

Abstract
Erythropoietin (EPO), a glycoprotein originally known for its important role in the stimulation of erythropoiesis, has recently been shown to have significant protective effects in animal models of kidney and intestinal ischemia-reperfusion injury (IRI). However, the mechanism underlying these protective effects remains unclear. The aim of the current study was to evaluate the effects of EPO on myocardial IRI and to investigate the mechanism underlying these effects. A total of 18 male Sprague Dawley rats were randomly divided into three groups, namely the sham, IRI-saline and IRI-EPO groups. Rats in the IRI-EPO group were administered 5,000 U/kg EPO intraperitoneally 24 h prior to the induction of IRI. IRI was induced by ligating the left descending coronary artery for 30 min, followed by reperfusion for 3 h. Pathological changes in the myocardial tissue were observed and scored. The levels of the proinflammatory cytokines, interleukin (IL)-6, IL-1β and tumor necrosis factor (TNF)-α, were evaluated in the serum and myocardial tissue. Furthermore, the effects of EPO on phosphoinositide 3-kinase/protein kinase B (PI3K/Akt) signaling and EPO receptor (EPOR) phosphorylation were measured. Pathological changes in the myocardial tissue, increased expression levels of TNF-α, IL-6 and IL-1β in the myocardium, and increased serum levels of these mediators, as a result of IRI, were significantly decreased by EPO pretreatment. The effects of EPO were found to be associated with the activation of PI3K/Akt signaling, which suppressed the inflammatory responses, following the initiation of EPOR activation by EPO. Therefore, EPO pretreatment was demonstrated to decrease myocardial IRI, which was associated with activation of EPOR, subsequently increasing PI3K/Akt signaling to inhibit the production and release of inflammatory mediators. Thus, the results of the present study indicated that EPO may be useful for preventing myocardial IRI.
Keywords: erythropoietin, myocardial ischemia-reperfusion injury, phosphoinositide 3-kinase/protein kinase B
Introduction
Myocardial ischemia-reperfusion injury (IRI) is a common pathophysiological event that occurs in a variety of clinical settings, including cardiopulmonary resuscitation, myocardial transplantation, trauma and shock (1,2). The molecular mechanisms underlying myocardial IRI are complex, specific details remain unclear. However, there is evidence to suggest that inflammation plays an important role in myocardial IRI (3,4). Therefore, suppressing inflammation may be an effective strategy for the treatment of myocardial IRI.
Phosphoinositide 3-kinase (PI3K) is composed of a catalytic subunit (p110) and a regulatory subunit (p85) (5); activation of the catalytic subunit relies on p85 phosphorylation. The activation of p85 by phosphorylation leads to the phosphorylation of protein kinase B (Akt), which subsequently results in the production of proinflammatory mediators, such as tumor necrosis factor (TNF)-α, interleukin (IL)-6 and IL-1β (6). However, further activation of PI3K signaling has been demonstrated to significantly suppress inflammation in IRI (7).
Erythropoietin (EPO) is a glycoprotein cytokine that is produced primarily in the kidneys (8). Previously, EPO has been widely used in clinical practice due to its ability to stimulate erythropoiesis (8,9). In addition to this well-known and widely recognized effect, EPO, has been shown to significantly attenuate inflammation in kidney and brain IRI through regulating the PI3K/Akt signaling pathway (10,11). However, the impact of EPO, and the associated PI3K/Akt signaling pathway, on myocardial IRI has not, to the best of our knowledge, been previously studied. Therefore, the aim of the present study was to establish a rat model of IRI in order to investigate this area further.
Materials and methods
Animals
A total of 18 male Sprague Dawley (SD) rats (6–8 weeks-old; weight, 200–250 g) were purchased from Hua Fukang Experimental Animal Center (Beijing, China). The rats were housed in a specific pathogen-free facility at 18–29°C on a 14 h light/10 h dark cycle. The rats were provided with standard laboratory chow and water ad libitum. Following a minimum acclimatization period of 7 days, the rats were randomly allocated to three groups (n=6 per group): i) IRI-saline group (IRI-SAL), in which the rats were administered intraperitoneal (IP) saline injections 24 h prior to IRI induction; ii) IRI-EPO group, in which the rats were administered EPO (5,000 U/kg, IP) 24 h prior to IRI induction; and iii) sham-operated group (Sham), in which the rats were subjected to the same surgical procedures without occlusion of the left descending coronary artery. The dosage of EPO was selected based on that of a previous study (12). The study was approved by the Institutional Animal Care and Use Committee at the West China Hospital of Sichuan University (Chengdu, China).
IRI induction in the heart
Rats were anesthetized by an IP injection of 1% pentobarbital sodium solution (6 ml/kg; Qiao Xing, Shanghai, China), and their body temperature was regulated by a lamp (Pang Kang, Shanghai, China) during the procedure. Myocardial IRI was induced as follows. The rats were placed in the supine position and an incision was made in the left side of the chest. The heart was exposed and exteriorized. A slipknot (4-0 silk; Ai De, Anhui, China) was placed around the left anterior descending coronary artery. After 30 min of ischemia, the slipknot was released and the myocardium was reperfused for 3 h. Rats in the Sham group underwent the same surgical procedure as the other groups, with the exception of coronary artery occlusion.
Histological analysis
After 3 h of reperfusion, the excised hearts were fixed with 10% formalin, embedded in paraffin and stained with hematoxylin and eosin. The hearts were visualized under a CX31 microscope (1,000x magnification; Olympus Corporation, Tokyo, Japan). Myocardial IRI was scored in the most obvious part of the lesion, according to previously published morphological criteria (13) as follows: 0, no damage; 1 (mild), interstitial edema and localized necrosis; 2 (moderate), widespread myocardial cell swelling and necrosis; 3 (severe), necrosis with contraction bands and compressed capillaries; and 4 (highly severe), diffuse necrosis with contraction bands, compressed capillaries and hemorrhage.
Western blot analysis
Myocardial samples (50 mg), were mechanically homogenized in 1 ml hypotonic buffer (200 µl HEPES, pH 7.5; 25 mmol/l magnesium chloride; 5 mmol/l dithiothreitol; 5 mmol/l phenylmethylsulfonyl fluoride; 2 mmol/l pepstatin A; 10 µg/ml leupeptin; 5 mmol/l EDTA; 10 µg/ml aprotinin; and 100 µl anti-phosphatases; Roche Diagnostics, Basel, Switzerland). Protein concentrations were determined using a bicinchoninic assay kit (Bi Yun-tian, Wuhan, China), according to the manufacturer's instructions. Samples of 80 µg protein were resolved using 12% SDS-PAGE, and transferred to polyvinylidene fluoride membranes (San Yin-Tan, Beijing). The membranes were blocked with 5% non-fat skim milk in tris-buffered saline solution containing Tween-20 (TBST; 10 mmol/l tris, pH 7.5; 140 mmol/l sodium chloride; 0.1% Tween-20) for 1 h at 37°C, washed, and incubated with the corresponding primary antibody in TBST containing 3% bovine serum albumin (Gibco Life Technologies, Carlsbad, CA, USA). The membranes were incubated for 12 hours with the following primary antibodies: Rabbit anti-EPOR (1:1,000, #41346), rabbit anti-phosphorylated (p)-EPOR (1:500, #BY-1424P), rabbit anti-p85 (1:1,000, #2883S), rabbit anti-p-p85 (1:500, #4292S), rabbit anti-Akt (1:1,000, #9037S) and rabbit anti-p-Akt (1:500, #8475S) (Cell Signaling Technology, Inc., Danvers, MA, USA) at 4°C. The membranes were then washed extensively with TBS, prior to being incubated with a horseradish peroxidase-conjugated immunoglobulin G secondary antibody (1:4,000; J010203; Jackson ImmunoResearch Laboratories, West Grove, PA, USA) for 1 h at 37°C. β-actin (1:3000, #4301; Abmart, Shanghai, China) was used for normalization of protein levels. The reactive bands were visualized using the Enhanced Chemiluminescence-Plus reagent (GE Healthcare, Piscataway, NJ, USA), according to the manufacturer's instructions. The density of each reactive band was quantified using the Labworks Image Acquisition platform, and its related analytical software (UVP, LLC, Upland, CA, USA).
ELISA analysis
Levels of the inflammatory mediators, TNF-α, IL-6 and IL-1β, in the serum were quantified using an ELISA kit specific to rats, according to the manufacturer's instructions (BioSource International Inc., Camarillo, CA, USA).
Quantitative polymerase chain reaction (PCR) analysis
Total RNA was isolated from the cardiac tissue using TRIzol® reagent, according to the manufacturer's instructions (Takara Bio, Inc., Otsu, Japan). In total, 4 µg RNA was reverse transcribed into cDNA using the PrimeScript RT Master Mix (Takara Bio, Inc.) as instructed. Quantitative PCR amplifications were conducted using the ABI 7500 system (Applied Biosystems Life Technologies, Foster City, CA, USA). PCR primers (Invitrogen Life Technologies, Carlsbad, CA, USA) for all the analyzed genes are shown in Table I. PCR was conducted as follows: Initial denaturation at 95°C for 30 sec, followed by 40 cycles of denaturation at 95°C for 5 sec, annealing at 60°C for 34 sec and elongation at 95°C for 15 sec. The mRNA expression for each gene was normalized against the values for β-actin, and the relative expression levels were calculated using the 2−ΔΔCt method, as previously reported (14).
Table I.
Primers used for quantitative polymerase chain reaction analysis.
| Gene | Species | Sense strand sequence | Anti-sense strand sequence |
|---|---|---|---|
| TNF-α | Rat | CTGAACTTCGGGGTGATCGG | GGCTTGTCACTCGAATTTTGAGA |
| IL-1β | Rat | AGCTTCCTTGTGCAAGTGTCT | GACAGCCCAGGTCAAAGGTT |
| IL-6 | Rat | CTGCAAGAGACTTCCATCCAG | AGTGGTATAGACAGGTCTGTTGG |
| β-actin | Rat | AGAGGGAAATCGTGCGTGAC | CAATAGTGATGACCTGGCCGT |
TNF, tumor necrosis factor; IL, interleukin.
Statistical analysis
SPSS software, version 12.0 (SPSS, Inc., Chicago, IL, USA) was used to perform all statistical analyses. All data are expressed as the mean ± standard error of the mean. All statistical comparisons were performed using one-way analysis of variance followed by Dunetts's test. In all cases, P<0.05 was considered to indicate a statistically significant difference.
Results
EPO attenuates IRI-induced pathological changes in the myocardial tissue
When compared with the rats in the Sham group, the pathological changes in the myocardial tissues from the rats in the IRI-SAL group were more marked (Fig. 1A and B). The changes included atrophy of the myocardial fibers, inflammatory cell infiltration, and coagulative and colliquative necroses. By contrast, the myocardial tissues from the rats in the IRI-EPO group (Fig. 1C) exhibited fewer pathological changes compared with the IRI-SAL group. Semi-quantitative assessment of the histological lesions revealed a significantly higher score in the IRI-SAL-treated rats as compared with the IRI-EPO-treated rats and Sham rats (Fig 1D).
Figure 1.

Histological analysis of the myocardial tissue. Representative hematoxylin and eosin (HE) staining results for the myocardial sections collected from the (A) Sham, (B) IRI-saline and (C) IRI-EPO rats. (D) Semi-quantitative analysis of the HE staining results for all the included rats. Three rats were analyzed in each group and the images are presented at x200 magnification. ***P<0.001, IRI-SAL vs. Sham; ###P<0.001, IRI-SAL vs. IRI-EPO. IRI, ischemia-reperfusion injury; EPO, erythropoietin.
EPO promotes IRI-induced EPOR activation
In order to address the mechanism underlying the EPO-mediated protection of rat myocardial tissue from IRI, EPOR expression and phosphorylation (activation) were examined. Neither the IRI-SAL or IRI-EPO procedures revealed a marked impact on EPOR expression (Fig. 2A, lower panel). However, the amount of p-EPOR observed in the rat myocardial tissue following IRI in the IRI-SAL group was significantly higher compared with that in the Sham rats (Fig. 2A, upper panel and Fig. 2B). Administration of EPO slightly promoted EPOR activation, as shown by the higher levels of p-EPOR (40% higher) detected in the IRI-EPO rats when compared with the IRI-SAL rats (Fig. 2B).
Figure 2.
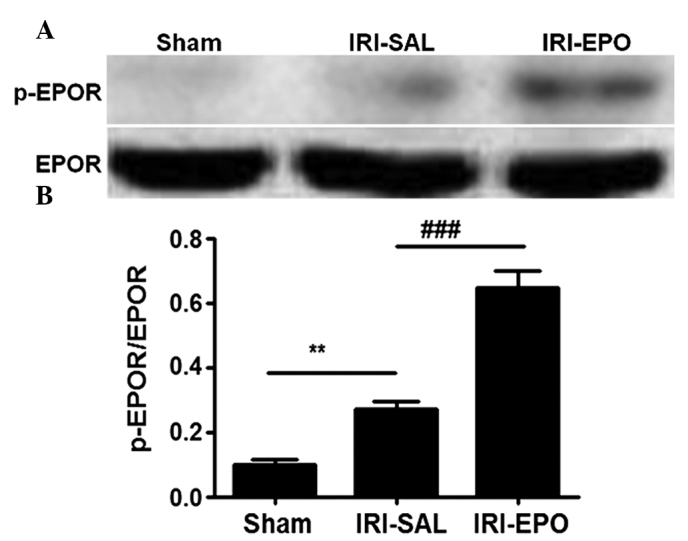
Pretreatment with EPO promotes EPOR activation. Western blot analysis was used to assess EPOR expression and activation by measuring the levels of total EPOR and p-EPOR. (A) Representative result for western blot analysis. (B) Semi-quantitative analysis of the six animals studied in each group. The relative expression levels of EPOR and p-EPOR in each group of rats were normalized against those of β-actin, and presented as a ratio between p-EPOR and EPOR. **P<0.01, IRI-SAL vs. Sham; ###P<0.001, IRI-EPO vs. IRI-SAL. EPO, erythropoietin; IRI, ischemia-reperfusion injury; EPOR, erythropoietin receptor; p-EPOR, phosphorylated EPOR.
EPOR activation further activates PI3K/Akt signaling
The PI3K/Akt pathway has been demonstrated to play an important role in myocardial injury (15), while EPO is able to regulate PI3K/Akt signaling (16). To investigate the contribution of the PI3K/Akt pathway in the rat model, the activity of the PI3K p85 regulatory subunit was examined. No statistically significant difference in the total p85 level was detected among the three groups of rats (Fig. 3A). However, IRI was shown to induce PI3K p85 regulatory subunit activation, as shown by the significantly higher levels of p-p85 in the IRI-SAL rats when compared with the sham rats. In addition, pretreatment with EPO promoted p85 activation, as manifested by the significantly higher levels of p-p85 in the IRI-EPO rats when compared with the IRI-SAL rats.
Figure 3.
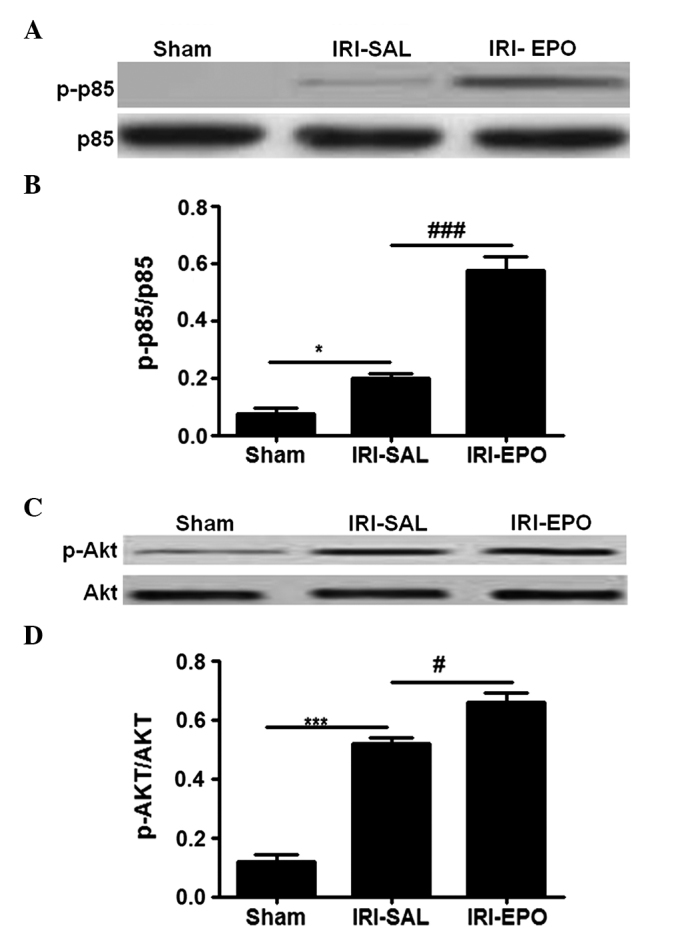
Administration of EPO activated the PI3K/Akt signaling pathway. Activation of p85 was assessed by measuring the levels of p-p85, while p-Akt was employed for evaluating Akt activation. (A) Representative western blot analysis showing the results for p85 and p-p85 (B) Quantitative analysis of p-p85 expression in all the treatment groups. (C) Representative western blot analysis showing the results for Akt. (D) Quantitative analysis of Akt activity in all the treatment groups. The relative activities for p85 and Akt were assessed by a ratio between the phosphorylated form and total form. Six rats were included in each study group. *P<0.05 and ***P<0.001, IRI-SAL vs. Sham; ###P<0.001 and #P<0.05, IRI-SAL vs. IRI-EPO. EPO, erythropoietin; IRI, ischemia-reperfusion injury; PI3K, phosphoinositide 3-kinase; Akt, protein kinase B; p-p85, phosphorylated p85; p-Akt, phosphorylated Akt.
Since p85 activation provides signals to phosphorylate Akt, Akt activity was examined. Similarly to p85, there was no statistically significant difference in the total levels of Akt among the three groups (Fig. 3C); however, the levels of p-Akt were significantly higher in the IRI-SAL rats when compared with the Sham rats. Furthermore, rats in the IRI-EPO group exhibited significantly higher levels of p-Akt when compared with the IRI-SAL rats (Fig. 3D). Thus, EPO pretreatment increased PI3K p85 activity, which subsequently increased Akt activation.
Activation of PI3K/Akt signaling attenuates inflammatory cytokine expression
The present study also investigated whether the activation of PI3K/Akt signaling attenuated the expression levels of inflammatory cytokines in the heart. The expression levels of TNF-α, IL-6 and IL-1β in the myocardial tissue following IRI were selectively analyzed using quantitative PCR. IRI-SAL treatment was shown to increase the expression of TNF-α by 13-fold (Fig. 4A), IL-6 by 3-fold (Fig. 4B) and IL-1β by 2.5-fold, as compared with the expression levels in the Sham rats. Administration of EPO inhibited IRI-induced TNF-α expression by 55% (Fig. 4A), IL-6 by 42% (Fig. 4B) and IL-1β by 30% (Fig. 4C) when compared with the expression levels in the IRI-SAL group. Thus, the data showed that activation of the PI3K/Akt signaling pathway by EPO significantly suppressed IRI-induced inflammatory cytokine expression in the myocardial tissue.
Figure 4.
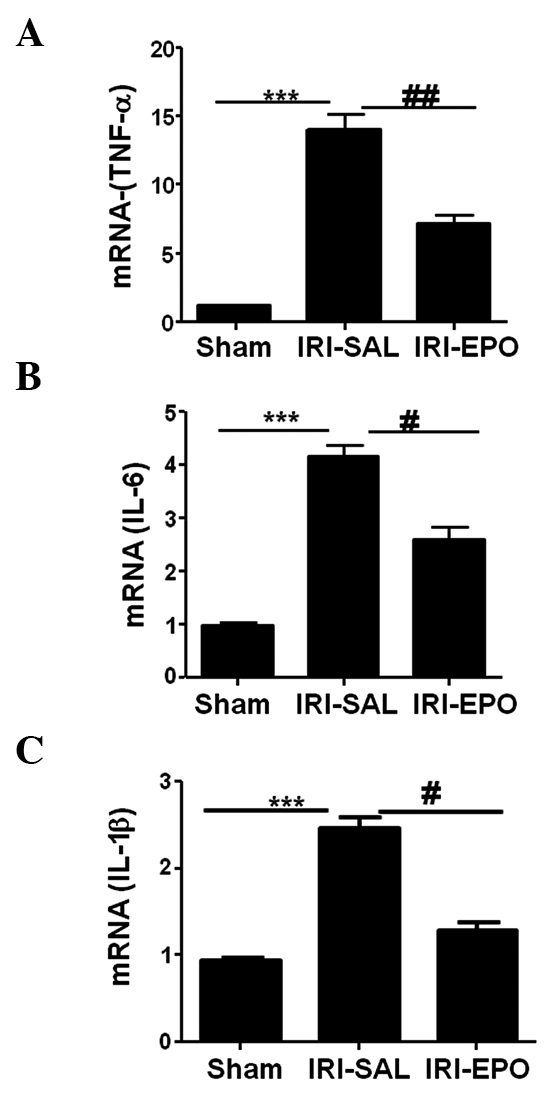
EPO treatment suppressed cytokine expression in the myocardium following IRI. Quantitative polymerase chain reaction was used to assess the expression of inflammatory cytokines in the myocardium. The expression levels for (A) TNF-α, (B) IL-6 and (C) IL-1β were selectively analyzed following IRI. ***P<0.001, IRI-SAL vs. Sham; ##P<0.01 and #P<0.05, IRI-SAL vs. IRI-EPO. Six rats were included in each group. EPO, erythropoietin; IRI, ischemia-reperfusion injury; TNF, tumor necrosis factor; IL, interleukin.
Activation of PI3K/Akt signaling suppresses inflammatory cytokine secretion
The expression levels of TNF-α, IL-6 and IL-1β were also analyzed in the serum following IRI. IRI-SAL treatment was demonstrated to increase the secretion of TNF-α by 4-fold (Fig. 5A), IL-6 by 2-fold (Fig. 5B) and IL-1β by 3-fold, as compared with the Sham rats. However, administration of EPO reduced the IRI-induced TNF-α levels by 50% (Fig. 5A), IL-6 levels by 30% (Fig. 5B) and IL-1β levels by 25% (Fig. 5C) when compared with the expression levels in the IRI-SAL group. Therefore, the data revealed that activation of the PI3K/Akt signaling pathway by EPO significantly suppressed the levels of IRI-induced inflammatory cytokines in the serum.
Figure 5.
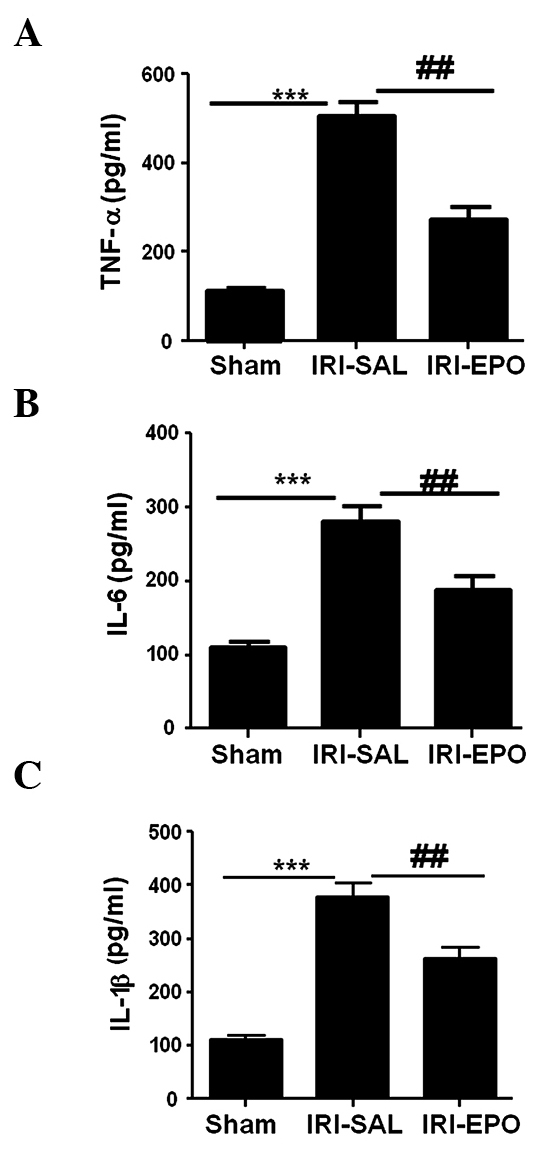
EPO treatment suppressed the levels of cytokines in the serum following myocardial IRI. An ELISA was used to assess the expression levels of inflammatory cytokines in the serum. The expression levels of (A) TNF-α, (B) IL-6 and (C) IL-1β were selectively analyzed following IRI. ***P<0.001, IRI-SAL vs. Sham; ###P<0.001 and ##P<0.01, IRI-SAL vs. IRI-EPO. Six rats were included in each group. EPO, erythropoietin; IRI, ischemia-reperfusion injury; TNF, tumor necrosis factor; IL, interleukin.
Discussion
EPO has been used for a number of years to promote red blood cell production in various clinical settings (8,9). In the present study, IRI was induced in rats by occluding the left descending coronary artery for 30 min and subsequently subjecting the heart to 3 h reperfusion. The effects of EPO on myocardial IRI were subsequently investigated. Histological analyses demonstrated significant pathological changes in the IRI group, as shown by marked atrophy of the myocardial fibers, inflammatory cell infiltration and coagulative and colliquative necroses.
Pretreatment of the rats with 5,000 U/kg EPO was shown to significantly attenuate the pathological changes associated with myocardial IRI. In agreement with the current results, previous studies have suggested that treatment of rats with EPO significantly decreases the pathological injury to the heart tissue that is induced by myocardial IRI (12,17,18). The present data indicate that EPO may be a novel agent for the prevention of myocardial IRI in clinical practice.
In order to determine the molecular mechanism through which EPO exerts protection against myocardial IRI in rats, the present study investigated the effect of EPO on EPOR activation. The results revealed that treatment with EPO did not affect EPOR expression; however, IRI-SAL treatment led to increased levels of p-EPOR. Pretreatment with EPO further increased EPOR activation, as shown by the higher levels of p-EPOR in the IRI-EPO rats when compared with the IRI-SAL rats. These observations may be the result of negative feedback pathways, which function to limit inflammatory responses and prevent a prolonged immune response following severe tissue injury.
A number of studies have shown that EPO ameliorates IRI in the kidneys and brain by suppressing inflammation (10,11). Therefore, the present study investigated the downstream pathways of EPOR activation that are associated with the regulation of inflammation, namely the PI3K/Akt signaling pathway. IRI-SAL treatment was shown to induce PI3K p85 regulatory subunit activation, as manifested by the significantly higher levels of p-p85 in the IRI-SAL rats when compared with the Sham rats. In addition, pretreatment of the rats with EPO further promoted p85 activation following IRI.
These results prompted the examination of Akt activation, since the phosphorylation of p85 can lead to Akt activation (5,19,20). Consistent with the aforementioned results, IRI-EPO treatment was shown to significantly increase Akt activity, as demonstrated by the higher levels of p-Akt expression in the IRI-EPO rats when compared with the IRI-SAL rats. To demonstrate that activation of the PI3K/Akt signaling pathway by EPO inhibited inflammatory responses, the expression levels of TNF-α, IL-6 and IL-1β were analyzed in the myocardium and serum following IRI. Rats pretreated with EPO exhibited significantly lower levels of TNF-α, IL-6 and IL-1β when compared with the IRI-SAL rats. Therefore, the current data suggest that EPO enhances EPOR activation, which subsequently leads to the activation of PI3K/Akt signaling and the suppression of inflammatory responses. This suppression protected the myocardium from IRI.
The duration of ischemia and the time point for measurements following reperfusion are critical factors in determining the severity of IRI in rats (21,22). Published studies have applied numerous variations in experimental conditions, including temperature and duration of ischemia. Furthermore, study results may vary depending on the strain of rats used (21–24). In the present study, Sprague-Dawley rats were used, 30 min of ischemia was applied and 3 h after reperfusion was selected as the time point for examining IRI. A similar degree of myocardial injury severity was observed in the current model, as compared with a previously published study (12). As previously mentioned, evidence for myocardial injury was strongly demonstrated by the pathological changes and infiltration of inflammatory cells. These data also support the hypothesis that IRI initiates a complex cascade of events, which results in myocardial injury characterized by histological changes and infiltration of inflammatory cells.
With regard to the ability of EPO pretreatment to prevent myocardial IRI, it is worth considering that EPOR activation by EPO may involve additional pathways other than PI3K/Akt, such as the mitogen-activated protein kinase cascade (24). Additional studies addressing the other potential pathways involved in EPOR activation following IRI are required to fully elucidate the mechanism of EPO.
In summary, the present study demonstrated that pretreatment of rats with EPO can provide protection against myocardial IRI. This protection is manifested through the amelioration of pathological changes to the myocardium and the suppression of inflammation. In the current study, EPO pretreatment decreased myocardial IRI, which was associated with the activation of EPOR, increased PI3K/Akt signaling activation and the inhibition of inflammatory mediator production. Therefore, the results indicated that EPO may be a potential therapy for the prevention of myocardial IRI in clinical practice.
Acknowledgements
This study was supported by grants from the Natural Sciences Fund of Hubei Province (no. 2012FFC115) and the Public Health Fund of Hubei Province (no. QJX2012-49).
References
- 1.Chen K, Li G, Geng F, et al. Berberine reduces ischemia/reperfusion-induced myocardial apoptosis via activating AMPK and PI3K-Akt signaling in diabetic rats. Apoptosis. 2014;19:946–957. doi: 10.1007/s10495-014-0977-0. [DOI] [PubMed] [Google Scholar]
- 2.Yan X, Qiu W, Jia B, et al. Myocardial protection by interferon-γ late preconditioning during cardiopulmonary bypass-associated myocardial ischemia-reperfusion in pigs. Oncol Rep. 2013;30:2145–2152. doi: 10.3892/or.2013.2707. [DOI] [PubMed] [Google Scholar]
- 3.Lassaletta AD, Elmadhun NY, Zanetti AV, et al. Rapamycin treatment of healthy pigs subjected to acute myocardial ischemia-reperfusion injury attenuates cardiac functions and increases myocardial necrosis. Ann Thorac Surg. 2014;97:901–907. doi: 10.1016/j.athoracsur.2013.09.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Du X, Hu X, Wei J. Anti-inflammatory effect of exendin-4 postconditioning during myocardial ischemia and reperfusion. Mol Biol Rep. 2014;41:3853–3857. doi: 10.1007/s11033-014-3252-0. [DOI] [PubMed] [Google Scholar]
- 5.Kim HJ, Joe Y, Kong JS, et al. Carbon monoxide protects against hepatic ischemia/reperfusion injury via ROS-dependent Akt signaling and inhibition of glycogen synthase kinase 3beta. Oxid Med Cell Longev. 2013;2013:306421. doi: 10.1155/2013/306421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hofmann BT, Jücker M. Activation of PI3K/Akt signaling by n-terminal SH2 domain mutants of the p85α regulatory subunit of PI3K is enhanced by deletion of its c-terminal SH2 domain. Cell Signal. 2012;24:1950–1954. doi: 10.1016/j.cellsig.2012.06.009. [DOI] [PubMed] [Google Scholar]
- 7.Chen L, Wei X, Hou Y, Liu X, et al. Tetramethylpyrazine analogue CXC195 protects against cerebral ischemia/reperfusion-induced apoptosis through PI3K/Akt/GSK3β pathway in rats. Neurochem Int. 2014;66:27–32. doi: 10.1016/j.neuint.2014.01.006. [DOI] [PubMed] [Google Scholar]
- 8.Palis J. Primitive and definitive erythropoiesis in mammals. Front Physiol. 2014;5:3. doi: 10.3389/fphys.2014.00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jelkmann W. Physiology and pharmacology of erythropoietin. Transfus Med Hemother. 2013;40:302–309. doi: 10.1159/000356193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang C, Zhao T, Lin M, et al. Helix B surface peptide administered after insult of ischemia reperfusion improved renal function, structure and apoptosis through beta common receptor/erythropoietin receptor and PI3K/Akt pathway in a murine model. Exp Biol Med (Maywood) 2013;238:111–119. doi: 10.1258/ebm.2012.012185. [DOI] [PubMed] [Google Scholar]
- 11.Zhang Q, Bian H, Li Y, Guo L, Tang Y, Zhu H. Preconditioning with the traditional Chinese medicine Huang-Lian-Jie-Du-Tang initiates HIF-1α-dependent neuroprotection against cerebral ischemia in rats. J Ethnopharmacol. 2014;154:443–452. doi: 10.1016/j.jep.2014.04.022. [DOI] [PubMed] [Google Scholar]
- 12.Liu X, Xie W, Liu P, et al. Mechanism of the cardioprotection of rhEPO pretreatment on suppressing the inflammatory response in ischemia-reperfusion. Life Sci. 2006;78:2255–2264. doi: 10.1016/j.lfs.2005.09.053. [DOI] [PubMed] [Google Scholar]
- 13.Meij JT, Sheikh F, Jimenez SK, et al. Exacerbation of myocardial injury in transgenic mice overexpressing FGF-2 is T cell dependent. Am J Physiol Heart Circ Physiol. 2002;282:H547–H555. doi: 10.1152/ajpheart.01019.2000. [DOI] [PubMed] [Google Scholar]
- 14.Zhang S, Lv JW, Yang P, et al. Loss of dicer exacerbates cyclophosphamide-induced bladder overactivity by enhancing purinergic signaling. Am J Pathol. 2012;181:937–946. doi: 10.1016/j.ajpath.2012.05.035. [DOI] [PubMed] [Google Scholar]
- 15.Liu H, Guo X, Chu Y, Lu S. Heart protective effects and mechanism of quercetin preconditioning on anti-myocardial ischemia reperfusion (IR) injuries in rats. Genes. 2014;545:149–155. doi: 10.1016/j.gene.2014.04.043. [DOI] [PubMed] [Google Scholar]
- 16.Jia Y, Mo SJ, Feng QQ, et al. EPO-Dependent activation of PI3K/AKT/FOXO3a signalling mediates neuroprotection in in vitro and in vivo models of Parkinson's disease. J Mol Neurosci. 2014;53:117–124. doi: 10.1007/s12031-013-0208-0. [DOI] [PubMed] [Google Scholar]
- 17.Jun JH, Jun NH, Shim JK, Shin EJ, Kwak YL. Erythropoietin protects myocardium against ischemia-reperfusion injury under moderate hyperglycemia. Eur J Pharmacol. 2014;745:1–9. doi: 10.1016/j.ejphar.2014.09.038. [DOI] [PubMed] [Google Scholar]
- 18.Lu MJ, Chen YS, Huang HS, Ma MC. Erythropoietin alleviates post-ischemic injury of rat hearts by attenuating nitrosative stress. Life Sci. 2012;90:776–784. doi: 10.1016/j.lfs.2012.04.012. [DOI] [PubMed] [Google Scholar]
- 19.He W, Wang Z, Zhou Z, et al. Lipopolysaccharide enhances Wnt5a expression through toll-like receptor 4, myeloid differentiating factor 88, phosphatidylinositol 3-OH kinase/AKT and nuclear factor kappa B pathways in human dental pulp stem cells. J Endod. 2014;40:69–75. doi: 10.1016/j.joen.2013.09.011. [DOI] [PubMed] [Google Scholar]
- 20.Zhang J, Yao Y, Xiao F, et al. Administration of dexamethasone protects mice against ichemia/reperfusion induced renal injury by suppressing PI3K/AKT signaling. Int J Clin Exp Pathol. 2013;6:2366–2375. [PMC free article] [PubMed] [Google Scholar]
- 21.Wang J, Li L, Jiang H. c-Cbl inhbition: A novel therapeutic approach for attenuating myocardial ischemia and reperfusion injury. Int J Cardiol. 2015;186:50–51. doi: 10.1016/j.ijcard.2015.03.254. [DOI] [PubMed] [Google Scholar]
- 22.Lin Y, Chen L, Li W, Fang J. Role of high-mobility group box-1 in myocardial ichemia/reperfusion injury and the effect of ethyl pyruvate. Exp Ther Med. 2015;9:1537–1541. doi: 10.3892/etm.2015.2290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yao HC, Yang LJ, Han QF, et al. Postconditioning with simvastatin decreases myocardial injury in rats following acute myocardial ischemia. Exp Ther Med. 2015;9:1166–1170. doi: 10.3892/etm.2015.2273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rafiee P, Shi Y, Su J, et al. Erythropoietin protects the infant heart against ischemia-reperfusion injury by triggering multiple signaling pathways. Basic Res Cardiol. 2005;100:187–197. doi: 10.1007/s00395-004-0508-1. [DOI] [PubMed] [Google Scholar]