

Abstract
Background
Renin-angiotensin system activation is a feature of many cardiovascular conditions. Activity of myocardial reduced nicotinamide adenine dinucleotide phosphate oxidase 2 (NADPH oxidase 2 or Nox2) is enhanced by angiotensin II (Ang II) and contributes to increased hypertrophy, fibrosis, and adverse remodeling. Recent studies found that Nox2-mediated reactive oxygen species production modulates physiological cardiomyocyte function.
Objectives
This study sought to investigate the effects of cardiomyocyte Nox2 on contractile function during increased Ang II activation.
Methods
We generated a cardiomyocyte-targeted Nox2-transgenic mouse model and studied the effects of in vivo and ex vivo Ang II stimulation, as well as chronic aortic banding.
Results
Chronic subpressor Ang II infusion induced greater cardiac hypertrophy in transgenic than wild-type mice but unexpectedly enhanced contractile function. Acute Ang II treatment also enhanced contractile function in transgenic hearts in vivo and transgenic cardiomyocytes ex vivo. Ang II–stimulated Nox2 activity increased sarcoplasmic reticulum (SR) Ca2+ uptake in transgenic mice, increased the Ca2+ transient and contractile amplitude, and accelerated cardiomyocyte contraction and relaxation. Elevated Nox2 activity increased phospholamban phosphorylation in both hearts and cardiomyocytes, related to inhibition of protein phosphatase 1 activity. In a model of aortic banding–induced chronic pressure overload, heart function was similarly depressed in transgenic and wild-type mice.
Conclusions
We identified a novel mechanism in which Nox2 modulates cardiomyocyte SR Ca2+ uptake and contractile function through redox-regulated changes in phospholamban phosphorylation. This mechanism can drive increased contractility in the short term in disease states characterized by enhanced renin-angiotensin system activation.
Key Words: angiotensin II, contraction, myocyte, NADPH oxidase
Abbreviations and Acronyms: Ang II, angiotensin II; [Ca2+]i, intracellular calcium ions; HF, heart failure; LV, left ventricular; NADPH, reduced nicotinamide adenine dinucleotide phosphate; NCX, sodium-calcium exchange; Nox2, NADPH oxidase 2; PKA, protein kinase A; PP1, protein phosphatase 1; RAS, renin-angiotensin system; ROS, reactive oxygen species; RyR2, ryanodine receptor; SERCA, sarcoplasmic reticulum Ca2+-ATPase; SR, sarcoplasmic reticulum
Physiological cardiac function requires precise regulation of intracellular calcium ions ([Ca2+]i) and excitation-contraction coupling. Aberrant cardiomyocyte Ca2+ handling contributes to contractile dysfunction, arrhythmia, hypertrophy, and cell death during heart failure (HF) (1). Reactive oxygen species (ROS) modulate cardiomyocyte [Ca2+]i fluxes through oxidation-reduction regulation of proteins involved in excitation-contraction coupling, both physiologically and in HF when ROS production is often increased (2). However, the sources of ROS and mechanisms of cardiomyocyte [Ca2+]i regulation remain elusive.
Among the sources of cellular ROS, reduced nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (Nox) proteins generate ROS as their primary function and are especially important in redox signaling. The tightly controlled and spatially confined production of low ROS levels by Nox enzymes is critical for their signaling function (3). The main cardiomyocyte Nox isoforms are Nox2 (also known as gp91phox) and Nox4; both occur as transmembrane heterodimers with a p22phox subunit. Nox4 is regulated mainly by its expression level and is involved in chronic cardiac stress responses (4). By contrast, Nox2 is acutely activated by agonists such as angiotensin II (Ang II) or mechanical forces in a process that involves association with p47phox, p67phox, p40phox, and Rac1 cytosolic subunits. Cardiac Nox2 protein levels and activity are elevated in experimental and human HF 5, 6. Previous studies using global Nox2-null mice, p47phox-null mice, and other models indicated that Nox2 is involved in the development of cardiac hypertrophy, fibrosis, myocyte apoptosis, and ventricular remodeling induced by Ang II, hemodynamic overload, or myocardial infarction 7, 8, 9, 10.
Recently, we reported a novel role of Nox2 in regulating physiological mechanosensitive cardiomyocyte function, dependent on the frequency and magnitude of cardiomyocyte stretch 11, 12. Physiological stretch acutely activated Nox2 in the sarcolemma and T-tubules, and the ROS that were generated enhanced sarcoplasmic reticulum (SR) ryanodine receptor (RyR2) Ca2+ release and excitation-contraction coupling. Although this mechanism may be beneficial physiologically, how elevated levels of Nox2 affect myocyte function in disease settings is poorly understood. Furthermore, discerning the influences of cardiomyocyte Nox2 versus Nox2 in other cell types (e.g., vascular, inflammatory) has hitherto been difficult.
Here, we generated a cardiomyocyte-targeted Nox2 transgenic mouse model to examine the effects of elevated Nox2 levels on myocyte function. Although Nox2 activation by chronic subpressor Ang II treatment induced greater hypertrophy in the transgenic heart, unexpectedly, in vivo cardiac function was enhanced with Ang II treatment. Single-cell studies demonstrated that increases in Nox2-mediated ROS production following Ang II stimulation promoted SR Ca2+-ATPase (SERCA) uptake of Ca2+, driving increased Ca2+ release and greater and more rapid cardiomyocyte contraction and relaxation. Investigation of the mechanism revealed a Nox2-dependent increase in phospholamban phosphorylation, which could be explained by an inhibition of protein phosphatase 1 (PP1) activity. These data identified a novel mechanism in which increased Nox2 activity can drive increased contractility during short-term Ang II stimulation.
Methods
Animal procedures were performed in accordance with institutional and national standards. Transgenic mice expressed human Nox2 complementary DNA under control of the mouse myosin light chain 2 promoter. Transgenic mice were compared with wild-type (WT) littermates. Left ventricular (LV) pressure–volume analyses were performed as previously described (4).
LV cardiomyocyte sarcomere length, [Ca2+]i transient, and voltage-clamp studies were performed as described 11, 12. SR load was quantified from measurements of the sodium-calcium exchange (NCX) current. SERCA function was estimated from the difference in decay rates of the systolic Ca2+ and caffeine-induced transients.
Myocardial Nox activity was assessed by lucigenin-enhanced chemiluminescence (9). Protein phosphatase activity was assayed using a malachite green assay, and PP1 activity was estimated by calyculin A inhibition (13).
Data are mean ± SE of the mean of at least 3 experiments. Comparisons were made by unpaired Student t test and 1-way or 2-way analysis of variance as appropriate, followed by Tukey post-hoc analysis. A p value <0.05 was considered significant.
For a more detailed methods summary, see the Online Appendix.
Results
Basal phenotype in Nox2 transgenic mice
NADPH oxidase activity is enhanced up to 2-fold in both human failing myocardium and experimental models of cardiac hypertrophy, with an increase in the levels of Nox2 predominantly in cardiomyocytes 5, 6, 10. To investigate the cardiomyocyte-specific effects of elevated Nox2, we generated mice with cardiomyocyte-targeted overexpression of Nox2. Transgenic mice were born in the expected Mendelian ratio and bred normally. These transgenic animals had approximately 5-fold higher Nox2 protein levels in the heart (Figure 1A), whereas levels of the Nox2 subunits p22phox, p40phox, p47phox, p67phox, and Rac1, as well as Nox4, were similar in transgenic and WT myocardium (Figure 1A, Online Figure 1). Nox2 colocalized with the T-tubule/Z-line marker titin Z and exhibited additional diffuse reticular staining in both transgenic and WT cells, but with significantly brighter fluorescence in transgenic cells (Figure 1B). Basal myocardial Nox activity was unaltered in transgenic mice but was approximately 50% higher after Ang II stimulation than in WT mice (Figure 1A), a similar range as that found in failing human myocardium 5, 6. The protein levels of endothelial nitric oxide synthase and phosphorylated (activated) endothelial nitric oxide synthase, which can modulate ROS effects, were similar in WT and transgenic hearts (Figure 1A, Online Figure 1). Levels of neuronal NOS and inducible NOS were also similar.
Figure 1.

Cardiomyocyte-Specific Overexpression of Nox2
(A) Protein expression in hearts of reduced nicotinamide adenine dinucleotide phosphate oxidase 2 (Nox2) transgenic (TG) mice and wild-type (WT) littermates is seen in representative immunoblots (left) and mean data (**p < 0.01; n = 3). Nox activity increased in TG versus WT hearts after angiotensin II (Ang II) stimulation (**p < 0.01; n = 4). (B) Confocal microscopy and immunostaining for Nox2 and the T-tubule/Z-line marker Titin Z in WT and TG cardiomyocytes. Lowermost panels show high-magnification images of myocytes. Scale bars: 10 μm and 4 μm, respectively. Fluorescence plot profile from region enclosed by dotted lines shows overlay of Titin Z and Nox2 immunostaining. Ctrl = control; nNOS = neuronal nitric oxide synthase; p-eNOS = phosphorylated endothelial nitric oxide synthase; phox = phagocyte oxidase; Rac1 = Ras-related C3 botulinum toxin substrate 1; t-eNOS = total endothelial nitric oxide synthase.
Basal cardiac structure and function assessed by echocardiography were unchanged in young adult and older (12-month-old) transgenic mice (Online Tables 1 and 2), indicating that a modest level of Nox2 overexpression was well tolerated by the heart in the absence of disease stimuli.
Effect of in vivo Ang II treatment
We first investigated the in vivo cardiac response to chronic subpressor Ang II stimulation (0.3 mg/kg/day for 2 weeks) in WT and transgenic mice. Neither group experienced a change in blood pressure (Online Figure 2). Transgenic mice showed increased interventricular diastolic septal thickness, higher heart/body weight ratio, and larger cardiomyocyte size than WT mice after 2 weeks of Ang II (Figures 2A and 2B, Online Figures 3A and 3D), indicating enhanced cardiac hypertrophy. However, there was no difference in interstitial myocardial fibrosis or myocyte apoptosis in transgenic versus WT hearts (Online Figure 4). Unexpectedly, transgenic mice had a significantly higher LV stroke volume and ejection fraction than WT mice after 2 weeks of Ang II, without changes in heart rate (Figure 2C, Online Figures 3B, 3C, and 3E), suggesting enhanced contractile function. To assess potential Ang II–induced increases in contractile function, independent of concomitant LV hypertrophy, we studied the acute in vivo effects of Ang II (1.5 mg/kg, intraperitoneal). Measurement of a relatively load-independent index of contractile performance, LV maximum dP/dt (dP/dtmax)/end-diastolic volume (14), confirmed that transgenic mice treated with Ang II developed a significant increase in contractility compared with WT mice (Figures 2D and 2F). This was independent of transient Ang II–induced changes in afterload as assessed by arterial elastance (Online Figure 5). Transgenic mice also showed a significant increase in LV dP/dtmin after acute Ang II treatment (Figures 2E and 2F), suggesting enhanced LV relaxation.
Figure 2.

Cardiac Hypertrophy and Function After Ang II Stimulation
Mean data for cardiac hypertrophy after 2 weeks of Ang II infusion in terms of heart/body weight (HW/BW) ratio (A) and histological myocyte cross-sectional area (CSA) (B). (C) Echocardiographic ejection fraction (EF) after 2 weeks of Ang II treatment. (D and E) Acute response to Ang II assessed by in vivo pressure-volume analyses. (F) Representative left ventricular (LV) dP/dt traces. *p < 0.05. **p < 0.01 versus respective basal. #p < 0.05. ##p < 0.01 versus WT/Ang II. dP/dt = rate of change of pressure; EDV = end-diastolic volume; other abbreviations as in Figure 1.
Changes in myocyte Ca2+ signaling and contractility
We next evaluated the [Ca2+]i transient and cell shortening in isolated LV cardiomyocytes from WT and transgenic hearts in the presence and absence of Ang II (1 μmol/l). Under basal conditions, there was no difference in contractility between groups (Figures 3A and 3B, Online Table 3). In the presence of Ang II, transgenic myocytes showed significant increases in contractility and the speed of contraction and relaxation (Figures 3A to 3D). Ang II exerted no significant effect on WT myocytes.
Figure 3.

Increased Cell Contractility and [Ca2+]i Transient in TG Cardiomyocytes
Examples of changes in sarcomere length (SL) (A) and intracellular calcium ion [Ca2+]i transient (E) in field-stimulated (1 Hz) WT and TG myocytes with or without Ang II stimulation (1 μmol/l for 20 min). Average values for SL changes (B) and contraction (C) or relaxation (D) time 10% to 90%. Mean data of [Ca2+]i transient amplitude (F) and rise (G) or decay (H) time 10% to 90%. *p < 0.05. **p < 0.01 versus respective basal. #p < 0.05. ##p < 0.01 versus WT/Ang II. ΔF/F0 = change in fluorescence intensity; other abbreviations as in Figure 1.
To investigate whether changes in [Ca2+]i handling contributed to the enhanced contractility of Ang II–stimulated transgenic myocytes, we monitored action potential–evoked steady state [Ca2+]i transients. Transgenic and WT cells showed similar global calcium handling in the absence of Ang II. Upon Ang II stimulation, however, transgenic myocytes demonstrated increased [Ca2+]i transient amplitude (Figures 3E and 3F) and faster [Ca2+]i transient decay (Figure 3H). There was no significant difference in the rise time of the [Ca2+]i transient (Figure 3G, Online Table 3).
We next tested whether these changes in cytosolic calcium handling and contractility depended on increased Nox2-derived ROS production. Either in the presence of N-acetyl cysteine, a general ROS scavenger, or following pre-incubation with gp91ds-tat, a peptide inhibitor specific for Nox2 (15), Ang II’s effect on transgenic myocyte contraction and relaxation velocities was fully blocked (Online Figure 6, Online Table 3). These results indicated that the measured effects in transgenic mice were indeed due to increased Nox2-derived ROS production.
SR Ca2+ load and SERCA function
SERCA2a controls both the rate of cytosolic Ca2+ removal and the degree of SR Ca2+ load ([Ca2+]SR), thereby representing a fundamental determinant of cardiac relaxation and contraction (16). The faster decay of the [Ca2+]i transient in transgenic myocytes could be explained by either enhanced SERCA pumping of Ca2+ into the SR or increased extrusion of Ca2+ across the sarcolemma, which occurs primarily via NCX. Enhanced SERCA function can also lead to elevated SR Ca2+ content, which in turn drives increased Ca2+ release from the SR with each heartbeat. To evaluate SERCA function and [Ca2+]SR, we performed voltage clamp studies. After a pre-pulse protocol to bring the cell to steady state, a rapid caffeine application was used to deplete SR Ca2+ stores. Under these conditions, Ca2+ released into the cytosol is removed by NCX, and thus integration of the NCX current following caffeine application was used as a precise measure of SR Ca2+ content. Ang II stimulation of transgenic myocytes significantly elevated SR calcium content (Figures 4A and 4B) but had no effect on [Ca2+]SR in WT myocytes. Assessment of SERCA function showed that Ang II significantly increased this in transgenic but not WT myocytes, consistent with an elevation in levels of [Ca2+]SR (Figure 4C).
Figure 4.

Increased SR Ca2+ Load and SERCA Function in TG Myocytes
(A) Representative traces of [Ca2+]i transient and decay by voltage clamp studies after a rapid application of caffeine to deplete sarcoplasmic reticulum (SR) Ca2+ stores. (B) Mean data for SR [Ca2+] load. (C) Average values of sarcoplasmic reticulum Ca2+-ATPase (SERCA) function by comparing the decay of the depolarization-induced Ca2+ transient with the decay of the caffeine-induced Ca2+ transient. *p < 0.05. **p < 0.01 versus TG basal. ##p < 0.01 versus WT/Ang II. Abbreviations as in Figure 1, Figure 3.
Taken together, these single-cell studies suggest that increased Nox2-derived ROS production following Ang II stimulation increases SERCA activity, causing more rapid removal of Ca2+ from the cytosol into the SR. This is evident by a faster decay of the Ca2+ transient and an increase in SR Ca2+ content, which drives increased Ca2+ release and stronger and more rapid cardiomyocyte contraction and relaxation, a similar pattern of change in contractile kinetics to that found in vivo after acute Ang II stimulation.
Changes in phospholamban phosphorylation
We next wished to identify the mechanism responsible for the Nox2-dependent increase in SERCA function and enhancement of excitation-contraction coupling. The levels of SERCA2a and total phospholamban were unaltered in transgenic compared with WT hearts at baseline and were unaffected by acute Ang II stimulation (Figure 5A). However, phospholamban phosphorylation at Ser16, the protein kinase A (PKA)–dependent site (17), was significantly elevated only in transgenic hearts after Ang II treatment (Figure 5A). There was no significant difference between groups in phospholamban phosphorylation at Thr17, the calcium calmodulin kinase II site (16) nor in RyR2 phosphorylation at 1 common PKA phosphorylation site (Ser2808) (18), either at baseline or after acute Ang II stimulation (Figure 5A, Online Figure 7). Furthermore, we found no changes in the phosphorylation of other PKA targets, such as the L-type calcium channel and troponin I (Online Figure 8A). The level of phospholamban Ser16 phosphorylation was also significantly elevated in transgenic mice treated with 2 weeks of Ang II infusion, again without changes in other PKA targets (Online Figures 8B and 9). Taken together, these results indicate that Ang II–stimulated Nox2 activity specifically increased phospholamban phosphorylation at the Ser16 PKA-dependent site without affecting other phosphorylation sites on this protein or affecting PKA targets in other subcellular compartments.
Figure 5.

Nox2-Dependent Regulation of Phospholamban Phosphorylation
(A) Western blots for SERCA2a, Ser16- and Thr17-phosphorylated (p) phospholamban (PLN), total (t) PLN, Ser2808-phosphorylated ryanodine receptor (RyR2), and total RyR2 in LV of TG and WT mice after acute Ang II stimulation or saline control (mean data on the right). (B) Effect of Nox2 overexpression, Nox2 knockdown (shNox2), control (beta-galactosidase [β-gal]), and reactive oxygen species inhibitor pegylated superoxide dismutase (peg-SOD) on protein levels of SERCA2a, total and phosphorylated PLN, and RyR2 in cardiomyocytes. (Representative blots on the left; mean data on the right.) *p < 0.05 versus TG/Ctrl or β-gal. #p < 0.05 versus WT/Ang II or Nox2 overexpression alone. §p < 0.05 versus Nox2/Ang II. n = 4. Abbreviations as in Figure 1, Figure 2, Figure 4.
To substantiate this finding, we undertook complementary experiments in cultured neonatal rat cardiomyocytes with overexpression or knockdown of Nox2 (Online Figure 10). Stimulation with Ang II (100 nmol/l for 24 h) significantly increased phospholamban phosphorylation at Ser16 in Nox2-overexpressing compared with beta-galactosidase–expressing control cells but had no effect on phospholamban phosphorylation at Thr17, RyR2 phosphorylation at Ser2808, or SERCA2a levels (Figure 5B, Online Figure 7B). The Ang II–stimulated increase in phospholamban Ser16 phosphorylation in Nox2-overexpressing cardiomyocytes was inhibited by the superoxide scavenger pegylated–superoxide dismutase (50 U/ml), indicating it was ROS mediated. No Ang II–stimulated increase in phospholamban Ser16 phosphorylation was found in cardiomyocytes in which endogenous Nox2 levels were depleted (Figure 5B). To confirm a role for endogenous Nox2 in modulating phospholamban phosphorylation in vivo, we studied the effects of chronic Ang II infusion administered at a dose (1.1 mg/kg/day for 2 weeks) that increased LV Nox2 protein levels by approximately 2-fold in WT mice (Online Figure 11). We found that phospholamban phosphorylation at Ser16 was enhanced in the Ang II–treated WT group but attenuated in Nox2-null mice (Online Figure 11).
Because we found a Nox2-dependent increase only in phospholamban phosphorylation at Ser16, without concomitant changes in other potentially redox-sensitive targets, we tested whether this effect might involve inhibition of a phosphatase that specifically regulates this site on phospholamban, analogous to redox signaling involving other protein targets 3, 19. The serine/threonine phosphatase PP1 was a likely target because it maintains a critical role in modulating phospholamban phosphorylation at Ser16 17, 20. The levels of the 3 main cardiac PP1 isoforms (i.e., PP1-alpha, PP1-beta, and PP1-gamma) were similar in WT and transgenic hearts under basal conditions or after Ang II treatment (Figure 6A, Online Figure 12). However, PP1 activity was significantly decreased after Ang II treatment in transgenic hearts compared with WT (Figure 6B).
Figure 6.

Nox2 Inhibits PP1 Activity After Ang II Stimulation
(A) Protein levels of protein phosphatase 1 (PP1) isoforms in LV of TG and WT mice. (B) PP1 activity in WT and TG LV. *p < 0.05 versus TG/Ctrl. #p < 0.05 versus WT/Ang II. Effects of protein kinase A inhibition (via H89 or Rp-cAMPS) on contraction (C) and relaxation (D) function in TG myocytes are shown. **p < 0.01 versus all other groups. Abbreviations as in Figure 1, Figure 2.
To confirm that Nox2-mediated modulation of contractile function is dependent on PKA activation, we studied the effects of 2 different PKA inhibitors, H89 and Rp-cAMPS. The enhanced Ang II–stimulated contractile state in transgenic myocytes was completely blocked by either inhibitor (Figures 6C and 6D). These inhibitors exerted no effect in WT myocytes (Online Figure 13).
Response to chronic pressure overload in transgenic mice
To evaluate increased Nox2 activity in a setting of more severe disease stress, we next subjected transgenic and WT mice to chronic abdominal aortic banding (3 weeks). Similar to Ang II treatment, this pressure overload induced greater hypertrophy in transgenic than WT mice (Online Figure 14A). However, compared with WT, transgenic mice trended toward poorer cardiac function (Online Figures 14B to 14D). Interestingly, immunoblot analyses showed that although transgenic/banded mice had higher levels of phospholamban phosphorylation at Ser16 than WT/banded mice, the levels were significantly reduced compared with respective sham controls (Online Figure 14E). SERCA2a protein levels were similar among groups (Online Figure 14E). The data therefore suggest that although Nox2-induced activation of phospholamban phosphorylation enhances short-term contractile function, in the setting of chronic overload stress, a sustained increase in cardiomyocyte Nox2 activity eventually leads to exaggerated cardiac remodeling.
Discussion
In this study, Ang II–stimulated Nox2-ROS production augmented cardiac contractile performance by increasing SERCA function and driving more rapid Ca2+ reuptake and enhanced Ca2+ release, thereby causing faster contraction and relaxation. These effects were attributable to inhibition of PP1 activity and increased phospholamban phosphorylation (Central Illustration). Although the chronic effects of Nox2 activation on cardiac hypertrophy and remodeling are well recognized, here we identified a novel mechanism through which enhanced Nox2 activation may acutely benefit cardiac contractile function.
Central Illustration.
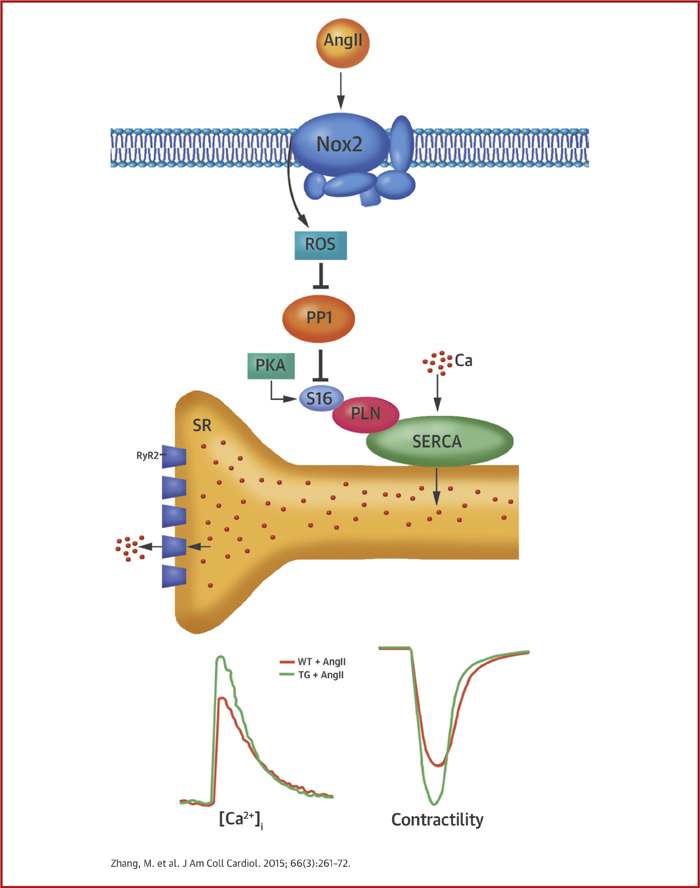
Nox2 and Myocyte Phospholamban Phosphorylation
Activation of reduced nicotinamide adenine dinucleotide phosphate oxidase 2 (Nox2) by angiotensin II (Ang II) leads to the inhibition of protein phosphatase 1 (PP1), thereby promoting an increase in protein kinase A (PKA)–dependent phospholamban (PLN) phosphorylation. This in turn promotes increased uptake of calcium ions (Ca2+) via the sarcoplasmic reticulum (SR) Ca2+ ATPase (SERCA), increased SR Ca2+ load, and increased Ca2+ release through ryanodine receptors (RyR2). The result is an increase in amplitude of Ca2+ transient and twitch contraction and faster contraction and relaxation in Nox2 transgenic (TG) compared with wild-type (WT) cardiomyocytes. Ca = calcium; [Ca2+]i = intracellular calcium concentration; ROS = reactive oxygen species; S16 = serine 16.
Myocardial ROS are generated in increased amounts during renin-angiotensin system (RAS) activation. The pathophysiological effects of ROS include those mediated by redox signaling (i.e., the specific, usually reversible, oxidation-reduction of components of cellular signaling pathways) (19). As dedicated ROS generators, Nox proteins are particularly important in redox signaling. Previous work showed that cardiomyocyte Nox2 expression and the level of myocardial Nox activity are significantly elevated in experimental models of hypertension and human chronic HF 5, 10, 21, 22, conditions associated with RAS activation. Studies using global Nox2-null mice found that Nox2-mediated redox signaling plays an important role in Ang II–mediated and post–myocardial infarction cardiac hypertrophy and adverse remodeling. Because Nox2 is expressed in multiple cell types (e.g., cardiomyocytes, endothelial cells, fibroblasts, inflammatory cells, neurons, renal cells), its cell-specific effects remain unclear. Here, we developed a new mouse model with a cardiomyocyte-targeted increase in Nox2 expression and definitively demonstrated that cardiomyocyte Nox2 is involved in the development of Ang II–induced hypertrophy in vivo. The increase in Ang II–stimulated NADPH oxidase activity in transgenic hearts was in a pathophysiologically relevant range 5, 6 and Nox2 overexpression was not accompanied by confounding changes in Nox4 levels. No significant phenotype was found in transgenic hearts in the absence of Ang II stimulation, even at age 12 months, consistent with the fact that Nox2 is quiescent unless activated by specific agonists.
In studying the effects of 2 weeks of subpressor Ang II treatment on cardiac function, we made the unanticipated finding that contractile function was enhanced in transgenic animals. To distinguish between the effects of cardiac hypertrophy and changes in contractile function, we assessed the acute in vivo response of transgenic mice to Ang II by invasive pressure-volume analysis and found that this was also significantly enhanced. This load-independent analysis suggests Ang II had intrinsic positive inotropic effects on transgenic myocardium, which was confirmed by studies in isolated cardiomyocytes. Therefore, increased cardiomyocyte Nox2 activation significantly augmented the acute contractile response to Ang II. The acute effects of Ang II on normal myocardium vary, with studies reporting positive (23), negative (24), or no inotropic effect (25), probably related to temporal, concentration-dependent, and species differences. We found minimal acute inotropic effects of Ang II in healthy, young adult mouse cardiomyocytes and intact hearts. However, when the capacity for Nox2-dependent ROS production was increased, as occurs in disease settings (10), Ang II had significant positive inotropic and lusitropic effects.
Changes in intracellular redox state may influence excitation-contraction coupling either through oxidative modifications of specific Ca2+ handling proteins (e.g., RyR2, SERCA2a) or via redox-mediated changes in activity of protein kinases that regulate Ca2+-handling proteins 2, 19. Such modifications have rarely been linked mechanistically to specific ROS sources. However, the recent finding of stretch-evoked Nox2-mediated regulation of RyR2 Ca2+ release in normal cardiomyocytes (termed “X-ROS signaling”) established a new paradigm for redox modulation of excitation-contraction coupling, by linking the ROS effect to specific stimulation of a distinct ROS source. In the current study, we found a conceptually similar but mechanistically quite different modulation in a disease setting. Here, the specific activation of Nox2 by Ang II resulted in increased SERCA activity driven by enhanced phospholamban phosphorylation and led to enhanced contraction and relaxation.
The mechanism of Nox2-mediated increase in SERCA activity was an increase in phospholamban phosphorylation at a PKA-dependent site. Interestingly, we did not find significant changes in the phosphorylation of other PKA targets, such as the L-type calcium channel and troponin I nor in the target of redox-activatable calcium-calmodulin kinase II on the phospholamban Thr17 site, suggesting a compartmentalized effect within the cardiac myocyte. An attractive mechanism for such an effect is the local redox inactivation of a phosphatase that regulates phospholamban phosphorylation, similar to mechanisms involved in redox enhancement of receptor phosphorylation 26, 27. The most likely candidate for such a mechanism was PP1, which specifically regulates phospholamban phosphorylation (20). Indeed, we found that PP1 activity was robustly inhibited in Ang II–treated transgenic mice, consistent with a mechanism in which Nox2 inactivates PP1, allowing for an increase in phospholamban phosphorylation. This mechanism was further confirmed when PKA inhibitors completely blocked Ang II–mediated enhancement of contractile function in transgenic myocytes.
The precise spatial interrelationship between Nox2, PP1, and phospholamban, or how PP1 is inhibited by Nox2, remains speculative at this stage. Previously, Nox2 was found to be located at the T-tubules (11); we observed a similar localization pattern in transgenic myocytes in the current study (i.e., a location that is relatively distant on the cellular scale of ROS signaling from phospholamban and SERCA at the network SR). It is possible that Nox2 inhibits PP1 locally at the junctional SR and this then transduces the signal to phospholamban at the network SR. Alternatively, there may be distinct subcellular pools of Nox2, with the effects on phospholamban phosphorylation potentially mediated by Nox2 at the network SR. Interestingly, immunostaining for Nox2 indicated both a T-tubule and a diffuse reticular pattern that could be consistent with the latter possibility.
A Nox2-mediated increase in contractile function during enhanced RAS activation may hold clinical and therapeutic implications. Whereas ROS-mediated effects are usually considered detrimental in disease settings, here we identified a novel mechanism that could maintain contractile function, at least in the relative short term. It is interesting to speculate whether the hypotensive effects observed upon initiating angiotensin-converting enzyme inhibitors may, at least in part, be related to loss of Nox2-mediated positive inotropic activity. To assess the impact of Nox2-dependent changes in contractile function in a more severe and chronic setting, we studied the effects of chronic abdominal aortic banding, an intervention that not only induces pressure overload but also increases RAS activation (28). Interestingly, although phospholamban phosphorylation was greater in transgenic compared with WT mice, as in the Ang II infusion model, cardiac remodeling and function were, if anything, worse in transgenic mice after aortic banding.
Study limitations
The inotropic effects of Nox2-mediated increases in SERCA function and SR Ca2+ release may be outweighed in longer-term settings of pressure overload cardiac hypertrophy, and this requires further study. Nevertheless, the current findings suggest that RAS activation may be accompanied by beneficial acute contractile effects related to increased myocardial Nox2 activation.
Conclusions
We identified a novel mechanism in which Nox2 modulates cardiomyocyte SR Ca2+ uptake and contractile function through redox-regulated changes in phospholamban phosphorylation. This mechanism can drive increased contractility, in the short term, in disease states characterized by enhanced RAS activation. A Nox2-mediated increase in contractile function during enhanced RAS activation may have clinical and therapeutic implications.
Perspectives.
COMPETENCY IN MEDICAL KNOWLEDGE: The impact of RAS activation on end-organ structure and function involves several signaling pathways, including the ROS-generating enzyme Nox2. The adverse cardiac remodeling associated with elevated levels of Ang II is partly mediated by activation of Nox2, but Nox2 activation can also enhance myocardial contractile function though effects on calcium transport.
TRANSLATIONAL OUTLOOK: Additional studies are needed to determine whether various drugs that inhibit the RAS exert different acute negative inotropic effects mediated by reduction in Nox2 activity.
Footnotes
This study was funded by the Foundation Leducq Transatlantic Network of Excellence Award and the British Heart Foundation (RG/13/11/30384, RE/13/2/30182). Dr. Hirsch is a cofounder of Kither Biotech. All other authors have reported that they have no relationships relevant to the contents of this paper to disclose. Drs. Zhang and Prosser contributed equally to this work.
Appendix
For a supplemental Methods section as well as figures and tables, please see the online version of this article.
Appendix
References
- 1.Luo M., Anderson M.E. Mechanisms of altered Ca2+ handling in heart failure. Circ Res. 2013;113:690–708. doi: 10.1161/CIRCRESAHA.113.301651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zima A.V., Blatter L.A. Redox regulation of cardiac calcium channels and transporters. Cardiovasc Res. 2006;71:310–321. doi: 10.1016/j.cardiores.2006.02.019. [DOI] [PubMed] [Google Scholar]
- 3.Brown D.I., Griendling K.K. Nox proteins in signal transduction. Free Radic Biol Med. 2009;47:1239–1253. doi: 10.1016/j.freeradbiomed.2009.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang M., Brewer A.C., Schröder K., et al. NADPH oxidase-4 mediates protection against chronic load-induced stress in mouse hearts by enhancing angiogenesis. Proc Natl Acad Sci U S A. 2010;107:18121–18126. doi: 10.1073/pnas.1009700107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Heymes C., Bendall J.K., Ratajczak P., et al. Increased myocardial NADPH oxidase activity in human heart failure. J Am Coll Cardiol. 2003;41:2164–2171. doi: 10.1016/s0735-1097(03)00471-6. [DOI] [PubMed] [Google Scholar]
- 6.Maack C., Kartes T., Kilter H., et al. Oxygen free radical release in human failing myocardium is associated with increased activity of Rac1-GTPase and represents a target for statin treatment. Circulation. 2003;108:1567–1574. doi: 10.1161/01.CIR.0000091084.46500.BB. [DOI] [PubMed] [Google Scholar]
- 7.Erickson J.R., Joiner M.L., Guan X., et al. A dynamic pathway for calcium-independent activation of CaMKII by methionine oxidation. Cell. 2008;133:462–474. doi: 10.1016/j.cell.2008.02.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Doerries C., Grote K., Hilfiker-Kleiner D., et al. Critical role of the NAD(P)H oxidase subunit p47phox for left ventricular remodeling/dysfunction and survival after myocardial infarction. Circ Res. 2007;100:894–903. doi: 10.1161/01.RES.0000261657.76299.ff. [DOI] [PubMed] [Google Scholar]
- 9.Bendall J.K., Cave A.C., Heymes C., Gall N., Shah A.M. Pivotal role of a gp91phox-containing NADPH oxidase in angiotensin II-induced cardiac hypertrophy in mice. Circulation. 2002;105:293–296. doi: 10.1161/hc0302.103712. [DOI] [PubMed] [Google Scholar]
- 10.Zhang M., Perino A., Ghigo A., Hirsch E., Shah A.M. NADPH oxidases in heart failure: poachers or gamekeepers? Antioxid Redox Signal. 2013;18:1024–1041. doi: 10.1089/ars.2012.4550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Prosser B.L., Ward C.W., Lederer W.J. X-ROS signaling: rapid mechano-chemo transduction in heart. Science. 2011;333:1440–1445. doi: 10.1126/science.1202768. [DOI] [PubMed] [Google Scholar]
- 12.Prosser B.L., Ward C.W., Lederer W.J. X-ROS signalling is enhanced and graded by cyclic cardiomyocyte stretch. Cardiovasc Res. 2013;98:307–314. doi: 10.1093/cvr/cvt066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim H.S., Song M.C., Kwak I.H., Park T.J., Lim I.K. Constitutive induction of p-Erk1/2 accompanied by reduced activities of protein phosphatases 1 and 2A and MKP3 due to reactive oxygen species during cellular senescence. J Biol Chem. 2003;278:37497–37510. doi: 10.1074/jbc.M211739200. [DOI] [PubMed] [Google Scholar]
- 14.Cingolani O.H.K., Kass D.A. Pressure-volume relation analysis of mouse ventricular function. Am J Physiol. 2011;301:H2198–H2206. doi: 10.1152/ajpheart.00781.2011. [DOI] [PubMed] [Google Scholar]
- 15.Rey F.E., Cifuentes M.E., Kiarash A., Quinn M.T., Pagano P.J. Novel competitive inhibitor of NAD(P)H oxidase assembly attenuates vascular O2− and systolic blood pressure in mice. Circ Res. 2001;89:408–414. doi: 10.1161/hh1701.096037. [DOI] [PubMed] [Google Scholar]
- 16.Mattiazzi A., Mundiña-Weilenmann C., Guoxiang C., Vittone L., Kranias E. Role of phospholamban phosphorylation on Thr17 in cardiac physiological and pathological conditions. Cardiovasc Res. 2005;68:366–375. doi: 10.1016/j.cardiores.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 17.Kranias E.G., Hajjar R.J. Modulation of cardiac contractility by the phospholamban/SERCA2a regulatome. Circ Res. 2012;110:1646–1660. doi: 10.1161/CIRCRESAHA.111.259754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wehrens X.H.T., Lehnart S.E., Reiken S., Vest J.A., Wronska A., Marks A.R. Ryanodine receptor/calcium release channel PKA phosphorylation: a critical mediator of heart failure progression. Proc Natl Acad Sci U S A. 2006;103:511–518. doi: 10.1073/pnas.0510113103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Burgoyne J.R., Mongue-Din H., Eaton P., Shah A.M. Redox signaling in cardiac physiology and pathology. Circ Res. 2012;111:1091–1106. doi: 10.1161/CIRCRESAHA.111.255216. [DOI] [PubMed] [Google Scholar]
- 20.Steenaart N.A.E., Ganim J.R., Di Salvo J., Kranias E.G. The phospholamban phosphatase associated with cardiac sarcoplasmic reticulum is a type 1 enzyme. Arch Biochem Biophys. 1992;293:17–24. doi: 10.1016/0003-9861(92)90359-5. [DOI] [PubMed] [Google Scholar]
- 21.Li J.M., Gall N.P., Grieve D.J., Chen M., Shah A.M. Activation of NADPH oxidase during progression of cardiac hypertrophy to failure. Hypertension. 2002;40:477–484. doi: 10.1161/01.hyp.0000032031.30374.32. [DOI] [PubMed] [Google Scholar]
- 22.Wang M., Zhang J., Walker S.J., Dworakowski R., Lakatta E.G., Shah A.M. Involvement of NADPH oxidase in age-associated cardiac remodeling. J Mol Cell Cardiol. 2010;48:765–772. doi: 10.1016/j.yjmcc.2010.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shen X., Cannell M.B., Ward M.L. Effect of SR load and pH regulatory mechanisms on stretch-dependent Ca2+ entry during the slow force response. J Mol Cell Cardiol. 2013;63:37–46. doi: 10.1016/j.yjmcc.2013.07.008. [DOI] [PubMed] [Google Scholar]
- 24.Palomeque J., Sapia L., Hajjar R.J., Mattiazzi A., Vila Petroff M. Angiotensin II-induced negative inotropy in rat ventricular myocytes: role of reactive oxygen species and p38 MAPK. Am J Physiol. 2006;290:H96–H106. doi: 10.1152/ajpheart.00324.2005. [DOI] [PubMed] [Google Scholar]
- 25.Lefroy D.C., Crake T., Del Monte F., et al. Angiotensin II and contraction of isolated myocytes from human, guinea pig, and infarcted rat hearts. Am J Physiol. 1996;270:H2060–H2069. doi: 10.1152/ajpheart.1996.270.6.H2060. [DOI] [PubMed] [Google Scholar]
- 26.Rhee S.G. Cell signaling. H2O2, a necessary evil for cell signaling. Science. 2006;312:1882–1883. doi: 10.1126/science.1130481. [DOI] [PubMed] [Google Scholar]
- 27.Tonks N.K. Redox redux: revisiting PTPs and the control of cell signaling. Cell. 2005;121:667–670. doi: 10.1016/j.cell.2005.05.016. [DOI] [PubMed] [Google Scholar]
- 28.Herzig T.C., Jobe S.M., Aoki H., et al. Angiotensin II type 1a receptor gene expression in the heart: AP-1 and GATA-4 participate in the response to pressure overload. Proc Natl Acad Sci U S A. 1997;94:7543–7548. doi: 10.1073/pnas.94.14.7543. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.