated.35,36,38−40
In the last study, the products resulting from base oxidation of dG in oligonucleotides of known sequence in single- (ODN1) and double-stranded (ODN2) contexts were determined, as well as products observed from λ-DNA. The product analysis was achieved using HF to hydrolyze the bases from the sugar–phosphate backbone. This approach prevented the quantification of sugar oxidation leading to base release. Because of this limitation, only base oxidation products are compared. Further, products resulting from oxidation of other nucleotides, leading to thymine glycol, for example, were observed but were not quantified; masses consistent with guanine cross-links with thymine and cytosine were observed, but their yields are not reported due to a lack of established extinction coefficients to obtain their yields (Figure S24). These limitations in product analysis prevent an ideal comparison; however, we can use these data to understand context-dependent product distributions leading to C5 products (2Ih and Z) vs C8 products (OG, Sp, and Gh) when iron-Fenton or X-ray mediated oxidations were conducted with Asc (2 mM) present under aerobic reaction conditions.
 |
The overall context-dependent trends were very similar for iron-Fenton and X-ray irradiation reactions (Figure 6 A and B). The relative yield of Z diminished dramatically when comparing oxidations between nucleoside to ODN and DNA contexts. In the duplex contexts no Z was observed, an observation similar to previous studies of the added reductant during the oxidations.52 The OG free base was observed in all contexts and comprised the smallest relative yield in the ODN and DNA contexts and largest yield in the nucleoside studies from both iron-Fenton and X-ray oxidations. The yield of Sp and Gh showed strong context-dependent yields, in which Sp was greatest in nucleoside and single-stranded contexts and Gh was observed to be greatest in duplex contexts. This observation is consistent with previous reports.8,43 Lastly, the relative yield of 2Ih remained nearly the same in all three contexts (∼40–50%, Figure 6A and B). Without complete mass balances for oxidations in the ODN and DNA contexts, it cannot be definitively stated that d2Ih is the major oxidation product of dG; nonetheless, d2Ih is clearly a major product observed from oxidation of dG in the nucleoside, single- and double-stranded ODN, and λ-DNA contexts. This observation supports the possibility of d2Ih formation in the cellular context.
Figure 6.
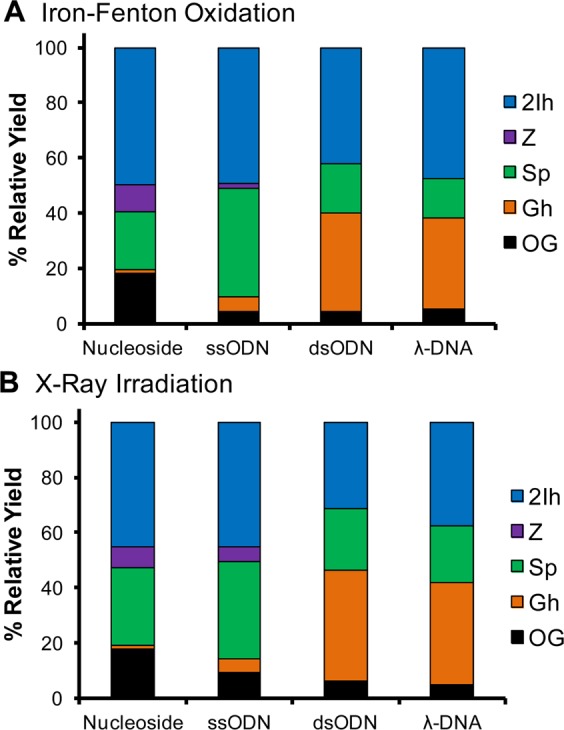
Context-dependent yields of dG base oxidation products from the iron-Fenton and X-ray irradiated samples. The ODN and λ-DNA distributions were obtained after HF hydrolysis of the oxidized samples. In the ODN and λ-DNA samples, products were also observed from oxidation of the other nucleotides. Because of the HF hydrolysis to liberate the oxidized free bases, the extent of sugar oxidation leading to free base could not be determined.
The current results are compared to literature studies that reported d2Ih as shown in Table 3. The Ball laboratory exclusively observed d2Ih when dG was exposed to the epoxidizing reagent dimethyldioxirane (DMDO), and they confirmed the structure of d2Ih via complementary NMR methods.37,63 The Meunier laboratory observed d2Ih (M+34) when dG was oxidized with Mn-TMPyP/KHSO5.36,61 Studies in the Karlin and Rokita laboratories observed a product mass in high yield consistent with d2Ih (M+34) when an oligodeoxynucleotide was allowed to react with a dicopper(II)-complex.38,64 In our laboratory, d2Ih was detected in high yield with the NiCR/KHSO5 system, and in studies utilizing the copper-mediated Fenton oxidation of dG.8,35 Further, the Bohme laboratory conducted the lead-mediated Fenton oxidation of dG to yield d2Ih as the major product.40 In all of the metal-catalyzed studies, the transition metal was proposed to play a role in the product-forming step leading to d2Ih. However, studies conducted in the Shafirovich laboratory observed d2Ih during CO3•– oxidations of dG, demonstrating that a transition metal was not required for d2Ih formation.39 In the current report, d2Ih was the major product of “free” HO•-mediated oxidation of dG with relevant amounts of reductant under aerobic conditions. The X-ray studies demonstrate once again that d2Ih can be formed under conditions that do not require the involvement of a transition-metal catalyst.
Table 3. Comparison of the Current Results to Other Reportsa.
| initial
reaction at C8 |
initial
reaction at C5 |
initial reaction at 2-deoxyribose | ||||
|---|---|---|---|---|---|---|
| products |
||||||
| oxidants | dOG | dSp/dGh | dIz/dZ | d2Ih | sugar | reference |
| HO• (radiolysis) | + | + | ++ | N.D. | + | (59,60) |
| CO3•– | + | + | + | ++ | + | (39) |
| Pb(II)/H2O2 | + | + | + | ++ | + | (40) |
| NiCR/KHSO5 | N.D. | + | N.D. | ++ | N.D. | (35) |
| DMDO | N.D. | N.D. | N.D. | ++ | N.D. | (37) |
| Cu(I)/H2O2 | + | + | + | ++ | + | (8) |
| Dicopper(II)-complex | N.D. | N.D. | N.D. | ++ | N.D. | (64) |
| Mn-TMPyP/KHSO5 | N.D. | + | + | ++ | + | (61) |
| Fe(II)-EDTA/H2O2/Asc or NAC | + | + | + | ++ | + | current study |
| X-ray/Asc or NAC | + | + | + | ++ | + | current study |
(++) a major product, (+) a minor product, (N.D.) not detected or not reported.
Conclusions
The current studies monitored oxidation products from dG resulting from HO• generated via the Fe(II)-mediated Fenton reaction or X-ray radiolysis of water. Products resulting from oxidation and initial reaction at the sugar, C5, or C8 carbons were quantified (Figures 2–5). Under aerobic conditions without a reductant, the major products observed include dZ, dSp, and sugar oxidation leading to Gua. Under anaerobic reaction conditions the major product observed was dSp. When reactions were conducted with cellularly relevant (mM) amounts of the reductant (Asc or NAC) and O2, the major product observed in all cases was d2Ih. The isomers of d2Ih are two-electron oxidation products of dG that have not been previously detected in oxidations with HO• generated by the Fe(II)-mediated Fenton reaction or X-ray radiolysis of H2O. The high yields of d2Ih observed and the observation that this lesion is highly prone to piperidine cleavage in a DNA oligomer, unlike dOG,65 lead to a hypothesis that oxidations in DNA imposed by electron transfer agents yield C5 oxidation products (i.e., d2Ih or dZ) and not the C8 oxidation product dOG.66 Oxidations in the context of single- and double-stranded ODNs and λ-DNA yielded d2Ih as a product of dG oxidation in significant yield that was competitive with the yield of dOG. More importantly, the current results highlight d2Ih as a major product observed in reactions that include relevant amounts of reductant in the presence of O2, suggesting that d2Ih should be investigated more closely for its biochemical properties.
Experimental Procedures
Fenton Reaction
A 200-μL solution of dG (3.0 mM, 0.6 μmoles, 0.16 mg) in a buffer (75.0 mM NaPi, pH 7.4) was mixed with Fe(II)/EDTA (0.10 mM, 0.02 μmoles, 0.008 mg), H2O2 (10.0 mM, 2 μmoles, 0.5 mg), and Asc or NAC (2.0 mM, 0.40 μmoles, 0.03 mg) and incubated for 1 h at 22 °C. The Fe(II)/EDTA complex was freshly made by mixing Fe(NH4)2(SO4)2 and Na2EDTA in a 1:2 ratio 30 min prior to reaction. The Fe(II)/EDTA complex was removed by column chromatography using ion-exchange resin prior to HPLC analysis following the method outlined below.
X-ray Irradiation
A 200-μL solution containing dG (3.0 mM, 0.6 μmoles, 0.16 mg) in buffer (75.0 mM NaPi, pH 7.4) and Asc or NAC (2.0 mM, 0.40 μmoles, 0.03 mg) was irradiated with an X-ray RS 2000 biological research irradiator source at 22 °C for variable times (1–30 min). The dose rate was 25 Gy/min.59 This solution was analyzed by the method outlined below.
Oligodeoxynucleotide and DNA Oxidations
Single-stranded ODN1 or double-stranded ODN2 were oxidized in 20 mM NaPi (pH 7.4) with 100 mM NaCl at 37 °C. Iron-Fenton oxidations were conducted in a 200-μL reaction volume with ODN (100 μM, 0.01 μmoles, 0.05 mg) to which were added Fe(II)EDTA (0.10 mM, 0.02 μmoles, 0.008 mg), Asc (2.0 mM, 0.40 μmoles, 0.03 mg), and H2O2 (1.0 mM, 0.2 μmoles, 0.05 mg); the reaction was allowed to proceed for 30 min. The stock solutions of Fe(II)EDTA, Asc, and H2O2 were all freshly prepared. To irradiate the ODNs, they were placed in the same buffer system as the iron-Fenton reaction followed by X-ray irradiation with a dose rate of 25 Gy/min for 30 min (total dose = 750 Gy). For the λ-DNA oxidations, everything was identical to the ODN oxidations with the exception that the λ-DNA (1 μM, 0.1 nmol, 0.03 mg) was different.
Hydrolysis of Oxidized Oligodeoxynucleotides and λ-DNA
The oxidized ODN or λ-DNA samples were hydrolyzed to the free bases for HPLC analysis. The hydrolysis was performed on the oxidized and lyophilized DNA by adding 50 μL of 70% HF in pyridine for 30 min at 37 °C. After the reaction, the excess HF was neutralized by adding 1 mL of ddH2O and 80 mg of CaCO3 to the sample. The insoluble salts were removed by centrifugation, and the supernatant was then lyophilized to dryness. Next, the lyophilized samples were dissolved in ddH2O and submitted to LC-MS and HPLC analysis as described for the nucleoside studies below.
Product Identification
Product identification was initially achieved by UPLC-ESI+-MS (100 mm × 2.1 mm, 1.7 μm) and UPLC-ESI+-MS with a Hypercarb column (100 mm × 2.1 mm, 5 μm). Then, each compound was HPLC purified for further structural analysis. The following masses were observed: Guam/z [M + H]+ calcd 152.1, found 152.1. OGm/z [M + H]+ calcd 168.1, found 168.1. dOGm/z [M + H]+ calcd 284.2, found 284.1. R and S diastereomers of cyclo-dGm/z [M + H]+ calcd 266.2, found 266.1. Ghm/z [M + H]+ calcd 158.1, found 158.1. (S)-d2Ih and (R)-d2Ih(42)m/z [M + H]+ calcd 302.3, found 302.1; ESI+-MS/MS m/z [M + H]+ lit.37 186, 158, and 141; found 186, 158, and 141. HRMS (ESI-TOF) m/z [M + Na]+ calcd for C5H7N5O3Na 208.0447, found 208.0449. The HRMS value was obtained on the free base 2Ih, due to the nucleoside’s instability toward acid. dGhm/z [M + H]+ calcd 274.3, found 274.1; HRMS (ESI-TOF) m/z [M + Na]+ calcd for C9H15N5O5Na 296.0971, found 296.0980. (S)-dSp and (R)-dSp(62)m/z [M + H]+ calcd 300.2, found 300.1; HRMS (ESI-TOF) m/z [M + Na]+ calcd for C10H13N5O6Na 322.0764, found 322.0761; ESI+-MS/MS m/z [M + H]+ lit.22 184, 156, 141, 113, 99, and 86; found 184, 156, 141, 113, 99, and 86. dZm/z [M + H]+ calcd 247.2, found 247.1; HRMS (ESI-TOF) m/z [M + Na]+ calcd for C8H14N4O5Na 269.0862, found 269.0870; ESI+-MS/MS m/z [M + H]+ lit.67 247, 203, and 131; found 247, 203, and 131 (Figures S3–S13, Supporting Information). The dGh diastereomers were characterized by NMR,44dZ was characterized by NMR,30cyclo-dG was characterized by X-ray crystallography,68 the dSp diastereomers have been characterized by X-ray crystallography69 and NMR,70 and the diastereomers of d2Ih have been previously characterized by NMR.37
Product Quantification
The oxidation products from the Fe(II)-Fenton reaction or X-ray irradiation reaction were quantified by C18 reversed-phase HPLC and Hypercarb HPLC. First, the reaction mixture was injected on a reversed-phase HPLC (250 mm × 4.6 mm, 5 μm) to quantify Gua, OG, dOG, and cyclo-dG. The void volume from this run was collected, lyophilized to dryness, and then dissolved with the Hypercarb column starting mobile phase (0.1% acetic acid). Next, the reconstituted void volume was injected on a Hypercarb column (150 mm × 4.6 mm, 5 μm) to quantify Gh, (R)-d2Ih, (S)-d2Ih,42dGh, (R)-dSp, (S)-dSp,62 and dZ. Product peaks were quantified by their absorbance intensity at 240 nm followed by normalization of these intensities by each compound’s extinction coefficient at 240 nm. The values for ε240 nm (ddH2O) are dG 14 080, dOG 14 300, cyclo-dG 14 080, Gua 14 080, OG 14 300,15dSp 3280,22d2Ih 2290,8,35dGh and Gh 2410,54 and dZ 1780.67 All values are in units of L mol–1 cm–1.
Acknowledgments
The authors are grateful to Dr. James Muller (University of Utah) for his assistance with mass spectrometry and to the National Institutes of Health for financial support (R01 CA090689).
Supporting Information Available
UPLC-ESI+-MS, HPLC-ESI+-MS, ESI+-MS/MS, reversed-phase HPLC, Hypercarb HPLC, UV–vis spectra, and product distribution tables. The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.joc.5b00689.
The authors declare no competing financial interest.
Supplementary Material
References
- Sies H. Klin. Wochenschr. 1991, 69, 965–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breen A. P.; Murphy J. A. Free Radic. Biol. Med. 1995, 18, 1033–1077. [DOI] [PubMed] [Google Scholar]
- Ames B. N.; Sheigenaga M. K.; Hagen T. M. Biochem. Biophys. Acta 1995, 1271, 165–170. [DOI] [PubMed] [Google Scholar]
- Burrows C. J.; Muller J. G. Chem. Rev. 1998, 98, 1109–1152. [DOI] [PubMed] [Google Scholar]
- Cadet J.; Douki T.; Gasparutto D.; Ravanat J.-L. Mutat. Res. 2003, 531, 5–23. [DOI] [PubMed] [Google Scholar]
- Walling C. Acc. Chem. Res. 1998, 31, 155–157. [Google Scholar]
- MacFaul P. A.; Wayner D. D. M.; Ingold K. U. A. Acc. Chem. Res. 1998, 31, 159–162. [Google Scholar]
- Fleming A. M.; Muller J. G.; Ji I.; Burrows C. J. Org. Biomol. Chem. 2011, 9, 3338–3348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadet J.; Douki T.; Ravanat J.-L. Acc. Chem. Res. 2008, 41, 1075–1083. [DOI] [PubMed] [Google Scholar]
- Pratviel G.; Meunier B. Chem.—Eur. J. 2006, 12, 6018–6030. [DOI] [PubMed] [Google Scholar]
- Gimisis T.; Cismas C. Eur. J. Org. Chem. 2006, 1351–1378. [Google Scholar]
- Pogozelski W. K.; Tullius T. D. Chem. Rev. 1998, 98, 1089–1108. [DOI] [PubMed] [Google Scholar]
- Steenken S.; Jovanovic S. V.; Bietti M.; Bernhard K. J. Am. Chem. Soc. 2000, 122, 2373–2374. [Google Scholar]
- Devasagayam T. P.; Steenken S.; Obendorf M. S.; Schulz W.; Sies H. Biochemistry 1991, 30, 6283–6289. [DOI] [PubMed] [Google Scholar]
- Henle E. S.; Luo Y.; Gassmann W.; Linn S. J. Biol. Chem. 1996, 271, 21177–21186. [PubMed] [Google Scholar]
- Grey C. E.; Adlercreutz P. J. Agric. Food Chem. 2006, 54, 2350–2358. [DOI] [PubMed] [Google Scholar]
- Belmadoui N.; Boussicault F.; Guerra M.; Ravanat J.-L.; Chatgilialoglu C.; Cadet J. Org. Biomol. Chem. 2010, 8, 3211–3219. [DOI] [PubMed] [Google Scholar]
- Pouget J.-P.; Frelon S.; Ravanat J.-L.; Testard I.; Odin F.; Cadet J. Radiat. Res. 2002, 157, 589–595. [DOI] [PubMed] [Google Scholar]
- Dizdaroglu M.; Rao G.; Halliwell B.; Gajewski E. Arch. Biochem. Biophys. 1991, 285, 317–324. [DOI] [PubMed] [Google Scholar]
- Douki T.; Martini R.; Ravanat J.-L.; Turesky R. J.; Cadet J. Carcinogenesis 1997, 18, 2385–2391. [DOI] [PubMed] [Google Scholar]
- Gedik C. M.; Collins A. FASEB J. 2005, 19, 82–84. [DOI] [PubMed] [Google Scholar]
- Luo W.; Muller J. G.; Rachlin E. M.; Burrows C. J. Org. Lett. 2000, 2, 613–616. [DOI] [PubMed] [Google Scholar]
- Slade P. G.; Hailer M. K.; Martin B. D.; Sugden K. D. Chem. Res. Toxicol. 2005, 18, 1140–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misiaszek R.; Crean C.; Geacintov N. E.; Shafirovich V. J. Am. Chem. Soc. 2005, 127, 2191–2200. [DOI] [PubMed] [Google Scholar]
- Niles J. C.; Wishnok J. S.; Tannenbaum S. R. Chem. Res. Toxicol. 2004, 17, 1510–1519. [DOI] [PubMed] [Google Scholar]
- Mangerich A.; Knutson C. G.; Parry N. M.; Muthupalani S.; Ye W.; Prestwich E.; Cui L.; McFaline J. L.; Mobley M.; Ge Z.; Taghizadeh K.; Wishnok J. S.; Wogan G. N.; Fox J. G.; Tannenbaum S. R.; Dedon P. C. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, E1820–E1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hailer M. K.; Slade P. G.; Martin B. D.; Sugden K. D. Chem. Res. Toxicol. 2005, 18, 1378–1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson P. T.; Delaney J. C.; Muller J. G.; Neeley W. L.; Tannenbaum S. R.; Burrows C. J.; Essigmann J. M. Biochemistry 2003, 42, 9257–9262. [DOI] [PubMed] [Google Scholar]
- Delaney J. C.; Neeley W. L.; Delaney J. C.; Essigmann J. M. Biochemistry 2007, 46, 1448–1455. [DOI] [PubMed] [Google Scholar]
- Cadet J.; Berger M.; Buchko G. W.; Joshi P. C.; Raoul S.; Ravanat J.-L. J. Am. Chem. Soc. 1994, 116, 7403–7404. [Google Scholar]
- Raoul S.; Berger M.; Buchko G. W.; Joshi P. C.; Morin B.; Weinfeld M.; Cadet J. J. Chem. Soc., Perkin Trans. 1996, 2, 371–381. [Google Scholar]
- Gasparutto D.; Ravanat J.-L.; Gérot O.; Cadet J. J. Am. Chem. Soc. 1998, 120, 10283–10286. [Google Scholar]
- Kino K.; Saito I.; Sugiyama H. J. Am. Chem. Soc. 1998, 120, 7373–7374. [Google Scholar]
- Kupan A.; Saulière A.; Broussy S.; Seguy C.; Pratviel G.; Meunier B. Chem. Bio. Chem. 2006, 7, 125–133. [DOI] [PubMed] [Google Scholar]
- Ghude P.; Schallenberger M. A.; Fleming A. M.; Muller J. G.; Burrows C. J. Inorg. Chem. Acta 2011, 369, 240–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vialas C.; Claparols C.; Pratviel G.; Meunier B. J. Am. Chem. Soc. 2000, 122, 2157–2167. [Google Scholar]
- Ye W.; Sangaiah R.; Degen D. E.; Gold A.; Jayaraj K.; Koshlap K. M.; Boysen G.; Williams J.; Tomer K. B.; Mocanu V.; Dicheva N.; Parker C. E.; Schaaper R. M.; Ball L. M. J. Am. Chem. Soc. 2009, 131, 6114–6123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L.; Murthy N. N.; Telser J.; Zakharov L. N.; Yap G. P. A.; Rheingold A. L.; Karlin K. D.; Rokita S. E. Inorg. Chem. 2006, 45, 7144–7159. [DOI] [PubMed] [Google Scholar]
- Rokhlenko Y.; Geacintov N. E.; Shafirovich V. J. Am. Chem. Soc. 2012, 134, 4955–4962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banu L.; Blagojevic V.; Bohme D. K. J. Phys. Chem. B 2012, 116, 11791–11797. [DOI] [PubMed] [Google Scholar]
- Meister A.; Anderson M. E. Annu. Rev. Biochem. 1983, 52, 711–760. [DOI] [PubMed] [Google Scholar]
- Fleming A. M.; Alshykhly O.; Orendt A. M.; Burrows C. J. Tetrahedron Lett. 2015, 56, 3191–3196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming A. M.; Muller J. G.; Dlouhy A. C.; Burrows C. J. J. Am. Chem. Soc. 2012, 134, 15091–15102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Y.; Muller J. G.; Luo W.; Mayne C. L.; Shallop A. J.; Jones R. A.; Burrows C. J. J. Am. Chem. Soc. 2003, 125, 13926–13927. [DOI] [PubMed] [Google Scholar]
- Extinction coefficients (ε240 nm) for dSp and d2Ih were assumed to be the same value; however, ring opening of the imidazole ring is anticipated to lead to a lower value of ε240 nm for d2Ih compared to the ring closed dSp structure. Support for this claim was taken from the TDDFT calculations we have conducted to determine the UV spectroscopic properties of d2Ih and dSp (see refs (42) and (62)). In these studies, UV–vis spectra were calculated with explicit and implicit solvation using the M06-2X functional and 6-311++G(2d,2p) basis set with implicit definition of water using the PCM, and these data allow a relative comparison of the ε240 nm between dSp and d2Ih to be made. These calculations identify that d2Ih absorbs light at ∼70% of dSp (see Figure S23).
- Crespo-Hernandez C. E.; Arce R. J. Photochem. Photobiol., B 2004, 73, 167–175. [DOI] [PubMed] [Google Scholar]
- Greenberg M. M. Acc. Chem. Res. 2012, 45, 588–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frelon S.; Douki T.; Favier A.; Cadet J. J. Chem. Soc., Perkin Trans. 1 2002, 2866–2870. [Google Scholar]
- Xiaoyun X.; Muller J. G.; Ye Y.; Burrows C. J. J. Am. Chem. Soc. 2008, 130, 703–709. [DOI] [PubMed] [Google Scholar]
- Solivio M. J.; Joy T. J.; Sallans L.; Merino E. J. J. Inorg. Biochem. 2010, 104, 1000–1005. [PubMed] [Google Scholar]
- Lee J.; Koo N.; Min D. B. Comp. Rev. Food Sci. Food Safety 2004, 3, 21–33. [DOI] [PubMed] [Google Scholar]
- Yu H.; Niles J. C.; Wishnok J. S.; Tannenbaum S. R. Org. Lett. 2004, 6, 3417–3420. [DOI] [PubMed] [Google Scholar]
- Steenken S.; Jovanovic S. V.; Bietti M.; Bernhard K. J. Am. Chem. Soc. 2000, 122, 2373–2374. [Google Scholar]
- Luo W.; Muller J. G.; Rachlin E. M.; Burrows C. J. Chem. Res. Toxicol. 2001, 14, 927–938. [DOI] [PubMed] [Google Scholar]
- Fleming A. M.; Burrows C. J. Chem. Res. Toxicol. 2013, 26, 593–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hailer M. K.; Slade P. G.; Martin B. D.; Sugden K. D. Chem. Res. Toxicol. 2005, 18, 1378–1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim K. S.; Cui L.; Taghizadeh K.; Wishnok J. S.; Chan W.; DeMott M. S.; Babu I. R.; Tannenbaum S. R.; Dedon P. C. J. Am. Chem. Soc. 2012, 134, 18053–18064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neeley W. L.; Essigman J. M. Chem. Res. Toxicol. 2006, 19, 491–505. [DOI] [PubMed] [Google Scholar]
- Douki T.; Spinelli S.; Ravanat J.-L.; Cadet J. J. Chem. Soc., Perkin Trans. 2 1999, 1875–1880. [Google Scholar]
- Cui L.; Ye W.; Prestwich E. G.; Wishnok J. S.; Taghizadeh K.; Dedon P. C.; Tannenbaum S. R. Chem. Res. Toxicol. 2013, 26, 195–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomaszewska A.; Mourgues S.; Guga P.; Nawrot B.; Pratviel G. Chem. Res. Toxicol. 2012, 25, 2505–2512. [DOI] [PubMed] [Google Scholar]
- Fleming A. M.; Orendt A. M.; He Y.; Zhu J.; Dukor R. K.; Burrows C. J. J. Am. Chem. Soc. 2013, 135, 18191–18204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye W.; Sangaiah R.; Degen D. E.; Gold A.; Jayaraj K.; Koshlap K. M.; Boysen G.; Williams J.; Tomer K. B.; Ball L. M. Chem. Res. Toxicol. 2006, 19, 506–510. [DOI] [PubMed] [Google Scholar]
- Li L.; Karlin K. D.; Rokita S. E. J. Am. Chem. Soc. 2005, 127, 520–521. [DOI] [PubMed] [Google Scholar]
- Fleming A. M.; Alshykhly O.; Zhu J.; Muller J. G.; Burrows C. J. Chem. Res. Toxicol. 2015, 28, 1292–1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanvah S.; Joseph J.; Schuster G. B.; Barnett R. N.; Cleveland C. L.; Landman U. Acc. Chem. Res. 2010, 43, 280–287. [DOI] [PubMed] [Google Scholar]
- Matter B.; Malejka-Giganti D.; Csallany A. S.; Tretyakova N. Nucleic Acids Res. 2006, 34, 5449–5460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H.; Das R. S.; Basu A. K.; Stone M. P. J. Am. Chem. Soc. 2011, 133, 20357–20368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckenroth B. E.; Fleming A. M.; Sweasy J. B.; Burrows C. J.; Doublié S. Biochemistry 2014, 53, 2075–2077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karwowski B.; Dupeyrat F.; Bardet M.; Ravanat J.-L.; Krajewski P.; Cadet J. Chem. Res. Toxicol. 2006, 19, 1357–1365. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.