

Abstract
Autophagy is an evolutionarily conserved process in eukaryotes that eliminates harmful components and maintains cellular homeostasis in response to a series of extracellular insults. However, these insults may trigger the downstream signaling of another prominent stress responsive pathway, the STAT3 signaling pathway, which has been implicated in multiple aspects of the autophagic process. Recent reports further indicate that different subcellular localization patterns of STAT3 affect autophagy in various ways. For example, nuclear STAT3 fine-tunes autophagy via the transcriptional regulation of several autophagy-related genes such as BCL2 family members, BECN1, PIK3C3, CTSB, CTSL, PIK3R1, HIF1A, BNIP3, and microRNAs with targets of autophagy modulators. Cytoplasmic STAT3 constitutively inhibits autophagy by sequestering EIF2AK2 as well as by interacting with other autophagy-related signaling molecules such as FOXO1 and FOXO3. Additionally, the mitochondrial translocation of STAT3 suppresses autophagy induced by oxidative stress and may effectively preserve mitochondria from being degraded by mitophagy. Understanding the role of STAT3 signaling in the regulation of autophagy may provide insight into the classic autophagy model and also into cancer therapy, especially for the emerging targeted therapy, because a series of targeted agents execute antitumor activities via blocking STAT3 signaling, which inevitably affects the autophagy pathway. Here, we review several of the representative studies and the current understanding in this particular field.
Keywords: autophagy, cancer, mitophagy, receptor tyrosine kinases, STAT3, targeted therapy
Abbreviations: ALK, anaplastic lymphoma receptor tyrosine kinase; ATF4, activating transcription factor 4; BNIP3, BCL2/adenovirus E1B 19kDa interacting protein 3; CNTF, ciliary neurotrophic factor; COX8, cytochrome c oxidase subunit VIII; ConA, concanavalin A; CTSB, cathepsin B; CTSL, cathepsin L; CuB, cucurbitacin B; CYCS, cytochrome c, somatic; EGF, epidermal growth factor; EIF2A, eukaryotic initiation factor 2A, 65kDa; EIF2AK2, eukaryotic translation initiation factor 2-α kinase 2; ER, endoplasmic reticulum; ETC, electron transport chain; FOXO1/3, forkhead box O1/3; HDAC3, histone deacetylase 3; HIF1A, hypoxia inducible factor 1, α subunit (basic helix-loop-helix transcription factor); IL6, interleukin 6; IMM, inner mitochondrial membrane; KDR, kinase insert domain receptor; LMP, lysosomal membrane permeabilization; MAPK1, mitogen-activated protein kinase 1; MAP1LC3A, microtubule-associated protein 1 light chain 3 α; miRNA, microRNA; mitoSTAT3, mitochondrial STAT3; MLS, mitochondrial localization sequence; MMP14, matrix metallopeptidase 14 (membrane-inserted); NDUFA13, NADH dehydrogenase (ubiquinone) 1 α subcomplex, 13; NES, nuclear export signal; NFKB1, nuclear factor of kappa light polypeptide gene enhancer in B-cells 1; NLS, nuclear localization signal; PDGFRB, platelet-derived growth factor receptor, β polypeptide; PRKAA2, protein kinase, AMP-activated, α 2 catalytic subunit; PTPN2, protein tyrosine phosphatase, non-receptor type 2; PTPN6, protein tyrosine phosphatase, non-receptor type 6; PTPN11, protein tyrosine phosphatase, non-receptor type 11; ROS, reactive oxygen species; RTK, receptor tyrosine kinases; SH2, src homology 2; STAT3, signal transducer and activator of transcription 3 (acute-phase response factor); VHL, von Hippel-Lindau tumor suppressor, E3 ubiquitin protein ligase; XPO1, exportin 1
Introduction
Autophagy is an evolutionarily conserved process that sequesters nonessential intracellular components for lysosomal degradation in response to a variety of stress stimuli, including nutrient or growth factor deprivation, hypoxia, reactive oxygen species (ROS), DNA damage, protein aggregates, damaged organelles, and intracellular microorganisms.1 Autophagy is generally categorized in 3 forms: macroautophagy, microautophagy, and chaperone-mediated autophagy. In this review, we focus on macroautophagy, which is generally referred to as ‘autophagy.’ During autophagy, nonessential cytoplasmic organelles or cytosolic components are sequestered by double-membrane vesicles named autophagosomes, which are delivered to and fuse with lysosomes for degradation and recycling, allowing cells to eliminate damaged or harmful components through catabolism and to maintain energy homeostasis.2 Autophagy is widely involved in the pathogenesis of many diseases, especially cancer. Under physiological conditions, autophagy has been proposed to function as a tumor suppressive mechanism for the removal or mitigation of harmful stimuli, including oxidative stress, inflammation and genome instability, which may lead to cellular transformation. However, in established tumor cells, which are constantly exposed to mutations, radiation, chemotherapy or targeted agents, autophagy may act as an additional means for survival.3 Although the exact role of autophagy is debatable in different tumor models or in different pathological conditions, certain tumors are proposed to be ‘autophagy addicted,’4 and numerous crosstalk occurs between autophagy and other cancer-promoting pathways.5
STAT3 (signal transducer and activator of transcription 3 [acute-phase response factor]) is a latent transcription factor that mediates extracellular signals such as cytokines and growth factors through interaction with polypeptide receptors at the cell surface.6 STAT3 protein becomes transcriptionally activated primarily by tyrosine phosphorylation. Activated STAT3 dimerizes, translocates to the nucleus, and binds to sequence-specific DNA elements for consequent transcription of target genes.7 STAT3 is expressed at a basal level in cells but rapidly increases through self-activation once stimulated because the STAT3 promoter contains a binding site for its own dimers.8,9 STAT3 is constitutively activated or is required to maintain the transformed phenotype in a majority of malignant tumors such as breast cancer, head and neck cancer, brain tumors, lung cancer, ovarian cancer, prostate cancer, pancreatic cancer, renal carcinoma, thyroid cancer, melanoma, myeloma, hepatocellular carcinoma, lymphomas, and leukemia.10,11 The constitutive activation of STAT3 on its own leads to fibroblast transformation, suggesting that STAT3 functions as an oncogene.12 Additionally, numerous cancer cell lines undergo growth arrest or apoptosis when treated with antisense or dominant negative constructs against STAT3.10,12 As the only embryonic lethal family member of the STAT family,13 STAT3 is a prominent nuclear transcription factor that affects the expression of more than 1,000 gene products.9 Recent literature has also endorsed the growing evidence of the role of STAT3 in the regulation of autophagy,2,4,14-16 a well-known stress-induced survival response in eukaryotes. However, the current data provide conflicting explanations of the mechanisms underlying this regulation, likely due to the different subcellular localizations of STAT3 protein. Differentially localized STAT3 is able to modulate autophagy both in a transcription-dependent and transcription-independent manner. We will now review the literature focusing on this novel understanding of STAT3 and its role in autophagy regulation.
The structure and subcellular localization of STAT3
Structure and functional domains of STAT3
STAT3 was first described as a transcriptional enhancer of acute phase genes that is activated by IL6 (interleukin 6).7 The STAT3 gene is located on chromosome 17q21 and encodes an 89-kDa protein. An alternative splicing transcript of the full-length STAT3/STAT3α encoding an 80-kDa protein exists and is known as STAT3B/STAT3β.17 Furthermore, a 72-kDa C-terminal-truncated form of STAT3 produced by the limited proteolysis of STAT3 during granulocytic differentiation also exists and is known as STAT3C/STAT3γ,18,19 and another 64-kDa truncated isoform known as STAT3δ that is expressed in the early stage of granulocytic differentiation has been described.20 The STAT3 protein shares similar functional domains with other STAT family members, including an amino-terminal coiled-coil domain beginning at residue 130, which enables protein-protein interactions, a central DNA-binding domain between residues 320 and 490, a linker domain extending from residues 490 to 580 that affects DNA-binding stability, a classic Src homology 2 (SH2) domain between residues 580 and 680, a tyrosine residue at 705 and a carboxyl transactivation domain, which has a serine phosphorylation site at Ser727 and is absent in the alternative splicing variant STAT3β6,21 (Fig. 1). Phosphorylation of the conserved Tyr705 residue between the SH2 and carboxyl transactivation domain leads to the homo- or hetero-dimerization of 2 STAT molecules by reciprocal phosphotyrosine interactions between the SH2 domains of 2 monomers. This dimerization alters the conformation of the STAT proteins to facilitate DNA binding.12 Notably, in addition to STAT3 dimers, STAT1 and STAT3 heterodimers also exist and may possess transcriptional potential that differs from the potential of STAT1 or STAT3.9,22,23 Additionally, the DNA-binding fold contains several β-sheets that are folded similarly to the sheets found in the DNA-binding domains of NFKB1 and TP53, which suggests that numerous crosstalk possibilities exist among these 3 important stress responsive pathways. For example, NFKB1 is activated through endoplasmic reticulum stress signals in starved cancer cells, resulting in the induction of IL6. Additionally, STAT3 is required for the induction of IL6 by NFKB1, and these proteins form identical nuclear complexes on proximal IL6 promoters.24 Moreover, the transcription factor TP53 piggybacks onto NFKB1-RELA and utilizes the κB motif at a cis-regulatory region to control MIR21 expression. STAT3 is also necessary for the TP53-RELA complex to associate with this cis-element and for MIR21 expression,25 whereas MIR21 targets BCL2.26
Figure 1.
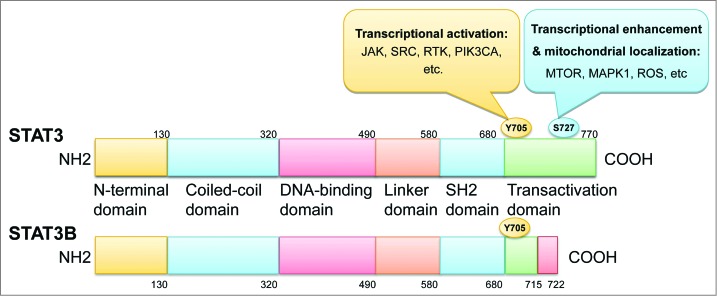
STAT3 structural domains and phosphorylation sites. The phosphorylation of Tyr705 by JAK, SRC, RTK, PIK3CA, and others activates the transcriptional ability of STAT3, whereas Ser727 phosphorylation in response to ROS or by MTOR, MAPK1, and others mediates the mitochondrial localization or enhances the transcriptional potential of STAT3.
Tyrosine phosphorylation of STAT3: the canonical STAT3 pathway
STAT3 transcriptional activity is primarily activated by the phosphorylation of a single tyrosine residue, Tyr705. Tyrosine phosphorylation of STAT3 can be directly catalyzed by receptor tyrosine kinases (RTKs) such as EGFR, KDR, and MET or by nonreceptor tyrosine kinases such as JAKs.17,27 The oncogene SRC also phosphorylates STAT3,28 and SRC may be the intermediate that facilitates STAT3 phosphorylation by RTKs. Once activated, STAT3 transcriptionally modulates a range of targeted genes, several of which even convey contradictory responses. Specifically, STAT3 stimulates proliferation through the upregulation of the antiapoptotic gene BCL2 in B cells, whereas STAT3 activation also leads to the downregulation of MYC and MYB and the induction of JUNB and IRF1 in monocytic cells, contributing to their terminal differentiation and growth arrest.27 Thus, STAT3-regulated genes (including both induced and repressed targets) have been reported in numerous cellular and organismal functions, including cell cycle progression, apoptosis, intermediate metabolism, inflammation, invasion, and angiogenesis.6,10,27,29
Unphosphorylated STAT3
Although the presence of the phosphotyrosine dimer is important in target gene activation, unphosphorylated STAT3 may drive a range of different gene expression. Yang and coworkers employed a Y705F STAT3 mutant, which cannot be phosphorylated on residue 705, and showed that the levels of many mRNAs were strongly affected by Y705F STAT3, including several well-known oncoproteins such as MRAS and MET.9 The exact mechanism underlying the transcriptional ability of unphosphorylated STAT3 is yet to be fully understood. However, one hypothesis suggests that the potential for protein-protein interactions is the primary mechanism, including the ability of STAT3 to interact with other transcription factors on target DNA and enhance transcription.9
Serine phosphorylation of STAT3
The literature suggests a modulatory role for Ser727 phosphorylation in STAT3 transcriptional activation, presumably through the enhanced recruitment of necessary transcriptional cofactors as occurs after the serine phosphorylation of STAT1.21,30,31 The enzymes that catalyze the phosphorylation of STAT3 on S727 are numerous, including MTOR and MAPK1.5,31,32 Studies have shown that STAT3 requires tyrosine and serine residues to be phosphorylated by independent protein kinase activities for the maximal activation of target gene transcription. Yokogami et al. showed that STAT3 Ser727 phosphorylation was inhibited by the MTOR inhibitor rapamycin during CNTF (ciliary neurotrophic factor) signaling and that the maximal activation of STAT3 in CNTF-stimulated neuroblastoma cells depends on serine phosphorylation.30
Translocation of STAT3 to the nucleus and mitochondria
The STAT3 protein primarily dwells in the cytoplasm, and nuclear localization of STAT3 is transient, lasting no more than several hours.6,7,12,27 Indeed, the activation of STAT3 is under tight control because the phosphorylated, active STAT3 dimers are regulated by nuclear and perinuclear tyrosine phosphatases such as PTPN6, PTPN11, and PTPN2.33 According to most literature, the nuclear localization of STAT3 requires phosphorylation and dimerization.5,7,12,17,27,34 However, Liu et al. reported that the nuclear import of STAT3 is mediated by a constitutively but not conditionally active nuclear localization signal (NLS) that requires the STAT3 coiled-coil domain to interact with a specific import carrier, importin-alpha3. This finding suggests that although phosphorylated dimers are required for STAT3 to bind to specific DNA target sites, nuclear import occurs constitutively and independently of tyrosine phosphorylation, and STAT3 proteins dynamically shuttle between cytoplasmic and nuclear compartments.35 However, these observations contradict several primary reports that STAT3 protein merely conditionally translocates into the nucleus in response to IL6.7 Thus, the conformational shift upon STAT3 dimerization may contribute to the access of the NLS. However, the diverse expression of nuclear transport proteins in various cell types may present different phenotypes. Reportedly, STAT3 nuclear export depends on nuclear export signal (NES) elements located within the DNA-binding domain, which can be recognized by XPO1 (exportin 1). The location of the NES sequences suggests that it is inaccessible to the nuclear receptor XPO1 when STAT3 is bound to DNA. Thus, only unbound STAT3 protein molecules can be actively exported from the nucleus after dephosphorylation.34,36
Apart from its well-described nuclear counterpart, recent evidence has also shown that a pool of mitochondrial STAT3 (mitoSTAT3) exists that executes transcription-independent functions. Normally, the pool of mitoSTAT3 is approximately one-tenth of the amount of STAT3 in the cytosol, yet increasing accumulation of mitoSTAT3 appears in response to various stimuli such as ischemia.37,38 The translocation of STAT3 into the mitochondria was reported to be regulated by NDUFA13, a component of the electron transport chain (ETC) complex I. Interestingly, NDUFA13 was found to colocalize and interact with Ser727-phosphorylated STAT3 in the mitochondria. As Wegrzyn and colleagues37 found, the immunoprecipitates of mitochondrial extracts yielded from mouse liver captured by a monoclonal antibody specifically targeted to the components of the ETC complex I contained STAT3 and NDUFA13. Using highly purified rat heart mitochondria in vitro, Tammineni et al.39 further discovered that STAT3 most likely resides in the inner mitochondrial membrane (IMM), facing toward the matrix. STAT3 import and translocation across the mitochondrial membrane requires membrane potential and, likely, energy. When STAT3 is imported alone, a small fraction of STAT3 associates with the IMM and matrix fractions. However, upon its co-import with NDUFA13, most of the STAT3 is recruited to the IMM. Thus, NDUFA13 may act as a chaperone to facilitate STAT3 localization into the mitochondrial membranes, especially the inner membrane, by a common membrane receptor. NDUFA13 most likely alters the topology of mitochondrial STAT3 and promotes its integration into complex I. The C terminus of STAT3, especially Ser727, is required for the NDUFA13-dependent import and integration of STAT3 into complex I because mutating serine 727 to alanine reduces the import of STAT3 into mitochondria, even in the presence of NDUFA13.39
STAT3 in autophagy
Autophagy is regulated by a network of intricate signaling pathways. The crosstalk between autophagy and other stress response pathways, including STAT3 signaling, may decide the survival or death of a cell. 2,5 Literature throughout the decade has implicated STAT3 in several steps of autophagy, from the assembly of autophagosomes to their maturation. Furthermore, the role of STAT3 is further complicated by its cellular localization because differentially localized STAT3 regulates autophagy in distinct ways (Fig. 2). This regulation also works in concert with other autophagy-related signaling molecules.
Figure 2.

Subcellular localization of STAT3 in the regulation of autophagy. STAT3 monomers in the cytosol are phosphorylated by Src or JAK kinase on Tyr705 and subsequently form STAT3 dimers, which are then shuttled into the cell nucleus and bind with specific DNA elements to transcriptionally activate or suppress target genes such as BCL2, BECN1, PIK3C3, CTSB, CTSL, PIK3R1/p55α, PIK3R1/p50α, and MIR17HG as well as HIF1A and BNIP3, which either inhibit or stimulate autophagy depending on the different cellular context or stimulus. In contrast, unphosphorylated cytoplasmic STAT3 sequesters EIF2AK2, FOXO1, and FOXO3. EIF2AK2 promotes autophagy by phosphorylating EIF2A, an endoplasmic reticulum stress-related protein that upregulates ATF4. FOXO1 and FOXO3 also positively modulate autophagy by transcriptionally activating a series of autophagy-related genes such as ULK2, BECN1, BNIP3, BNIP3L, PIK3C3, ATG12, ATG4B, and MAP1LC3A. STAT3 monomers also translocate to the mitochondria and interact with complexes I and II of the ETC to repress ROS production. ROS induces autophagy by activating HIF1A, possibly by modulating the transcriptional ability of STAT3.
Regulation of autophagy by nuclear STAT3
STAT3 is the main transcriptional enhancer of several autophagy-related genes in the nucleus, and this activity contributes to a range of anti- vs. pro-autophagic functions of STAT3 in autophagy, which we will discuss separately below (Table 1). The anti-autophagic function primarily lies in its ability to disrupt the formation of the BECN1/PIK3C3 complex. One of its core components, BECN1, like other proapoptotic BH3-only proteins (such as BAX, BAD, BNIP3, and BCL2L11/BIM), contains a BH3 domain that may interact with the antiapoptotic BCL2 family members (such as BCL2, BCL2L1, and MCL1). In this regard, the antiapoptotic family members are important negative regulators of BECN1, whereas members of the proapoptotic BH3-only proteins displace BECN1 to retain its proautophagic activity.2
Table 1.
Nuclear STAT3-regulated genes in autophagy
| Target genes | Anti/pro-autophagy | Mechanisms | Reference |
|---|---|---|---|
| BCL2 family members | anti- | STAT3 transcriptionally activates BCL2 and MCL1 expression, which leads to autophagy inhibition | 41,42 |
| BECN1 | anti- | STAT3 directly binds to the promoter region of BECN1 and represses its transcription by recruiting HDAC3 | 43 |
| PIK3C3 | anti- | STAT3 downregulates PIK3C3, which is an essential component of the BECN1-PIK3C3 complex | 44 |
| PIK3R1/p55α, PIK3R1/p50α | anti- | STAT3 upregulates the expression of PIK3R1/p55α and PIK3R1/p50α, which compete with PIK3R1/p85α to inhibit autophagy. | 46 |
| CTSB, CTSL | possibly anti- | STAT3 upregulates CTSB and CTSL, which enhances cell death by LMP; LMP impairs the autophagy flux. | 45 |
| HIF1A | pro- in hypoxia anti- in normoxia. | STAT3 transcriptionally activates and stabilizes HIF1A, which induces autophagy in hypoxia and represses autophagy in normoxia. | 47,48,50 |
| BNIP3 | pro- | STAT3 phosphorylation upregulates BNIP3 expression; BNIP3 is a pro-autophagic BH3-only protein. | 51 |
Nuclear STAT3 has the ability to transcriptionally activate BCL2, BCL2L1, and MCL1.11,12,40 Upon activation, nuclear STAT3 upregulates BCL2 expression and consequently leads to autophagy inhibition, and the dominant negative inhibition of STAT3 typically induces the upregulation of autophagy. Feng et al.41 showed that the biguanide metformin downregulates STAT3 activity and BCL2 expression and induces autophagy both in vitro and in vivo. A similar report by Tai and colleagues42 showed that sorafenib, a small molecular inhibitor of KDR and PDGFRB, activates autophagy in a dose- and time-dependent manner in multiple HCC cell lines through the downregulation of phospho-STAT3 (pSTAT3) and the subsequent reduction of MCL1. Conversely, the ectopic expression of MCL1 reverses the effect of sorafenib on autophagy.
The findings of Miao et al.43 indicate that BECN1 may also be a direct transcriptional target of STAT3 because IL6 treatment significantly promotes STAT3 phosphorylation while reducing BECN1 mRNA and protein levels in AGS and NCI-H1650 cells. A significant reduction of BECN1 occurs in cells transfected with constitutively activated STAT3, and a dominant-interfering STAT3 mutant (STAT3Y705F) increases BECN1 mRNA and protein levels in AGS and NCI-H1650 cells.43 Further studies revealed that STAT3 directly binds to the promoter region of BECN1 and represses its transcription through the recruitment of HDAC3 (histone deacetylase 3).43
In addition, PIK3C3 is also regulated by STAT3 tyrosine phosphorylation. Yamada et al.44 found that increased STAT3Y705 phosphorylation is associated with the downregulation of PIK3C3. The expression of the STAT3Y705F mutant markedly increases autophagic flow in the WT tibialis anterior muscle, along with a marked induction of PIK3C3 protein expression, suggesting the possible indirect regulation of PIK3C3 protein levels by nuclear STAT3.
One crucial stage of autophagy is the degradation of autophagosome, which involves fusion with lysosome to form autolysosome and degradation of the inner membrane together with its luminal contents. Thus functional lysosomes are indispensable for the progression of autophagic flux. STAT3 plays an important role in lysosomal-mediated programmed cell death during post-lactational regression (involution) of the mammary gland.45 A recent study found that autophagy is strikingly induced at 24 h of involution, whereas STAT3 inhibits this prosurvival autophagy that occurs concomitantly with lysosomal-mediated programmed cell death. STAT3 suppresses autophagy in mammary gland cells by directly upregulating the PIK3R1/p55α and PIK3R1/p50α (phosphoinositide-3-kinase, regulatory subunit 1 [α], p55α and p50α isoforms), which function as inhibitors of PIK3R1/p85α-mediated autophagy.46 In addition, STAT3 transcriptionally enhances the expression of CTSB (cathepsin B) and CTSL (cathepsin L), possibly through the downregulation of Serpina3g, a known target of NFKB1 in memory T cells.45 STAT3 can inhibit the transcriptional activity of NFKB1 by binding to NFKB1 and RELA, subsequently downregulating its target genes, including Serpina3g. Thus STAT3-mediated upregulation of CTSB and CTSL enhances cell death by lysosomal membrane permeabilization (LMP) during mammary gland involution,45 whereas LMP impairs autophagy because the autophagy flux requires functional lysosomes.46
As described above, nuclear STAT3 executes anti-autophagic functions by upregulating negative regulators of autophagy such as BCL2, BCL2L1, MCL1, PIK3R1/p55α, and PIK3R1/p50α, or by downregulating essential autophagy genes such as BECN1 and PIK3C3. However, STAT3 also executes its pro-autophagic function by modulating the hypoxic expression of HIF1A (hypoxia inducible factor 1, α subunit [basic helix-loop-helix transcription factor]) and BNIP3.
The regulation of HIF1A by STAT3 is 2-fold. On the one hand, STAT3 transcriptionally upregulates HIF1A;47 on the other hand, STAT3 interacts with the C-terminal domain of HIF1A and stabilizes the protein from ubiquitination mediated by VHL (von Hippel-Lindau tumor suppressor, E3 ubiquitin protein ligase).48 Interestingly, HIF1A may also modulate autophagy in a context-dependent manner. HIF is a heterodimer of a constitutive β subunit and an oxygen-regulated α subunit that is stabilized and expressed when oxygen concentrations decline below a threshold of 5%. Upon moderate hypoxia (1%-3%), HIF activates the transcription of BNIP3 and BNIP3L, genes encoding 2 BH3-only proteins that disrupt the inhibitory interaction between BECN1 and BCL2 in favor of autophagy induction.2,49 On the other hand, under normoxic conditions, the report by Yuan et al.50 demonstrates that the stable overexpression of HIF1A reverses the induction of autophagy by the JAK2-STAT3 inhibitor cucurbitacin I in U251 cells.
In the case of BNIP3 regulation by STAT3, research performed by Pratt and Annabi51 showed that concanavalin A (ConA) induces autophagy, evidenced by the formation of acidic vacuoles and the induction of BNIP3 expression. However, the genetic or pharmaceutical inhibition of STAT3 abrogates ConA-induced BNIP3 expression and subsequent autophagy, indicating that STAT3 phosphorylation potentially upregulates autophagy via increased BNIP3 expression. Notably, no other autophagosome biomarkers have been examined except for the acidic vacuolar organelles. Thus, further study is required to validate that the acidic vacuolar organelles induced by ConA are autophagosomes and, if so, that this accumulation of autophagosomes represents the induction of autophagy rather than a failure to clear autophagosomes.
Furthermore, STAT3 has been reported to regulate the expression of a series of microRNAs (miRNAs) that target autophagy-related genes, which may be another possible mechanism for nuclear STAT3 regulation of autophagy. For example, STAT3 upregulates MIR17HG (miR-17-92 cluster host gene [non-protein coding]) through a highly conserved STAT3 binding site in the promoter region,52 and members of the MIR17HG cluster have been reported to target the autophagy-related genes ULK1, BECN1, and BCL2L11.53-55 Several miRNAs that have been transcriptionally modulated by STAT3 and have been reported to target autophagy pathways are summarized in Table 2. 26,52-64 However, the role of miRNAs in the regulation of STAT3-mediated autophagy has not been well established, and this need to be further investigated.
Table 2.
STAT3 regulated microRNAs that target autophagy pathway
Regulation of autophagy by cytoplasmic STAT3
Nuclear STAT3 regulates autophagy in a multifaceted manner. For example, STAT3 inhibits autophagy by transcriptionally activating BCL2 or stimulates autophagy by upregulating and stabilizing HIF1A under hypoxia. However, as previously mentioned, to effectively transduce extracellular signals, a large fraction of STAT3 protein resides in the cytoplasm, where the main autophagy process occurs. A frequently cited report by the Kroemer group14 unraveled a novel mechanism to explain how cytoplasmic STAT3 inhibits autophagy by inhibiting EIF2AK2 (eukaryotic translation initiation factor 2-α kinase 2) activity. In a library screen for autophagy inducers, a series of pharmacological inhibitors of STAT3 were identified as potent activators of autophagy. Small interfering RNA (siRNA)-mediated knockdown of STAT3 also showed that decreasing the total STAT3 level induces autophagy. Further investigation revealed that cytoplasmic STAT3 regulates autophagy in a more direct manner. The SH2 domain of cytoplasmic STAT3 exhibits a conformational fold that resembles the C terminus of the EIF2AK2 substrate EIF2A (eukaryotic initiation factor 2A, 65kDa), and interacts with the catalytic domain of EIF2AK2, thus inhibiting EIF2AK2 enzymatic activity and preventing it from phosphorylating EIF2A, a known autophagy activator. Wild-type STAT3 and nonphosphorylatable STAT3Y705F transfected into U2OS cells inhibit baseline as well as starvation-induced EIF2A phosphorylation, further confirming this hypothesis.14 The phosphorylation of EIF2A mediates general translational arrest and promotes the selective transactivation of stress response genes. Cells carrying a nonphosphorylatable EIF2A (S51A) mutant fail to induce autophagy in starvation conditions, suggesting that EIF2A phosphorylation of serine 51 plays a major role in autophagy regulation.65 Currently, how EIF2A phosphorylation contributes to autophagy initiation has not been thoroughly described. One speculation is that EIF2A phosphorylation may affect the endoplasmic reticulum in a manner that promotes the physical formation of the phagophore. Another possibility is that EIF2A phosphorylation stimulates autophagy via its effects on the transactivation of autophagy genes. EIF2A phosphorylation stimulates the selective translation of the ATF4 transcription factor while suspending general translation, which subsequently stimulates MAP1LC3A expression.66 This revelation suggests that cytoplasmic STAT3 functions as a negative regulator of autophagy in a transcription-independent manner.
In addition to its direct effect on EIF2AK2 activity, cytoplasmic STAT3 also interacts with the autophagy-related proteins FOXO1 and FOXO3. Following its dephosphorylation, FOXO3 translocates into the nucleus and upregulates multiple autophagy-related genes such as ULK2, BECN1, PIK3C3, BNIP3, BNIP3L, ATG12, ATG4B, and MAP1LC3A.67 Oh et al. 68 showed that active FOXO1 and FOXO3 reside exclusively in the nucleus of naive T cells, whereas inactive phosphorylated FOXO1 and FOXO3 are most abundant in activated T cells, sequestered in their cytoplasm in association with unphosphorylated STAT3. When the JAK-STAT3 pathway is activated by IL6 or IL10, FOXO1 and FOXO3 rapidly relocalize into the nucleus in response to STAT3 phosphorylation, and the accumulation of FOXO1 and FOXO3 in the nuclei coincides with the increased expression of target genes. STAT3 inhibitors abrogate the cytokine-induced translocation of FOXO1 and FOXO3 into the nucleus.
As depicted above, cytoplasmic STAT3 sequesters EIF2AK2, FOXO1, and FOXO3 under normal conditions, suggesting an inhibitory role. However, upon immediate STAT3 activation, EIF2AK2, FOXO1, and FOXO3 are released from cytoplasmic anchorage and mediate the proautophagic response through the phosphorylation of EIF2A, FOXO1, and FOXO3 through translocation to the nucleus and upregulation of autophagy-related gene products. Thus, a proautophagic signaling pathway may also lie within STAT3 activation.
Regulation of autophagy by mitochondrial STAT3
Complementing the roles of nuclear and cytoplasmic STAT3, the aforementioned translocation of STAT3 to the mitochondria may play a critical role in autophagy regulation. Recent discoveries unveiled a novel role for STAT3 translocated to the mitochondria in regulating the activity of the electron transport chain, which represents a major source of ROS production.38 Wegrzyn et al.37 reconstituted STAT3 into STAT3−/− B cells with a retrovirus expressing STAT3. Oxidative phosphorylation in mitochondria significantly decreased in the STAT3−/− pro-B cells (40% decrease of complex I activity and 85% decrease of complex II activity in STAT3−/− cells compared with WT pro-B cells); however, the expression of STAT3 restored the activities of complexes I and II. To confirm that the defects were due to a lack of mitoSTAT3, a STAT3 construct containing the COX8 (cytochrome c oxidase subunit VIII) mitochondrial localization sequence (MLS) fused to the N terminus of STAT3 (MLS-STAT3) was generated. The MLS places the protein of interest in the inner mitochondrial membrane. Mutations in either Tyr705 or the DNA-binding domain of MLS-STAT3 restore the activity of complexes I and II. Interestingly, the phospho-mimicking mutant MLS-STAT3Y705F,S727D reconstitutes the activities of complexes I and II, whereas the dominant negative mutant MLS-STAT3Y705F,S727A is ineffective, suggesting that serine but not tyrosine phosphorylation is important for the activity of STAT3 in the mitochondria and that STAT3 most likely exerts its actions as a monomer. Mitochondria from the hearts of stat3flox/flox/cre (stat3−/−) and Stat3flox/flox (wild-type) mice were assayed for complex I and II activities.37 ETC assays confirmed that stat3−/− heart mitochondria have defects in complexes I and II but have normal complex III activity. Using transgenic mice generated with cardiomyocyte-restricted expression of STAT3 that was targeted to the mitochondria with a MLS and containing mutations in the DNA-binding domain (MLS-STAT3E), Szczepanek et al.69 discovered that ischemia increases the release of H2O2 in WT mitochondria respiring on glutamate and malate, whereas no such effect is observed in mitochondria expressing MLS-STAT3E. Additionally, the maximal capacity of complex I to produce H2O2 after ischemia, established by inhibiting complex I with rotenone and supplying glutamate and malate as a substrate, is decreased in mitochondria expressing MLS-STAT3E compared with WT mitochondria. Thus, at this point, STAT3 is considered to interact with complexes I and II of the ETC to modulate their activities through protein-protein interactions or post-translational modifications, and the loss of STAT3 results in a significant increase in ROS.70 Several studies have implicated mitochondrial ROS in the induction of autophagy, especially the selective autophagic degradation of mitochondria, also known as mitophagy.71 Thus, the mitochondrial translocation of STAT3 inevitably suppresses autophagy induced by oxidative stress and effectively preserves mitochondria from mitophagy. This phenomenon supports the prosurvival features of STAT3 because MLS-STAT3 expression inhibits CYCS (cytochrome c, somatic) release by preventing the opening of the mitochondrial permeability transition pore during ischemia.69
Implication of STAT3-regulated autophagy in cancer therapeutics
The last decade marked a revolutionary improvement for targeted treatments and chemotherapy agents directed against numerous oncogenic pathways, especially for agents targeting receptor tyrosine kinases. Autophagy is well known for its prosurvival phenotype in numerous diseases, including cancer. Additionally, autophagy-related chemoresistance and targeted therapy resistance have been extensively studied in recent years.3,72,73 Currently, several agents targeting the JAK-STAT pathway have been under investigation in clinical trials for cancer treatment, and the same attention must be paid to the resistances provoked by the upregulation of autophagy.
Tyrosine kinases are the primary oncogenic pathways in numerous malignancies. Many targeted therapies focus their efforts on blocking these kinases and their corresponding pathways. However, these interferences inevitably trigger the activation of bypassing mechanisms, including autophagy. A wide range of RTKs phosphorylate STAT3 directly or via SRC kinase, and several studies have proven that blocking kinases such as KDR, PDGFRB, and ALK results in enhanced autophagy by subsequent inhibition of the STAT3 pathway (Table 3).14,41,42,50,74-83 Currently, several JAK-STAT inhibitors are being examined in clinical trials for cancer therapy. For example, in a phase II clinical trial to treat metastatic pancreatic cancer, ruxolitinib together with capecitabine showed a benefit for overall survival and progression-free survival compared with a placebo plus capecitabine used in second-line therapy, promoting the application of JAK-STAT inhibitors in cancer therapy.84 However, similar to other targeted therapies, acquired resistance causes problems for the further application of such agents, among which is the secondary mutation JAK2V617F in JAK kinase. 85 A number of other factors also need to be considered, including the upregulation of autophagy. In the research conducted by Yuan et al.86 human malignant glioma U251 and A172 cells were treated with a STAT3 inhibitor, WP1066, or a short hairpin RNA plasmid targeting STAT3 to suppress the activation of STAT3 signaling. STAT3 inhibition enhances the radiosensitivity of glioma cells; however, WP1066 or shSTAT3 treatment and ionizing radiation exposure induce the lipidation of MAP1LC3A and increase BECN1 expression. Furthermore, the combination of ionizing radiation exposure and STAT3 inhibition trigger a pronounced increase of autophagy flux. The inhibition of autophagy by 3-methyladenine or ATG5 siRNA strengthens the radiosensitizing effects of STAT3 inhibition, and the apoptosis of more cells is induced by the cotargeting of autophagy and STAT3 signaling, indicating a prosurvival role of STAT3 inhibition-induced autophagy. Our recent work also indicated that the autophagy inhibitor static induces cytoprotective autophagy in hepatic tumor cells and xenografts, whereas cotargeting of the autophagy pathway sensitizes the antitumor effect of STAT3 inhibition (unpublished data).
Table 3.
Targeted and chemical agents that induced autophagy via the STAT3 pathway
| Agent | Target gene | Tumor type | Mechanism | References |
|---|---|---|---|---|
| Sorafenib | KDR, PDGFRB, RAF | Prostate cancer, Glioma, hepatocellular carcinoma | Downregulation of pSTAT3 and MCL1 expression | 42,74,75 |
| Crizotinib | ALK fusion gene | anaplastic large cell lymphoma | Downregulation of pSTAT3 and BCL2 family | 76 |
| Stattic | STAT3 | Osteosarcoma | Blockage of the interaction between cytoplasmic STAT3 and EIF2AK2 | 14 |
| Cucurbitacin I | JAK2 | Glioblastoma | Downregulation of pSTAT3 and HIF1A | 50 |
| Metformin | PRKAA2? | esophageal squamous cell carcinoma | Downregulation of pSTAT3 and BCL2 | 41 |
| Cucurbitacin B (CuB) | ? | melanoma | Downregulation of pSTAT3 and BCL2 | 77 |
| Nexrutine | STAT3? | pancreatic cancer | Modulation of STAT3 and decrease of ROS generation | 78 |
| Obatoclax | PTPN6 | hepatocellular carcinoma | Downregulation of pSTAT3 and MCL1 | 79,80 |
| Niclosamide | ? | myeloma | Inhibition of pSTAT3 and upregulation of autophagy | 81 |
| Arsenic trioxide | STAT3? | Glioblastoma | Inhibition of pSTAT3 and upregulation of autophagy | 82 |
| Concanavalin A | MMP14 | Glioblastoma | Upregulation of pSTAT3 and BNIP3 | 83 |
Issues and Perspectives
The regulation of autophagy by STAT3 primarily features prosurvival characteristics regardless of its subcellular localization. The translocation of STAT3 affects both the early-phase autophagic flux and the prolonged autophagic response. As a well-recognized stress responsive nuclear factor, STAT3 responds to a panel of autophagy inducers, including hypoxia, ROS, growth factor deprivation, and a number of targeting agents. For example, during growth factor deprivation, the absence of RTK or SRC phosphorylation represses the tyrosine phosphorylation of STAT3 and its target gene BCL2; however, the expression of BECN1 and PIK3C3 are elevated. All of these effects accommodate the formation of the BECN1-PIK3C3 complex, initiating autophagy. As a consequence, the amount of total STAT3 may also be reduced because STAT3 transcription is self-activated, rendering the pro-autophagic proteins EIF2AK2, FOXO1, and FOXO3 free to phosphorylate EIF2A or to upregulate autophagy-related genes in promoting cytoprotective autophagy. In the case of hypoxia, the low oxygen concentration results in the hypoxic expression of HIF1A, which leads to STAT3 phosphorylation. In turn, pSTAT3 contributes to HIF1A transcriptional activation and stabilization, followed by the transcription of the BH3-only proteins BNIP3 and BNIP3L. Meanwhile, ROS production is increased in mitochondria, which mediates the mitochondrial translocation of STAT3. mitoSTAT3 stabilizes and protects complexes I and II of the ETC from hypoxic insult, consequently reducing the ROS level and preserving the mitochondria from ROS-induced mitophagy as well as preventing the release of CYCS due to mitochondrial outer membrane permeabilization, thereby avoiding mitochondrial-dependent apoptosis. However, many questions remain that require further study. For example, as a prominent nuclear factor, STAT3 reportedly regulates the expression of more than 1,000 gene products. As described in this paper, not all of these products modulate autophagy in the same direction. Although most of this modulation occurs in a context-dependent manner, the mechanism underlying how specific stimuli drive STAT3 signaling to accommodate or restrain autophagy has not been fully explained. The regulation of autophagy by cytoplasmic STAT3 involves multiple protein-protein interactions, but the different affinities of autophagy-related molecules to cytoplasmic STAT3 remain unclear, and whether these proteins comprise a large interactome or bind to different partners in response to different signals remains puzzling. Notably, a number of studies have contributed to the revelation of the role of STAT3 in autophagy. However, several of the recent studies have not distinguished between the transcriptional and nontranscriptional functions of STAT3 signaling in autophagy. Thus, future studies may shed greater insights into the role of STAT3 if they address these differences.
Further study of the role of STAT3 in autophagy may provide us with an in-depth understanding of its physiology as well as with a novel strategy for cancer therapeutics. Although the existing data do not bode well for an eminent outcome for the clinical application of autophagy inhibition in cancer treatment, the cotargeting of the autophagic pathway may prove beneficial in specific types of cancer. Presently, most of the STAT3-targeted agents are in early trials, with only a few reports of autophagy-related resistance. However, in an attempt to raise the efficacy of these agents, due consideration is needed to anticipate the resistance caused by the upregulation of cellular autophagy. To date, autophagy inhibition has been shown to be an efficacious addition to several cytotoxic agents and targeted therapeutics in multiple preclinical models.3 Currently, combination treatments using targeted agents and autophagy inhibitors are in trials for clinical application. The latest reports have shown that the autophagy inhibitor hydroxychloroquine is tolerable and potentially effective in combination with the MTOR-targeted agent temsirolimus87 and the proteasome inhibitor bortezomib88 in phase I trials and may be a valid complement to STAT3-targeted therapy. Nevertheless, the systemic inhibition of autophagy introduces another problem involving the impairment of the toxicity tolerance of noncancerous cells. Autophagy in normal cells represents a defense mechanism, and reports have demonstrated that the activation of autophagy protects hepatocytes against chemical-induced hepatotoxicity.89 Thus, the rational selection of an autophagy-targeted subpopulation or the development of a more targeted delivery method remain as challenges for further studies.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
This work is supported by grants from the National Natural Science Foundation of China (grant number 81272593) to H Pan and the National Natural Science Foundation of China (grant number 81372621) to W Han.
References
- 1. Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell 2008; 132:27-42; PMID:18191218; http://dx.doi.org/ 10.1016/j.cell.2007.12.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kroemer G, Marino G, Levine B. Autophagy and the integrated stress response. Mol Cell 2010; 40:280-93; PMID:20965422; http://dx.doi.org/ 10.1016/j.molcel.2010.09.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Janku F, McConkey DJ, Hong DS, Kurzrock R. Autophagy as a target for anticancer therapy. Nat Rev Clinical Oncol 2011; 8:528-39; PMID:21587219; http://dx.doi.org/ 10.1038/nrclinonc.2011.71 [DOI] [PubMed] [Google Scholar]
- 4. Maycotte P, Gearheart CM, Barnard R, Aryal S, Mulcahy Levy JM, Fosmire SP, Hansen RJ, Morgan MJ, Porter CC, Gustafson DL, et al. . STAT3-Mediated Autophagy Dependence Identifies Subtypes of Breast Cancer Where Autophagy Inhibition Can Be Efficacious. Cancer Res 2014; PMID:24590058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Pietrocola F, Izzo V, Niso-Santano M, Vacchelli E, Galluzzi L, Maiuri MC, Kroemer G. Regulation of autophagy by stress-responsive transcription factors. Semin Cancer Biol 2013; 23:310-22; PMID:23726895; http://dx.doi.org/ 10.1016/j.semcancer.2013.05.008 [DOI] [PubMed] [Google Scholar]
- 6. Levy DE, Darnell JE, Jr. Stats: transcriptional control and biological impact. Nat Rev Mol Cell Biol 2002; 3:651-62; PMID:12209125; http://dx.doi.org/ 10.1038/nrm909 [DOI] [PubMed] [Google Scholar]
- 7. Akira S, Nishio Y, Inoue M, Wang XJ, Wei S, Matsusaka T, Yoshida K, Sudo T, Naruto M, Kishimoto T. Molecular cloning of APRF, a novel IFN-stimulated gene factor 3 p91-related transcription factor involved in the gp130-mediated signaling pathway. Cell 1994; 77:63-71; PMID:7512451; http://dx.doi.org/ 10.1016/0092-8674(94)90235-6 [DOI] [PubMed] [Google Scholar]
- 8. Narimatsu M, Maeda H, Itoh S, Atsumi T, Ohtani T, Nishida K, Itoh M, Kamimura D, Park SJ, Mizuno K, et al. . Tissue-specific autoregulation of the stat3 gene and its role in interleukin-6-induced survival signals in T cells. Mol Cell Biol 2001; 21:6615-25; PMID:11533249; http://dx.doi.org/ 10.1128/MCB.21.19.6615-6625.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yang J, Chatterjee-Kishore M, Staugaitis SM, Nguyen H, Schlessinger K, Levy DE, Stark GR. Novel roles of unphosphorylated STAT3 in oncogenesis and transcriptional regulation. Cancer Res 2005; 65:939-47; PMID:15705894 [PubMed] [Google Scholar]
- 10. Bromberg J. Stat proteins and oncogenesis. J Clin Investigat 2002; 109:1139-42; PMID:11994401; http://dx.doi.org/ 10.1172/JCI0215617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bowman T, Garcia R, Turkson J, Jove R. STATs in oncogenesis. Oncogene 2000; 19:2474-88; PMID:10851046; http://dx.doi.org/ 10.1038/sj.onc.1203527 [DOI] [PubMed] [Google Scholar]
- 12. Bromberg JF, Wrzeszczynska MH, Devgan G, Zhao Y, Pestell RG, Albanese C, Darnell JE, Jr. Stat3 as an oncogene. Cell 1999; 98:295-303; PMID:10458605; http://dx.doi.org/ 10.1016/S0092-8674(00)81959-5 [DOI] [PubMed] [Google Scholar]
- 13. Takeda K, Noguchi K, Shi W, Tanaka T, Matsumoto M, Yoshida N, Kishimoto T, Akira S. Targeted disruption of the mouse Stat3 gene leads to early embryonic lethality. Proc Natl Acad Sc U S A 1997; 94:3801-4; PMID:9108058; http://dx.doi.org/ 10.1073/pnas.94.8.3801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Shen S, Niso-Santano M, Adjemian S, Takehara T, Malik SA, Minoux H, Souquere S, Marino G, Lachkar S, Senovilla L, et al. . Cytoplasmic STAT3 represses autophagy by inhibiting PKR activity. Mol Cell 2012; 48:667-80; PMID:23084476; http://dx.doi.org/ 10.1016/j.molcel.2012.09.013 [DOI] [PubMed] [Google Scholar]
- 15. Du Toit A. Autophagy: STAT3 maintains order. Nat Rev Mol Cell Biol 2012; 13:754; PMID:23151659; http://dx.doi.org/ 10.1038/nrm3472 [DOI] [PubMed] [Google Scholar]
- 16. Yokoyama T, Kondo Y, Kondo S. Roles of mTOR and STAT3 in autophagy induced by telomere 3' overhang-specific DNA oligonucleotides. Autophagy 2007; 3:496-8; PMID:17617738; http://dx.doi.org/ 10.4161/auto.4602 [DOI] [PubMed] [Google Scholar]
- 17. Zhong Z, Wen Z, Darnell JE, Jr. Stat3: a STAT family member activated by tyrosine phosphorylation in response to epidermal growth factor and interleukin-6. Science 1994; 264:95-8; PMID:8140422; http://dx.doi.org/ 10.1126/science.8140422 [DOI] [PubMed] [Google Scholar]
- 18. Kato T, Sakamoto E, Kutsuna H, Kimura-Eto A, Hato F, Kitagawa S. Proteolytic conversion of STAT3alpha to STAT3gamma in human neutrophils: role of granule-derived serine proteases. J Biol Chem 2004; 279:31076-80; PMID:15145953; http://dx.doi.org/ 10.1074/jbc.M400637200 [DOI] [PubMed] [Google Scholar]
- 19. Chakraborty A, Tweardy DJ. Granulocyte colony-stimulating factor activates a 72-kDa isoform of STAT3 in human neutrophils. J Leukoc Biol 1998; 64:675-80; PMID:9823774 [DOI] [PubMed] [Google Scholar]
- 20. Hevehan DL, Miller WM, Papoutsakis ET. Differential expression and phosphorylation of distinct STAT3 proteins during granulocytic differentiation. Blood 2002; 99:1627-37; PMID:11861277; http://dx.doi.org/ 10.1182/blood.V99.5.1627 [DOI] [PubMed] [Google Scholar]
- 21. Wen Z, Zhong Z, Darnell JE, Jr. Maximal activation of transcription by Stat1 and Stat3 requires both tyrosine and serine phosphorylation. Cell 1995; 82:241-50; PMID:7543024; http://dx.doi.org/ 10.1016/0092-8674(95)90311-9 [DOI] [PubMed] [Google Scholar]
- 22. Khatib H, Huang W, Mikheil D, Schutzkus V, Monson RL. Effects of signal transducer and activator of transcription (STAT) genes STAT1 and STAT3 genotypic combinations on fertilization and embryonic survival rates in Holstein cattle. J Dairy Sci 2009; 92:6186-91; PMID:19923622; http://dx.doi.org/ 10.3168/jds.2009-2439 [DOI] [PubMed] [Google Scholar]
- 23. Kodama H, Fukuda K, Pan J, Makino S, Baba A, Hori S, Ogawa S. Leukemia inhibitory factor, a potent cardiac hypertrophic cytokine, activates the JAK/STAT pathway in rat cardiomyocytes. Circulat Res 1997; 81:656-63; PMID:9351438; http://dx.doi.org/ 10.1161/01.RES.81.5.656 [DOI] [PubMed] [Google Scholar]
- 24. Yoon S, Woo SU, Kang JH, Kim K, Shin HJ, Gwak HS, Park S, Chwae YJ. NF-kappaB and STAT3 cooperatively induce IL6 in starved cancer cells. Oncogene 2012; 31:3467-81; PMID:22105366; http://dx.doi.org/ 10.1038/onc.2011.517 [DOI] [PubMed] [Google Scholar]
- 25. Choy MK, Movassagh M, Siggens L, Vujic A, Goddard M, Sanchez A, Perkins N, Figg N, Bennett M, Carroll J, et al. . High-throughput sequencing identifies STAT3 as the DNA-associated factor for p53-NF-kappaB-complex-dependent gene expression in human heart failure. Genome Med 2010; 2:37; PMID:20546595; http://dx.doi.org/ 10.1186/gm158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wu Z, Lu H, Sheng J, Li L. Inductive microRNA-21 impairs anti-mycobacterial responses by targeting IL-12 and Bcl-2. FEBS letters 2012; 586:2459-67; PMID:22710123; http://dx.doi.org/ 10.1016/j.febslet.2012.06.004 [DOI] [PubMed] [Google Scholar]
- 27. Levy DE, Lee CK. What does Stat3 do? J Cclin Investigat 2002; 109:1143-8; PMID:11994402; http://dx.doi.org/ 10.1172/JCI0215650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yu CL, Meyer DJ, Campbell GS, Larner AC, Carter-Su C, Schwartz J, Jove R. Enhanced DNA-binding activity of a Stat3-related protein in cells transformed by the Src oncoprotein. Science 1995; 269:81-3; PMID:7541555; http://dx.doi.org/ 10.1126/science.7541555 [DOI] [PubMed] [Google Scholar]
- 29. Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer 2009; 9:798-809; PMID:19851315; http://dx.doi.org/ 10.1038/nrc2734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yokogami K, Wakisaka S, Avruch J, Reeves SA. Serine phosphorylation and maximal activation of STAT3 during CNTF signaling is mediated by the rapamycin target mTOR. Curr Biol 2000; 10:47-50; PMID:10660304; http://dx.doi.org/ 10.1016/S0960-9822(99)00268-7 [DOI] [PubMed] [Google Scholar]
- 31. Laplante M, Sabatini DM. Regulation of mTORC1 and its impact on gene expression at a glance. J Cell Scie 2013; 126:1713-9; PMID:23641065; http://dx.doi.org/ 10.1242/jcs.125773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chung J, Uchida E, Grammer TC, Blenis J. STAT3 serine phosphorylation by ERK-dependent and -independent pathways negatively modulates its tyrosine phosphorylation. Mol Cell Biol 1997; 17:6508-16; PMID:9343414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kim DJ, Tremblay ML, Digiovanni J. Protein tyrosine phosphatases, TC-PTP, SHP1, and SHP2, cooperate in rapid dephosphorylation of Stat3 in keratinocytes following UVB irradiation. PloS One 2010; 5:e10290; PMID:20421975; http://dx.doi.org/ 10.1371/journal.pone.0010290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bhattacharya S, Schindler C. Regulation of Stat3 nuclear export. J Clin Investig 2003; 111:553-9; PMID:12588893; http://dx.doi.org/ 10.1172/JCI15372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Liu L, McBride KM, Reich NC. STAT3 nuclear import is independent of tyrosine phosphorylation and mediated by importin-alpha3. Proc Natl Acad Sc U S A 2005; 102:8150-5; PMID:15919823; http://dx.doi.org/ 10.1073/pnas.0501643102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. McBride KM, McDonald C, Reich NC. Nuclear export signal located within theDNA-binding domain of the STAT1transcription factor. EMBO J 2000; 19:6196-206; PMID:11080165; http://dx.doi.org/ 10.1093/emboj/19.22.6196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wegrzyn J, Potla R, Chwae YJ, Sepuri NB, Zhang Q, Koeck T, Derecka M, Szczepanek K, Szelag M, Gornicka A, et al. . Function of mitochondrial Stat3 in cellular respiration. Science 2009; 323:793-7; PMID:19131594; http://dx.doi.org/ 10.1126/science.1164551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Szczepanek K, Lesnefsky EJ, Larner AC. Multi-tasking: nuclear transcription factors with novel roles in the mitochondria. Trend Cell Biol 2012; 22:429-37; PMID:22705015; http://dx.doi.org/ 10.1016/j.tcb.2012.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Tammineni P, Anugula C, Mohammed F, Anjaneyulu M, Larner AC, Sepuri NB. The import of the transcription factor STAT3 into mitochondria depends on GRIM-19, a component of the electron transport chain. J Biol Chem 2013; 288:4723-32; PMID:23271731; http://dx.doi.org/ 10.1074/jbc.M112.378984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Fukada T, Hibi M, Yamanaka Y, Takahashi-Tezuka M, Fujitani Y, Yamaguchi T, Nakajima K, Hirano T. Two signals are necessary for cell proliferation induced by a cytokine receptor gp130: involvement of STAT3 in anti-apoptosis. Immunity 1996; 5:449-60; PMID:8934572; http://dx.doi.org/ 10.1016/S1074-7613(00)80501-4 [DOI] [PubMed] [Google Scholar]
- 41. Feng Y, Ke C, Tang Q, Dong H, Zheng X, Lin W, Ke J, Huang J, Yeung SC, Zhang H. Metformin promotes autophagy and apoptosis in esophageal squamous cell carcinoma by downregulating Stat3 signaling. Cell Ddeath Dis 2014; 5:e1088; PMID:24577086; http://dx.doi.org/ 10.1038/cddis.2014.59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tai WT, Shiau CW, Chen HL, Liu CY, Lin CS, Cheng AL, Chen PJ, Chen KF. Mcl-1-dependent activation of Beclin 1 mediates autophagic cell death induced by sorafenib and SC-59 in hepatocellular carcinoma cells. Cell Death Dis 2013; 4:e485; PMID:23392173; http://dx.doi.org/ 10.1038/cddis.2013.18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Miao LJ, Huang FX, Sun ZT, Zhang RX, Huang SF, Wang J. Stat3 inhibits Beclin 1 expression through recruitment of HDAC3 in nonsmall cell lung cancer cells. Tumor Biol 2014; 35:7097-103; PMID:24760274; http://dx.doi.org/ 10.1007/s13277-014-1961-6 [DOI] [PubMed] [Google Scholar]
- 44. Yamada E, Bastie CC, Koga H, Wang Y, Cuervo AM, Pessin JE. Mouse skeletal muscle fiber-type-specific macroautophagy and muscle wasting are regulated by a Fyn/STAT3/Vps34 signaling pathway. Cell Rep 2012; 1:557-69; PMID:22745922; http://dx.doi.org/ 10.1016/j.celrep.2012.03.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kreuzaler PA, Staniszewska AD, Li W, Omidvar N, Kedjouar B, Turkson J, Poli V, Flavell RA, Clarkson RW, Watson CJ. Stat3 controls lysosomal-mediated cell death in vivo. Nat Cell Biol 2011; 13:303-9; PMID:21336304; http://dx.doi.org/ 10.1038/ncb2171 [DOI] [PubMed] [Google Scholar]
- 46. Pensa S, Lloyd-Lewis B, Sargeant TJ, Resemann HK, Kahn CR, Watson CJ. Signal transducer and activator of transcription 3 and the phosphatidylinositol 3-kinase regulatory subunits p55alpha and p50alpha regulate autophagy in vivo. FEBS J 2014; 281:4557-67; PMID:25205393; http://dx.doi.org/ 10.1111/febs.13035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Jung JE, Lee HG, Cho IH, Chung DH, Yoon SH, Yang YM, Lee JW, Choi S, Park JW, Ye SK, et al. . STAT3 is a potential modulator of HIF-1-mediated VEGF expression in human renal carcinoma cells. FASEB J 2005; 19:1296-8; PMID:15919761 [DOI] [PubMed] [Google Scholar]
- 48. Jung JE, Kim HS, Lee CS, Shin YJ, Kim YN, Kang GH, Kim TY, Juhnn YS, Kim SJ, Park JW, et al. . STAT3 inhibits the degradation of HIF-1alpha by pVHL-mediated ubiquitination. Exp Mol Med 2008; 40:479-85; PMID:18985005; http://dx.doi.org/ 10.3858/emm.2008.40.5.479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Mazure NM, Pouyssegur J. Hypoxia-induced autophagy: cell death or cell survival? Curr Opin Cell Biol 2010; 22:177-80; PMID:20022734; http://dx.doi.org/ 10.1016/j.ceb.2009.11.015 [DOI] [PubMed] [Google Scholar]
- 50. Yuan G, Yan SF, Xue H, Zhang P, Sun JT, Li G. Cucurbitacin I Induces Protective Autophagy in Glioblastoma in Vitro and in Vivo. J Biol Chem 2014; 289:10607-19; PMID:24599950; http://dx.doi.org/ 10.1074/jbc.M113.528760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Pratt J, Annabi B. Induction of autophagy biomarker BNIP3 requires a JAK2/STAT3 and MT1-MMP signaling interplay in Concanavalin-A-activated U87 glioblastoma cells. Cell Signal 2014; 26:917-24; PMID:24462646; http://dx.doi.org/ 10.1016/j.cellsig.2014.01.012 [DOI] [PubMed] [Google Scholar]
- 52. Brock M, Trenkmann M, Gay RE, Michel BA, Gay S, Fischler M, Ulrich S, Speich R, Huber LC. Interleukin-6 modulates the expression of the bone morphogenic protein receptor type II through a novel STAT3-microRNA cluster 17/92 pathway. Circulat Res 2009; 104:1184-91; PMID:19390056; http://dx.doi.org/ 10.1161/CIRCRESAHA.109.197491 [DOI] [PubMed] [Google Scholar]
- 53. Wu H, Wang F, Hu S, Yin C, Li X, Zhao S, Wang J, Yan X. MiR-20a and miR-106b negatively regulate autophagy induced by leucine deprivation via suppression of ULK1 expression in C2C12 myoblasts. Cell Signal 2012; 24:2179-86; PMID:22781751; http://dx.doi.org/ 10.1016/j.cellsig.2012.07.001 [DOI] [PubMed] [Google Scholar]
- 54. Chatterjee A, Chattopadhyay D, Chakrabarti G. miR-17-5p downregulation contributes to paclitaxel resistance of lung cancer cells through altering beclin1 expression. PloS One 2014; 9:e95716; PMID:24755562; http://dx.doi.org/ 10.1371/journal.pone.0095716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Spaccarotella E, Pellegrino E, Ferracin M, Ferreri C, Cuccuru G, Liu C, Iqbal J, Cantarella D, Taulli R, Provero P, et al. . STAT3-mediated activation of microRNA cluster 17∼92 promotes proliferation and survival of ALK-positive anaplastic large cell lymphoma. Haematologica 2014; 99:116-24; PMID:23975180; http://dx.doi.org/ 10.3324/haematol.2013.088286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Rokavec M, Oner MG, Li H, Jackstadt R, Jiang L, Lodygin D, Kaller M, Horst D, Ziegler PK, Schwitalla S, et al. . IL-6R/STAT3/miR-34a feedback loop promotes EMT-mediated colorectal cancer invasion and metastasis. J Clin Investigat 2014; 124:1853-67; PMID:24642471; http://dx.doi.org/ 10.1172/JCI73531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Li L, Yuan L, Luo J, Gao J, Guo J, Xie X. MiR-34a inhibits proliferation and migration of breast cancer through down-regulation of Bcl-2 and SIRT1. Clin Exp Med 2013; 13:109-17; PMID:22623155; http://dx.doi.org/ 10.1007/s10238-012-0186-5 [DOI] [PubMed] [Google Scholar]
- 58. Cai Y, Chen H, Jin L, You Y, Shen J. STAT3-dependent transactivation of miRNA genes following Toxoplasma gondii infection in macrophage. Parasit Vectors 2013; 6:356; PMID:24341525; http://dx.doi.org/ 10.1186/1756-3305-6-356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Nguyen HT, Dalmasso G, Muller S, Carriere J, Seibold F, Darfeuille-Michaud A. Crohn's disease-associated adherent invasive Escherichia coli modulate levels of microRNAs in intestinal epithelial cells to reduce autophagy. Gastroenterol 2014; 146:508-19; PMID:24148619; http://dx.doi.org/ 10.1053/j.gastro.2013.10.021 [DOI] [PubMed] [Google Scholar]
- 60. Xu S, Xu Z, Liu B, Sun Q, Yang L, Wang J, Wang Y, Liu H. LIFRalpha-CT3 induces differentiation of a human acute myelogenous leukemia cell line HL-60 by suppressing miR-155 expression through the JAK/STAT Pathway. Leuk Res 2014; 38:1237-44; PMID:25092123; http://dx.doi.org/ 10.1016/j.leukres.2014.07.004 [DOI] [PubMed] [Google Scholar]
- 61. Wan G, Xie W, Liu Z, Xu W, Lao Y, Huang N, Cui K, Liao M, He J, Jiang Y, et al. . Hypoxia-induced MIR155 is a potent autophagy inducer by targeting multiple players in the MTOR pathway. Autophagy 2014; 10:70-9; PMID:24262949; http://dx.doi.org/ 10.4161/auto.26534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Zhou X, Ren Y, Liu A, Han L, Zhang K, Li S, Li P, Li P, Kang C, Wang X, et al. . STAT3 inhibitor WP1066 attenuates miRNA-21 to suppress human oral squamous cell carcinoma growth in vitro and in vivo. Oncol Rep 2014; 31:2173-80; PMID:24676554 [DOI] [PubMed] [Google Scholar]
- 63. Bao W, Wang HH, Tian FJ, He XY, Qiu MT, Wang JY, Zhang HJ, Wang LH, Wan XP. A TrkB-STAT3-miR-204-5p regulatory circuitry controls proliferation and invasion of endometrial carcinoma cells. Mol Cancer 2013; 12:155; PMID:24321270; http://dx.doi.org/ 10.1186/1476-4598-12-155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Mikhaylova O, Stratton Y, Hall D, Kellner E, Ehmer B, Drew AF, Gallo CA, Plas DR, Biesiada J, Meller J, et al. . VHL-regulated MiR-204 suppresses tumor growth through inhibition of LC3B-mediated autophagy in renal clear cell carcinoma. Cancer cell 2012; 21:532-46; PMID:22516261; http://dx.doi.org/ 10.1016/j.ccr.2012.02.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Kouroku Y, Fujita E, Tanida I, Ueno T, Isoai A, Kumagai H, Ogawa S, Kaufman RJ, Kominami E, Momoi T. ER stress (PERK/eIF2alpha phosphorylation) mediates the polyglutamine-induced LC3 conversion, an essential step for autophagy formation. Cell Death Differ 2007; 14:230-9; PMID:16794605; http://dx.doi.org/ 10.1038/sj.cdd.4401984 [DOI] [PubMed] [Google Scholar]
- 66. Milani M, Rzymski T, Mellor HR, Pike L, Bottini A, Generali D, Harris AL. The role of ATF4 stabilization and autophagy in resistance of breast cancer cells treated with Bortezomib. Cancer Res 2009; 69:4415-23; PMID:19417138; http://dx.doi.org/ 10.1158/0008-5472.CAN-08-2839 [DOI] [PubMed] [Google Scholar]
- 67. Mammucari C, Milan G, Romanello V, Masiero E, Rudolf R, Del Piccolo P, Burden SJ, Di Lisi R, Sandri C, Zhao J, et al. . FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metabol 2007; 6:458-71; PMID:18054315; http://dx.doi.org/ 10.1016/j.cmet.2007.11.001 [DOI] [PubMed] [Google Scholar]
- 68. Oh HM, Yu CR, Dambuza I, Marrero B, Egwuagu CE. STAT3 protein interacts with Class O Forkhead transcription factors in the cytoplasm and regulates nuclear/cytoplasmic localization of FoxO1 and FoxO3a proteins in CD4(+) T cells. J Biol Chem 2012; 287:30436-43; PMID:22761423; http://dx.doi.org/ 10.1074/jbc.M112.359661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Szczepanek K, Chen Q, Derecka M, Salloum FN, Zhang Q, Szelag M, Cichy J, Kukreja RC, Dulak J, Lesnefsky EJ, et al. . Mitochondrial-targeted Signal transducer and activator of transcription 3 (STAT3) protects against ischemia-induced changes in the electron transport chain and the generation of reactive oxygen species. J Biol Chem 2011; 286:29610-20; PMID:21715323; http://dx.doi.org/ 10.1074/jbc.M111.226209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Szczepanek K, Chen Q, Larner AC, Lesnefsky EJ. Cytoprotection by the modulation of mitochondrial electron transport chain: the emerging role of mitochondrial STAT3. Mitochondrion 2012; 12:180-9; PMID:21930250; http://dx.doi.org/ 10.1016/j.mito.2011.08.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Scherz-Shouval R, Elazar Z. ROS, mitochondria and the regulation of autophagy. Trend Cell Biol 2007; 17:422-7; PMID:17804237; http://dx.doi.org/ 10.1016/j.tcb.2007.07.009 [DOI] [PubMed] [Google Scholar]
- 72. Wei Y, Zou Z, Becker N, Anderson M, Sumpter R, Xiao G, Kinch L, Koduru P, Christudass CS, Veltri RW, et al. . EGFR-Mediated Beclin 1 Phosphorylation in Autophagy Suppression, Tumor Progression, and Tumor Chemoresistance. Cell 2013; 154:1269-84; PMID:24034250; http://dx.doi.org/ 10.1016/j.cell.2013.08.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Rubinsztein DC, Codogno P, Levine B. Autophagy modulation as a potential therapeutic target for diverse diseases. Nat Rev Drug Discov 2012; 11:709-30; PMID:22935804; http://dx.doi.org/ 10.1038/nrd3802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Siegelin MD, Raskett CM, Gilbert CA, Ross AH, Altieri DC. Sorafenib exerts anti-glioma activity in vitro and in vivo. Neurosci Lett 2010; 478:165-70; PMID:20470863; http://dx.doi.org/ 10.1016/j.neulet.2010.05.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Lian JQ, Ni ZH, Dai XF, Su C, Smith AR, Xu L, He FT. Sorafenib Sensitizes (-)-Gossypol-Induced Growth Suppression in Androgen-Independent Prostate Cancer Cells via Mcl-1 Inhibition and Bak Activation. Mol Cancer Therapeut 2012; 11:416-26; PMID:22188816; http://dx.doi.org/ 10.1158/1535-7163.MCT-11-0559 [DOI] [PubMed] [Google Scholar]
- 76. Hamedani FS, Cinar M, Mo Z, Cervania MA, Amin HM, Alkan S. Crizotinib (PF-2341066) induces apoptosis due to downregulation of pSTAT3 and BCL-2 family proteins in NPM-ALK(+) anaplastic large cell lymphoma. Leuk Res 2014; 38:503-8; PMID:24486291; http://dx.doi.org/ 10.1016/j.leukres.2013.12.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Ouyang DY, Zhang YT, Xu LH, Li JJ, Zha QB, He XH. Histone deacetylase inhibitor valproic acid sensitizes B16F10 melanoma cells to cucurbitacin B treatment. Acta Biochim Biophys Sin 2011; 43:487-95; PMID:21628505; http://dx.doi.org/ 10.1093/abbs/gmr032 [DOI] [PubMed] [Google Scholar]
- 78. Gong J, Munoz AR, Chan D, Ghosh R, Kumar AP. STAT3 down regulates LC3 to inhibit autophagy and pancreatic cancer cell growth. Oncotarget 2014; 5:2529-41; PMID:24796733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Su JC, Tseng PH, Hsu CY, Tai WT, Huang JW, Ko CH, Lin MW, Liu CY, Chen KF, Shiau CW. RFX1-dependent activation of SHP-1 induces autophagy by a novel obatoclax derivative in hepatocellular carcinoma cells. Oncotarget 2014; 5:4909-19; PMID:24952874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Su JC, Liu CY, Tai WT, Chen KF, Shiau CW. Obatoclax analogue SC-2001 induced autophagy through SHP1/STAT3 pathway in hepatocelluar carcinoma. Cancer Res 2013; 73:3447; http://dx.doi.org/ 10.1158/1538-7445.AM2013-3447 [DOI] [Google Scholar]
- 81. Khanim FL, Merrick BA, Giles HV, Jankute M, Jackson JB, Giles LJ, Birtwistle J, Bunce CM, Drayson MT. Redeployment-based drug screening identifies the anti-helminthic niclosamide as anti-myeloma therapy that also reduces free light chain production. Blood Cancer J 2011; 1:e39; PMID:22829072; http://dx.doi.org/ 10.1038/bcj.2011.38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Carmignani M, Volpe AR, Aldea M, Soritau O, Irimie A, Florian IS, Tomuleasa C, Baritchii A, Petrushev B, Crisan G, et al. . Glioblastoma stem cells: a new target for metformin and arsenic trioxide. J Biol Regulat Homeost Agents 2014; 28:1-15; PMID:24750786 [PubMed] [Google Scholar]
- 83. Pratt J, Annabi B. Induction of autophagy biomarker BNIP3 requires a JAK2/STAT3 and MT1-MMP signaling interplay in Concanavalin-A-activated U87 glioblastoma cells. Cellular Signal 2014; 26:917-24; PMID:24462646; http://dx.doi.org/ 10.1016/j.cellsig.2014.01.012 [DOI] [PubMed] [Google Scholar]
- 84. Kaddis N, Saif MW. Second-line treatment for pancreatic cancer. JOP 2014; 15:344-7; PMID:25076339 [DOI] [PubMed] [Google Scholar]
- 85. Quintas-Cardama A, Verstovsek S. Molecular pathways: Jak/STAT pathway: mutations, inhibitors, and resistance. Clin Cancer Res 2013; 19:1933-40; PMID:23406773; http://dx.doi.org/ 10.1158/1078-0432.CCR-12-0284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Yuan X, Du J, Hua S, Zhang H, Gu C, Wang J, Yang L, Huang J, Yu J, Liu F. Suppression of autophagy augments the radiosensitizing effects of STAT3 inhibition on human glioma cells. Exp Cell Rese 2015; 330:267-76; PMID:25220423; http://dx.doi.org/24991838 10.1016/j.yexcr.2014.09.006 [DOI] [PubMed] [Google Scholar]
- 87. Rangwala R, Chang YC, Hu J, Algazy KM, Evans TL, Fecher LA, Schuchter LM, Torigian DA, Panosian JT, Troxel AB, et al. . Combined MTOR and autophagy inhibition: Phase I trial of hydroxychloroquine and temsirolimus in patients with advanced solid tumors and melanoma. Autophagy 2014; 10:1391-402; PMID:24991838; http://dx.doi.org/ 10.4161/auto.29119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Vogl DT, Stadtmauer EA, Tan KS, Heitjan DF, Davis LE, Pontiggia L, Rangwala R, Piao S, Chang YC, Scott EC, et al. . Combined autophagy and proteasome inhibition: A phase 1 trial of hydroxychloroquine and bortezomib in patients with relapsed/refractory myeloma. Autophagy 2014; 10:1380-90; PMID:24991834; http://dx.doi.org/ 10.4161/auto.29264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Ni HM, Bockus A, Boggess N, Jaeschke H, Ding WX. Activation of autophagy protects against acetaminophen-induced hepatotoxicity. Hepatology 2012; 55:222-32; PMID:21932416; http://dx.doi.org/ 10.1002/hep.24690 [DOI] [PMC free article] [PubMed] [Google Scholar]