

Abstract
Hypoxia has long been known to serve as a stimulus for cell cycle arrest. Hypoxia-mediated cell cycle arrest is mediated through the actions of HIF1α (hypoxia inducible factor 1, α subunit [basic helix-loop-helix transcription factor]), which has a nontranscriptional role as an inhibitor of MCM (minichromosome maintenance complex component) helicase activity. We identified chaperone-mediated autophagy as a pathway for selective degradation of HIF1α through lysosomes prior to the onset of DNA replication. CDK2 (cyclin-dependent kinase 2) mediates degradation of HIF1α at the G1/S transition, whereas CDK1 (cyclin-dependent kinase 1) increases HIF1α levels and transcriptional activity prior to the onset of G1 phase. Lysosomal inhibitors induce cell cycle arrest, which is recovered by knockdown of HIF1α and EPAS1/HIF2α. These findings establish lysosomes as essential regulators of cell cycle progression through the degradation of HIF1α.
Keywords: chaperone-mediated autophagy, cell cycle progression, cyclin-dependent kinases, DNA replication, hypoxia-inducible factor
Abbreviations: CDK, cyclin-dependent kinase; HIF, hypoxia-inducible factor; MCM, minichromosome maintenance complex component.
Hypoxia is a physiological stimulus that induces adaptive changes in metabolism, angiogenesis, erythropoiesis, autophagy, and cell proliferation. Many of these changes are mediated through the activity of the hypoxia-inducible factors (HIFs). HIF-1 is a heterodimeric transcription factor composed of HIF1α and ARNT/HIF-1β subunits. The HIF1α subunit is subject to oxygen-dependent hydroxylation, which targets the protein for ubiquitination and proteasomal degradation by the VHL (von Hippel-Lindau tumor suppressor, E3 ubiquitin protein ligase) complex. HIF-2α is regulated in a similar manner, although the HIF-2α protein has a more limited tissue distribution and in some cases has distinct functions from HIF1α. In addition to the oxygen-dependent degradation pathway, we have identified an oxygen-independent and lysosomal-dependent pathway for HIF1α degradation through chaperone-mediated autophagy.
The mechanisms by which HIF1α mediates cell cycle arrest have been controversial. Several groups have reported that hypoxia leads to an upregulation of the cyclin-dependent kinase (CDK) inhibitors CDKN1A/p21 and CDKN1B/p27. We identified a nontranscriptional role for the HIF1α protein as an inhibitor of the MCM helicase. The HIF1α protein binds to the MCM proteins and the helicase loading partner CDC6, maintaining the complex in a loaded but inactive state.
The identification of the nontranscriptional role for HIF1α as an inhibitor of DNA replication raises the question of how cells can proliferate under hypoxia at all. Cancer cells in particular simultaneously possess high HIF1α levels and rapid rates of cell division. These cells are relatively resistant to the effects of hypoxia on the cell cycle. We speculated that a pathway exists to couple HIF1α levels to particular phases of the cell cycle.
As the CDK proteins have major roles in orchestrating specific events within the cell cycle, we examined interactions between the CDK proteins and HIF1α. CDK1 is activated by its CCND (cyclin D) binding partner at the G2/M phase, whereas the CDK2 protein is activated by either CCNE (cyclin E) or CCNA (cyclin A) at the G1/S phase transition or S phase, respectively. We identified physical interactions through co-immunoprecipitation experiments of both CDK1 and CDK2 with HIF1α.
CDK1 activity, induced by overexpression of CDK1 or CCND, leads to an increase in HIF1α protein levels and target gene expression. Inhibition of CDK1 activity, through either genetic or pharmacological means, had the opposite effect. In contrast, CDK2 overexpression or overexpression of either the CCNE or CCNA binding partners leads to a decrease in HIF1α levels (Fig. 1). The effects of both CDK1 and CDK2 were abolished in the presence of lysosomal inhibitors, but maintained in the presence of proteasome or hydroxylase inhibitors. Thus, CDK activity serves to couple HIF1α levels to particular phases of the cell cycle through lysosomal degradation.
Figure 1.
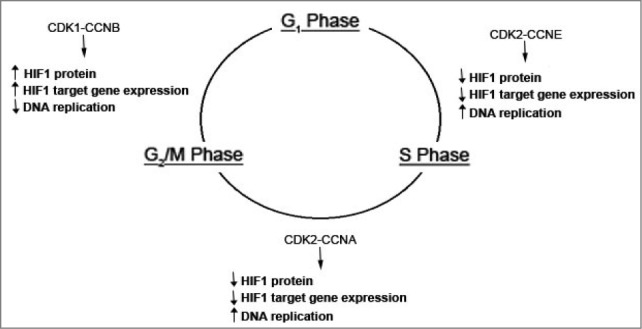
Model for regulation of HIF1α by the cell cycle. Prior to G1 phase, CDK1 is activated by binding to its CCNB/cyclin B partner. This leads to a block in lysosomal degradation of HIF1α and increased HIF-1 target gene expression and inhibition of DNA replication. During the G1/S phase transition and during S phase, CDK2 activity mediated by binding to either CCNE/cyclin E or CCNA leads to an increase in lysosomal degradation of HIF1α allowing DNA replication to proceed. In cancer cell lines, CDK2 activity leads to a concurrent upregulation of HIF-1 transcriptional activity and target gene expression.
Interestingly, we found that cancer cell lines were able to dissociate the transcriptional and nontranscriptional effects of HIF1α. Mouse embryonic fibroblasts from either Ccne1/2 knockout mice or Cdk2 knockout mice have enhanced induction of HIF1α protein and increased induction of HIF-1 target genes in comparison to mouse embryonic fibroblasts derived from wild-type littermates, consistent with a role for CDK2 activity in promoting HIF1α degradation. However, although overexpression of CDK2 or its CCNE or CCNA binding partners decreases HIF1α levels in cancer cell lines, this is associated with an increase rather than a decrease in HIF-1 transcriptional activity. The mechanism by which this occurs is unclear, but indicates that cancer cells are able to dissociate the antiproliferative and nontranscriptional effects of the HIF1α protein from its transcriptional effects on angiogenesis, metabolism, and metastasis.
These data also clarify mechanisms by which cancer cell lines maintain different sensitivities to the effect of hypoxia on the cell cycle. Previous work suggested that the HIF-2α protein serves to promote cell proliferation, in contrast to HIF1α, with the relative balance between HIF1α and HIF-2α determining the effect of hypoxia on cell proliferation. However, in certain cell lines, overexpression of EPAS1 inhibits, rather than promotes, cell proliferation and HIF-2α can bind the MCM helicase proteins in a similar fashion as HIF1α. We showed that treatment with lysosomal inhibitors leads to cell cycle arrest in a hepatocellular carcinoma cell line that is resistant to the effect of hypoxia on cell proliferation. Importantly, this effect is abrogated in cell lines with concurrent knockdown of HIF1α and HIF-2α. These results demonstrate that, at least in certain cancer cell lines, autophagy is used to degrade HIF1a and HIF-2α to permit DNA replication, thereby promoting cell proliferation. This is consistent with recent work in the field demonstrating the importance of chaperone-mediated autophagy in tumor development. Importantly, these data also demonstrate that the effect of the lysosome on cell proliferation is largely mediated through the degradation of HIF1α and HIF-2α subunits.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.