

Abstract
The study investigated the reversal of multidrug resistance (MDR) and the biodistribution of nanoparticles (NPs) that target leukemia cells in a nude mice model via a surface-bound transferrin (Tf). The cytotoxic cargo of daunorubicin (DNR) and tetrandrine (Tet) was protected in the NPs by an outer coat composed of polyethylene glycol (PEG)-poly-l-lysine (PLL)-poly(lactic-co-glycolic acid) (PLGA) NPs. Injection of DNR-Tet-Tf-PEG-PLL-PLGA NPs into nude mice bearing MDR leukemia cell K562/A02 xenografts was shown to inhibit tumor growth, and contemporaneous immunohistochemical analysis of tumor tissue showed the targeted NPs induced apoptosis in tumor cells. Targeted tumor cells exhibited a marked increase in Tf receptor expression, with noticeable decreases in P-glycoprotein, MDR protein, and nuclear factor κB, as assessed by quantitative real-time polymerase chain reaction and Western blot analysis. Moreover, the concentration of DNR was shown to increase in plasma, tumor tissue, and major organs. Flow cytometry analysis with a near-infrared fluorescent (NIRF) dye, NIR797, was used to study the effectiveness of Tf as a targeting group for leukemia cells, a finding that was supported by NIRF imaging in tumor-bearing nude mice. In summary, our studies show that DNR-Tet-Tf-PEG-PLL-PLGA NPs provide a specific and effective means to target cytotoxic drugs to MDR tumor cells.
Keywords: PEG-PLL-PLGA nanoparticles, transferrin, tetrandrine, multidrug resistance
Introduction
The most comprehensive treatment for leukemia, a common hematologic malignancy, is based on combination chemotherapy. Chemotherapeutic drugs, while effective in killing tumor cells, are nonetheless nonselective in their targeting of tumor cells. Troublingly, the dosage of chemotherapeutics used in conventional treatments results in significant off target side effects in normal tissues and organs that are poorly tolerated by the patient. A major challenge in chemotherapy, therefore, is to improve the targeting of drugs to the tumor cells. Transferrin (Tf) is a single-chain iron-transporting glycoprotein, its membrane receptor, Tf receptor (TfR), mediates intracellular iron uptake and is involved in the regulation of cell growth.1 TfR is expressed at high levels in human tumor cells, including leukemia, lung cancer, and breast cancer.2 Significant for its application in molecular targeting, the binding of Tf to TfR on the external surface of tumor cells is ten to100 times more effective than in normal cells.3 This feature has been exploited for drug delivery, most often by labeling the surface of the drug carrier with Tf, which is recognized by, and actively transported into, tumor cells.
Although the majority of patients receiving chemotherapy can achieve remission, for others, the therapy may be ineffective due to multidrug resistance (MDR).4 MDR is often correlated with the high expression of P-glycoprotein (P-gp) on the surface of resistant tumor cells. P-gp encoded by the MDRl gene is a member of the ATP-binding cassette (ABC) transporter superfamily.5 It is an energy-dependent transporter discharge pump that removes foreign drugs from the cell to confer drug resistance.6,7 Tetrandrine (Tet) is an isoquinoline alkaloid extracted from the root of menispermaceous plants tetrandra (Stephania tetrandra S. Moore). Tet is a natural nonselective calcium channel blocker and is also known to function as a calmodulin antagonist. Previous studies have demonstrated that Tet binds to P-gp and can thereby reverse drug resistance in P-gp-expressing tumor cells. Tet blocks the transport activity of P-gp and prevents the entrapped chemotherapy drug to be pumped out of the tumor cells.8 Nuclear factor kappaB (NF-κB) is also linked to tumor progression, invasion, metastasis, and MDR, and Tet is known to inhibit NF-κB activity in K562/A02 cells (resistant human chronic myeloid leukemia cell line) as a natural antitumor drug.7,8
Nanoparticles (NPs), defined as having a diameter between 1 and 1,000 nm, are widely used in drug delivery systems.9 Typically, one or more cytotoxic drug is encapsulated within or bound to a semisynthetic biodegradable polymer taking the form of a nanosphere or nanocapsule. The ideal nano drug delivery system (NDDS) would reach the tumor cell via a molecular-targeted delivery, where it would release its cytotoxic cargo to the affected cell or across the cell membrane. NDDS should also exhibit low to zero off target effects.10
Here, we introduce a new type of NDDS that is composed of biocompatible and biodegradable polymers, namely, polyethylene glycol (PEG), poly-l-lysine (PLL), and poly(lactic-co-glycolic acid) (PLGA). We describe the synthesis and characterization of PEG-PLL-PLGA NPs (abbreviated as NPs) and show how manipulation of specific reaction conditions can be used to generate uniform NPs harboring high levels of the anticancer drug daunorubicin (DNR) and Tet. Targeted delivery of PEG-PLL-PLGA drug-loaded vehicles to target cells and tissue is realized by covalently linking Tf to the NPs’ surface (Tf-PEG-PLL-PLGA). We will use Tet to block the transport activity of P-gp, and thereby allow DNR to enter and remain in MDR cancer cells, which will increase their sensitivity to the chemotherapeutic agent. Finally, we demonstrate the therapeutic effect of Tf-modified NPs in cell and animal models of leukemia, and highlight their favorable biodistribution, pharmacokinetic features, and therapeutic effects in mice. In summary, our studies demonstrate the potential of Tf-PEG-PLL-PLGA (abbreviated as Tf-NPs) as an NDDS for the clinical therapy of specific cancers.
Materials and methods
Main materials and apparatus
RPMI-1640 was purchased from Gibco Chemical Co. (Carlsbad, CA, USA), fetal bovine serum (FBS) from Wisent Inc. (Montreal, QC, Canada), PLGA (50:50, lactic acid:glycolic acid) with carboxylic acid ends of molecular weight (MW) 30 kDa from Evonik Industries (Frankfurt, Germany), and PEG (MW 6 kDa), PLL (MW 4 kDa), 4-(dimethylamino) pyridine, anhydrous N,N-dimethylformamide hydrous (DMF), N,N′-dicyclohexylcarbodiimide (DCC), and N,N′-carbonyldiimidazole (CDI) from the Aladdin Company (Shanghai, People’s Republic of China). DNR and Tet were purchased from Jinan Huifengda Chemical Co., Ltd. (Jinan, Shandong, People’s Republic of China). Tf was purchased from Sigma–Aldrich Co. (St Louis, MO, USA); hematoxylin–eosin dye kit from KeyGen Biotech Co., Ltd. (Nanjing, Jiangsu, People’s Republic of China); Streptavidin–peroxidase (SP) immunohistochemical detection kit from Beijing Zhongshan Biotechnology Co., Ltd. (Beijing, People’s Republic of China); TRIzol reagent from Invitrogen Co., Ltd. (Carlsbad, CA, USA); and monoclonal antibodies to caspase-3, Bax, Bcl-2, survivin, TfR, P-gp, MRP (MDR protein), and NF-κB from Santa Cruz Biotechnology Inc (Santa Cruz, CA, USA). NIR797-isothiocyanate (MW 880.14 Da) was purchased from Sigma–Aldrich Co. The transmission electron microscopic (TEM) images was obtained by a JEM-2100, JEOL Ltd., Tokyo, Japan. Flow cytometry analysis was done by BD FACSCalibur (BD Biosciences, San Jose, CA, USA). Cells were imaged using a confocal microscope (Carl Zeiss LSM 710, Zeiss, Oberkochen, Germany), and labeled cells in tumors were imaged in nude mice within a Caliper IVIS (in vivo imaging system, PerkinElmer, Waltham, MA, USA). All used reagents were of analytical grade.
Preparation of DNR-Tet-Tf-PEG-PLL-PLGA NPs
DNR-Tet-Tf-PEG-PLL-PLGA (abbreviated as DNR-Tet-Tf-NPs) was prepared in the School of Pharmacy (Nanjing University of Technology, Jiangsu, People’s Republic of China). The PEG-PLL-PLGA NPs were synthesized as previously reported using a double-emulsion method.11–14 According to the optimization condition in our previous study,13,14 ethylenediamine as the macroinitiator protected by nitrogen was added to induce the N-carboxy-(Nε-benzyloxy-carbonyl)-l-lysine anhydride ring opening polymerization in anhydrous DMF (Lys(Z)-NCA) to prepare PLL-carbobenzoxy (CBZ).15 PLGA and PLL–CBZ were dissolved in anhydrous DMF under a nitrogen atmosphere, and the dried products PLGA-PLL-CBZ were dissolved in a sufficient amount of 33% hydrogen bromide, and stirred for 2 hours; the off-white polymer was dried under vacuum for 12 hours to produce the deprotected PLGA-PLL.16 PEG was dissolved in dioxane, to which CDI was added at room temperature, to synthesize the activated PEG. The solution remaining in the dialysis tube was freeze-dried for 3 days to produce activated PEG-CDI.17 Then using an excess of activated CDI-PEG-CDI with PLGA-PLL side primary amino chain reaction, the process of polymer dissolution and precipitation was repeated three times until the unconjugated PEG was removed, eventually forming PEG-PLL-PLGA.18 The drug (DNR, Tet, and NIR797 dye) was directly added into the inner aqueous solution to prepare the drug-loaded PEG-PLL-PLGA. PEG-PLL-PLGA was resuspended in phosphate-buffered saline (PBS) using ultrasound treatment for 30 seconds. Tf was added (Tf/PEG-PLL-PLGA molar ratio 8:1) and stirred at room temperature for 3 hours to conjugate with the surface of DNR-Tet-Tf-NPs via CDI of PEG. Thereafter, DNR-Tet-Tf-NPs were extracted and the disperse system was centrifuged for 30 minutes (4°C, 1,500 rpm). The morphological characteristics of NPs were observed under TEM. The average diameter, polydispersity index, and zeta potential were measured by laser particle size analyzer and zeta potential analyzer. Meanwhile, drug loading efficiency and encapsulation efficiency were calculated by the following equations:19
Cell line and culture
K562/A02 cells (resistant human chronic myeloid leukemia cell line), was established by stepwise selection with an increasing concentration of Adriamycin, provided by the Institute of Hematology, Chinese Academy of Medical Sciences. The K562/A02 cell proliferation was maintained at a final concentration of adriamycin 1 mg/mL. K562/A02 cells were grown in 10% fetal calf serum in RPMI-1640 medium, placed in 37°C, 5% CO2 humidified incubator, and passaged every 2 or 3 days.8
MDR leukemia xenograft model in nude mice
BALB/c-nu mice were purchased from Shanghai National Center for Laboratory Animals (Shanghai, People’s Republic of China), aged 5 weeks, weight 18–22 g, half male and half female. They were maintained in specific pathogen-free conditions. All animal experiments were evaluated and approved by the Animal and Ethics Review Committee of Southeast University (Nanjing, Jiangsu, People’s Republic of China). A total of 36 nude mice were injected subcutaneously in the right forelimb with 1×107 K562/A02 cells/0.2 mL per nude mouse, and the injected site developed grain-sized tumors. When the tumor size reached 75–150 mm3, the mice were randomly divided into six groups: Group a, control group, mice were treated with 0.9% normal saline 0.2 mL; Group b, DNR; Group c, DNR and Tet; Group d, DNR-NPs; Group e, DNR-Tet-NPs; Group f, DNR-Tet-Tf-NPs. These nude mice were injected intravenously with the respective treatments on every other day (day 2, 4, 6, 8, 10, 12, 14 for seven times). Based on the drug loading efficiency, the DNR concentration was 1 mg/kg in each group.20
Tumor growth measurement and inhibition rate
The size of the tumor length (a) and width (b) were measured using a digital caliper. Tumor volume (V/mm3) was calculated using the formula V =1/2 (a × b2). The change in tumor size among different groups was recorded as the relative tumor volume (RTV) using the formula RTV = VX/V1, where VX and V1 represent the volumes on day X and the first day of tumor treatment. The antitumor effect of tumor inhibition rate is defined as the inhibitory rate, which is calculated using the formula inhibitory rate (%) = (1–average experimental group RTV/average control group RTV) ×100%.20
Histopathological examination
After a specific treatment, the mice were sacrificed by cervical dislocation. Main organs, including heart, liver, spleen, lung, kidney, and tumor tissues isolated from each group were fixed in 4% paraformaldehyde solution, embedded in paraffin, and stained with hematoxylin–eosin, and the structure and internal organization were observed by microscopy.20
Immunohistochemistry analysis
Tumor tissue was cut into 5 μm slices, and the expression of Bax, Bcl-2, caspase-3, and survivin proteins were detected by an immunohistochemical staining SP method. Positive cells were counted within five randomly selected horizons for each slice at a magnification of 400×. Positive cells were scored as follows: <10% as negative, “-”; 3%–10% as low expression, “+”; 30%–50% as moderate expression, “2+”; positive cells >50% as strong expression, “3+”; the latter three categories represent positive expression of marker proteins.21
Quantitative real-time PCR
Total RNA was extracted from fresh tumor tissue using TRIzol reagent (Invitrogen). The following gene sequences were designed and synthesized: glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (103 bp) sense primer: 5′-ACAACTTTGGTATCGTGGAAGG-3′, antisense primer: 5′-GCCATCACGCCACAGTTTC-3′; P-gp (99 bp) sense primer: 5′-GGTCAGTGTTG ATGGACAGG-3′, antisense primer: 5′-GTGGTGGCAAACA ATACAGG-3′; NF-κB (87 bp) sense primer: 5′-TCAAGAT CTGCCGAGTGAAC-3′, antisense primer: 5′-CCTCTTTCTGCACCTTGTCA-3′; MRP1 (94 bp) sense primer: 5′-GACCATGAATGTGCAGAAGG-3′, antisense primer: 5′-GGATCTTCGTCTTCCTCAGC-3′; TfR (90 bp) sense primer: 5′-GAAACGCTGTTCAGAAACCA-3′, antisense primer: 5′-GTCAATGTCCCAAACGTCAC-3′. Quantitative real-time polymerase chain reaction (PCR) was performed by monitoring the increase of fluorescence of SYBR Green (Toyobo, Osaka, Japan), with the ABI prism 7300 sequence detection system (Applied Biosystems, Grand Island, NY, USA). The relative mRNA expression levels were calculated by the comparative Ct (ΔΔCt) method, normalized with the average expression of GAPDH.22
Western blot analysis
The total proteins were extracted from each group; the protein concentration was measured by the Bradford method. Proteins subjected to 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) were transferred from the protein gel to polyvinylidene difluoride membrane. Skimmed milk (5%) was used as a blocking agent (1 hour incubation) for the membrane and was followed by an incubation of TfR, P-gp, MRP, and NF-κB monoclonal antibodies overnight at 4°C. After a washing cycle, the membrane was incubated with peroxidase-labeled secondary antibody for 2 hours at room temperature. The protein band was detected using an enhanced chemiluminescence detection system (Amersham, Buckinghamshire, UK).22
Determination of DNR concentration by high-performance liquid chromatography
The tissues of heart, liver, kidney, lung, spleen, tumor, and plasma from nude mice were mixed with a defined volume of 100 mg/L standard solution of DNR, to achieve final concentrations of 0.5, 1, 5, 10, and 20 mg/L. The concentration of DNR in each tissue was calculated using high-performance liquid chromatography (HPLC) for subsequent determination of its distribution in tissues from the different groups.23
NIR797-labeled NPs in vitro and in vivo
On the basis of toxicity results from our previous studies,13,14 we plated 4×105/mL cells in six-well plates and incubated with 1.0 μg/mL equivalents of DNR in NIR797-labeled NPs for 12 and 36 hours. After washing the plated cells twice with cold PBS (1,000 rpm, centrifuge for 5 minutes), 500 μL of the binding buffer was added, followed by 5 μL annexin V-FITC at room temperature, and protected from light for 15 minutes. The autofluorescence of DNR and the annexin V-FITC measure of apoptosis were measured in the fluorescein emission channel. NIR797-labeled NPs were detected in the fluorescence-3 (FL3) channel of FACS (Calibur, BD Biosciences).
The distribution of Tf-modified NPs was imaged by the confocal microscope as follows. The cells were cultured at a density of 2×105/mL in six-well plates. DNR-NPs and DNR-Tf-NPs were added to each well at 1 μg/mL, and after overnight incubation, the cells were washed twice with cold PBS. Cells were imaged to record the DNR intrinsic fluorescence (excitation wavelength of 488 nm, emission wavelength of 515 nm).
The cells were divided into three groups: control group, no drug; group a, NIR797-labeled DNR-NPs (DNR 1 μg/mL); group b, NIR797-labeled DNR-Tf-NPs (DNR 1 μg/mL). Tumor-bearing mice randomly divided into the Tf-targeted group and non-Tf-targeted group were injected via the tail vein with NIR797-labeled DNR-NPs (DNR 2 mg/kg) and NIR797-labeled DNR-Tf-NPs (DNR 2 mg/kg). Mice were anesthetized at 1, 2, 4, 8, 24, 48, and 72 hours, and the near-infrared (NIR) emission from the NIR797 of NPs was recorded by the Caliper IVIS (PerkinElmer).24
Statistical analysis
All the quantitative data were described as means ± standard deviation (SD) and analyzed using the SPSS software (version 15.0; SPSS Inc., Chicago, IL, USA). The F-test was used for significance testing, and P<0.05 was considered to be statistically significant.
Results
Characteristics of NIR797-labeled NPs
The surface morphology of PEG-PLL-PLGA NPs was spherical shape, smooth surface, and no obvious aggregates phenomenon, observed by TEM (Figure 1A). The polydispersity index was 0.079±0.022 (n=3), and the average diameter was approximately 185±13 nm, detected by laser particle size analyzer. The zeta potential was −20.3±1.4 mV, measured by zeta potential analyzer (Figure 1B). Figure 1C described the schematic representation of NIR797-labeled NPs. Wavelength of NIR797-labeled NPs was detected in the FL3 channel. The relative fluorescence intensities (RFI = FI-treated group/FI-control group) of NIR797 in K562 cells treated with NIR797-labeled DNR-NPs and NIR797-labeled DNR-Tf-NPs were 32.97±1.66 and 39.89±1.58, respectively. Compared to the control group (Figure 1D), the two groups obviously displayed NIR797 wavelength (P<0.05). The wavelength of NIR797-labeled NPs was in this range (NIR797 excitation wavelength, 795 nm; emission wavelength, 817 nm), indicating that the NPs successfully encapsulated the dye NIR797.
Figure 1.

Characterization of NIR797-labeled NPs.
Notes: (A) Transmission electron microscopic images, (B) particle size distribution, (C) schematic diagram, (D) fluorescence intensity.
Abbreviations: Tf, transferrin; PEG, polyethylene glycol; PLL, poly-l-lysine; PLGA, poly(lactic-co-glycolic acid); NIR, near-infrared; DNR, daunorubicin; NPs, nanoparticles; SSC-H, side scatter light histogram; FL3-H, fluorescent light 3 histogram.
Tumor growth inhibition
All the mice injected via the tail vein for 14 days were alive; no adverse reactions were observed during the whole treatment. As shown in Figure 2A and B, tumor volumes of control group and DNR group were larger than in DNR-Tet group, suggesting that Tet as P-gp inhibitor can reverse drug resistance. The RTVs in the DNR-NPs group and DNR-Tet-NPs group were much lower than in other groups (P<0.05) after treatment, as shown in Figure 2C. Furthermore, the inhibition rates in DNR-NPs group, DNR-Tet-NPs group, and DNR-Tet-Tf-NPs group were 40.63%±3.55%, 50.33%±4.06%, and 64.31%±4.89%, respectively (Figure 2D). It was apparent that the tumor growth was more effectively controlled in DNR-Tet-Tf-NPs group than in the other five groups (P<0.05). These results indicated that targeted antitumor effect and reversal of MDR function can be achieved with DNR-Tet-Tf-NPs of our new delivery system.
Figure 2.

Photographs showing the appearance of tumor body in tumor-bearing nude mice (A) and tumor size (B) at the end of treatment, relative tumor volume (C), and tumor inhibition rate (D) after the treatment.
Notes: (a) Control, (b) DNR, (c) DNR and Tet, (d) DNR-NPs, (e) DNR-Tet-NPs, (f) DNR-Tet-Tf-NPs.
Abbreviations: DNR, daunorubicin; Tet, tetrandrine; NPs, nanoparticles; Tf, transferrin; d, day.
Histopathological examination of tumor tissue
In Figure 3, tumor cells exhibit disorder, different size, appeared more mitotic figures, heterogeneous and nuclear large stained, and increased angiogenesis were observed in tumor tissues. The microscopic structural difference of each group was mainly in fibrous necrosis. The heart, liver, spleen, lung, and kidney of DNR-Tet-Tf-NPs group had no significant pathological changes compared to control group. In DNR-Tet-Tf-NPs group, there were no inflammatory cells in the heart tissue, no liver cell degeneration necrosis, no hyperplasia, atrophy, and hyaline degeneration in the spleen tissue, no pulmonary edema, and the glomerular volume was normal. Moreover, no chemotherapeutic drug-induced histological changes and tumor metastasis could be seen in DNR-Tet-Tf-NPs group. These observations demonstrate that Tf conjugated to the surface of DNR-Tet-NPs can increase the transport of DNR to the tumor site and avoid the toxicity of other major organs.
Figure 3.

Histopathological examination of organs and tumor tissues in K562/A02 xenograft model after the treatment (hematoxylin–eosin staining, ×200).
Notes: (a) Control, (b) DNR, (c) DNR and Tet, (d) DNR-NPs, (e) DNR-Tet-NPs, (f) DNR-Tet-Tf-NPs.
Abbreviations: DNR, daunorubicin; Tet, tetrandrine; NPs, nanoparticles; Tf, transferrin.
Apoptosis role of Bax, caspase-3, Bcl-2, and survivin in vivo
Expression of Bax, caspase-3, Bcl-2, and survivin was detected by immunohistochemistry, as shown in Figure 4. Immunohistochemical staining showed brown fine particles for positive expression, mainly located in the cell membrane and the cytoplasm of tumor cells; the positive cells exhibited diffuse or focal distribution. As for the expression of Bax, caspase-3 in the DNR-Tet-NPs and DNR-Tet-Tf-NPs groups compared to the control and DNR groups, positive cells markedly increased. However, the opposite result was observed in Bcl-2 and survivin expression; positive cells obviously decreased. The results displayed the strongest Bax and caspase-3 expression but the weakest Bcl-2 and survivin expression in DNR-Tet-Tf-NPs group.
Figure 4.

Expression of Bax, caspase-3, Bcl-2, and survivin in tumor tissues after the treatment (immunohistochemistry, ×400).
Notes: (a) Control, (b) DNR, (c) DNR and Tet, (d) DNR-NPs, (e) DNR-Tet-NPs, (f) DNR-Tet-Tf-NPs.
Abbreviations: DNR, daunorubicin; Tet, tetrandrine; NPs, nanoparticles; Tf, transferrin.
mRNA and protein expression of TfR, P-gp, MRP, and NF-κB
Quantitative real-time PCR and Western blot were used to detect the expression of TfR, P-gp, MRP, and NF-κB in the tumor tissues of nude mice (Figure 5). In comparison with the control group, transcription of P-gp, MRP, and NF-κB mRNA were downregulated and TfR was upregulated to some extent in DNR, DNR and Tet, DNR-NPs, DNR-Tet-NPs, and DNR-Tet-Tf-NPs groups. As expected, we found that the level of TfR mRNA was much higher in the DNR-Tet-Tf-NPs group than the other groups, and there was a significant difference (P<0.05), levels of P-gp, MRP, NF-κB mRNA expression were obviously reduced in the DNR-Tet-Tf-NPs groups than other groups (P<0.05). Interestingly, there was the same tendency in the protein expression treated with these groups.
Figure 5.

Expression of TfR, P-gp, MRP, NF-κB mRNAs (A) and proteins (B, C) after the treatment.
Notes: (a) Control, (b) DNR, (c) DNR and Tet, (d) DNR-NPs, (e) DNR-Tet-NPs, (f) DNR-Tet-Tf-NPs. *P<0.05 when compared to DNR-Tet-NPs.
Abbreviations: DNR, daunorubicin; Tet, tetrandrine; NPs, nanoparticles; Tf, transferrin; P-gp, P-glycoprotein; MRP, multidrug resistance protein; NF-κB, nuclear factor kappaB; TfR, Tf receptor; GAPDH, glyceraldehyde 3-phosphate dehydrogenase.
In vitro and in vivo results of NIR797-labeled NPs
The apoptotic rates of the groups were assessed by flow cytometry (Figure 6): the control group for 12 and 36 hours, DNR-NPs group for 12 hours, DNR-Tf-NPs group for 12 hours, DNR-NPs group for 36 hours, DNR-Tf-NPs group for 36 hours were 0.02%±0.01%, 73.63%±2.35%, 79.50%±2.21%, 83.25%±3.37%, and 84.53%±3.48%, respectively. Compared to the control group, other groups can significantly increase the apoptotic rate (P<0.05), the apoptotic rate could be found in 12 hours and lasted up to 36 hours.
Figure 6.

The apoptosis assay on K562 cells after the treatment with NIR797-labeled NPs.
Note: *P<0.05 when compared to control group.
Abbreviations: SSC-H, side scatter light histogram; FL1-H, fluorescent light 1 histogram; AV, annexin V-FITC; DNR, daunorubicin; NPs, nanoparticles; Tf, transferrin; NIR, near-infrared.
As the results in Figure 7 indicate, DNR fluorescence (orange fluorescence) in NIR797-labeled DNR-NPs group and NIR797-labeled DNR-Tf-NPs group diffused in the nucleus and cytoplasm of K562 cells, indicating that drug-loaded NPs were endocytosed. Furthermore, necrosis and apoptotic cells in NIR797-labeled DNR-Tf-NPs group was much more than in NIR797-labeled DNR-NPs group, and the better antitumor effect was due to the targeted ability of Tf-modified NPs.
Figure 7.
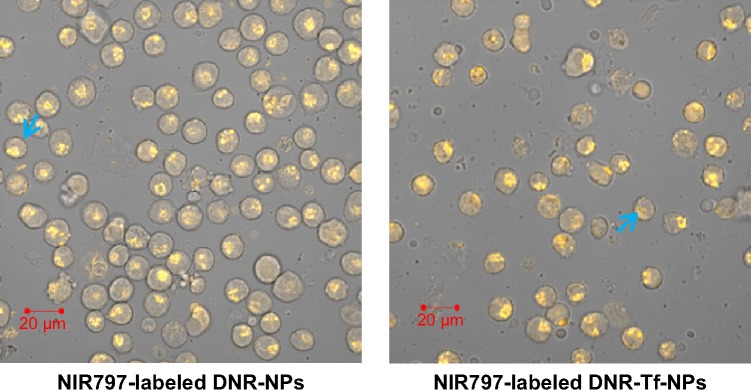
K562 cells observed by digital confocal microscope.
Note: The blue arrow demonstrates drug-loaded NPs were endocytosed in necrosis and apoptotic cells.
Abbreviations: NIR, near-infrared; DNR, daunorubicin; Tf, transferrin; NPs, nanoparticles.
To visualize the biodistribution of NPs in K562 tumor-bearing mice, a near-infrared fluorescent (NIRF) dye, NIR797, was labeled to PEG-PLL-PLGA NPs. NIR fluorescence signals are clearly observed in K562 cells (Figure 8A); the signal was stronger in NIR797-labeled DNR-Tf-NPs than in NIR797-labeled DNR-NPs. In vivo results are as follows (Figure 8B): the fluorescence signal of NIR797-labeled DNR-Tf-NPs at 1 hour mainly was in the liver area, and the fluorescence signal began to appear in the tumor site after the injection at 24 hours. As time increased, the tumor fluorescence signal became stronger and the fluorescence signal of the liver began to weaken; the noticeable signal reached the tumor location after the injection at 72 hours. The fluorescence signal of NIR797-labeled DNR-NPs was consistent with Tf-targeted group, but the tumor fluorescence signal was weaker than Tf-targeted group. Through the Caliper IVIS of the major organs and part of tumors, a large number of NIR797-labeled NPs were mainly visualized in the tumor site. The NPs showed certain targeting features, but NIR797-labeled DNR-Tf-NPs in nude mice exhibited apparently better targeting effect (Figure 8B). Moreover, in the HPLC experiment, the DNR concentration in tumor tissue was higher in DNR-Tet-Tf-NPs group than in DNR-Tet-NPs group, which indicated that DNR-Tet-Tf-NPs entered the tumor site (P<0.05). However, the concentrations of DNR in heart, liver, spleen, lung, kidney, and plasma between the two groups were not significant (P>0.05). NPs were combined with DNR, Tet, and Tf to demonstrate that DNR-Tet-Tf-NPs could efficiently overcome multidrug-resistant leukemia and increase its targeting property.
Figure 8.

NIRF images of K562 cells in vitro (A) and in tumor-bearing mice (B) after the treatment.
Notes: (a) NIR797-labeled DNR-NPs, (b) NIR797-labeled DNR-Tf-NPs. The blue arrow shows the DNR concentration in tumor tissue.
Abbreviations: NIRF, near-infrared fluorescence; DNR, daunorubicin; NPs, nanoparticles; Min, minimum; Max, maximum; h, hour; Tf, transferrin.
Discussion
Nowadays, the main cause of tumor chemotherapy failure is MDR. Studies have primarily focused on the NPs applications: NPs as carriers of chemotherapeutic drugs to reverse MDR.25,26 Pharmacokinetic studies have displayed that NPs can significantly increase the intracellular concentration of anticancer drugs and still maintain a certain concentration of drug after drug withdrawal to achieve better therapeutic effect.10,27 Many traditional Chinese medicines such as Tet and gambogic acid (GA) have antitumor effects,8,22,28 and could reverse drug resistance, kill tumor cells, and improve immune function. In our study, tumor growth was also significantly slowed in the groups treated with NPs. PEG-PLL-PLGA NPs as NDDS are capable of concentrating drugs selectively in the tumor tissue through systemic circulation, improving efficacy, and reducing toxicity of the drugs. In addition, the inhibition rate in DNR-Tet-NPs group was higher than in DNR-NPs group, suggesting that Tet has MDR reversal effect on K562/A02.
Recent studies have found that TfR expression was markedly increased in tumor, could be as tumor antigens and targeted diagnostic treatment markers. Targeting the TfR has been shown to be effective in delivering chemotherapeutic drugs.29 Tf/TfR-mediated tumor active targeting pathway as the transport of anticancer drugs is highly efficient and well targeted.30 In our experiment, intravenously administered DNR-Tet-Tf-NPs dramatically inhibited the growth of transplantable tumor. DNR-Tet-Tf-NPs have properties to act as NDDS to improve drug concentration in cells; furthermore, Tf-modified NPs has been explored as a target to deliver therapeutics into cancer cells due to their increased expression on malignant cells and accessibility on the cell surface.
Apoptosis is regulated by genes. A variety of oncogenes and tumor suppressor genes are involved in the regulation of apoptosis; the proto-oncogene Bcl-2 is the most important gene of them.31 Bcl-2 and Bax are a pair of positive and negative regulators of gene; Bax induced apoptosis, while Bcl-2 inhibited apoptosis.32 Caspase-3 may promote apoptosis, while survivin is the strongest inhibitor of apoptosis, also promoting cell proliferation, which can directly inhibit the activity of caspase-3 and caspase-7, thereby blocking the apoptotic process.33,34 These results of apoptosis phenomenon in the mice treated with DNR-Tet-NPs and DNR-Tet-Tf-NPs by immunohistochemistry observation showed that NPs combined with Tet can decrease Bcl-2 and survivin protein expression, and increase Bax and caspase-3 protein expression; the difference was significant compared to the control group.
P-gp overexpressed in multidrug-resistant cancer could induce the chemotherapy drugs to pump from the cells.4 Many researchers have shown that MDR protein 1 (MRP1) is the most studied in the MRP family. MRPl and P-gp have many similarities in their structures and functions. It is also a kind of energy-dependent drug pump.25,35 NF-κB directs the role to imbalance the expression of apoptosis protein and antiapoptotic protein. Inhibitory effect of NF-κB-mediated phagocytosis is its antiapoptotic mechanisms.36,37 Data from quantitative real-time PCR and Western blot indicated that TfR upregulation and P-gp, MRP, NF-κB downregulation led to the inhibition of leukemia cell proliferation and invasion in tumor xenografts. We supposed that the functionalized DNR-Tet-Tf-NPs can promote apoptosis, reverse drug resistance, and target antitumor effect. No toxicity was found in the histopathological study; DNR-Tet-Tf-NPs can directly deliver into the tumor, reduce the dose of chemotherapy drugs, and avoid the side effects of cardiac toxicity and other major organs.
Currently, the NIRF imaging technique is widely used for cancer molecular imaging research; it has a more sensible and longer visualization for tracking in vivo.24 Drug loaded distribution of molecular imaging NPs modified by Tf can be detected using the normalized NIRF images. Our previous studies have demonstrated PEG-PLL-PLGA NPs with high efficiency and low toxicity.13,14 Thus, in the present study, we wanted to demonstrate the potential synergistic effects of DNR-Tet-NPs and Tf in leukemia cells. Evidenced by NIR imaging experiments, drug accumulation at the tumor site of DNR-Tet-Tf-NPs group was obviously higher than in the DNR-Tet-NPs group, suggesting that Tf-modified NPs have targeted antitumor effect. DNR-Tet-Tf-NPs reduced the drug side effects of the main organs, achieved high efficiency and low toxicity of therapeutic effect. Interestingly, we not only took visible molecular imaging NPs but also used quality HPLC to demonstrate its active targeting capacity. These results are in agreement with HPLC study; DNR-Tet-NPs modified by Tf increase the active targeting capacity. PEG-PLL-PLGA NPs present a convenient, prolonged, targeted, and effective NDDS with the ability of loading DNR, Tet, and Tf.
Conclusion
In summary, our research successfully fabricated the targeted DNR-Tet-Tf-NPs DDS for overcoming MDR in tumors. The finding demonstrates that the active targeting of Tf-modified NDDS could increase the effectiveness of chemotherapy, reduce the side effects, and reverse MDR, which may pave the way for better cancer treatment.
Acknowledgments
We thank Professor Gerard Marriott of the Department of Bioengineering, University of California, Berkeley, for his help in the improvement of the paper. This research was supported by the National Key Basic Research Program 973 of the People’s Republic of China (Number 2010CB732404), the National Nature Science Foundation of the People’s Republic of China (Numbers 81170492, 81370673), and the National High Technology Research and Development Program 863 Projects of the People’s Republic of China (Number 2012AA022703).
Footnotes
Disclosure
The authors report no conflicts of interests in this work.
References
- 1.Daniels TR, Delgado T, Rodriguez JA, Helguera G, Penichet ML. The transferrin receptor part I: biology and targeting with cytotoxic antibodies for the treatment of cancer. Clin Immunol. 2006;121:144–158. doi: 10.1016/j.clim.2006.06.010. [DOI] [PubMed] [Google Scholar]
- 2.Daniels TR, Delgado T, Helguera G, Penichet ML. The transferrin receptor part II: targeted delivery of therapeutic agents into cancer cells. Clin Immunol. 2006;121:159–176. doi: 10.1016/j.clim.2006.06.006. [DOI] [PubMed] [Google Scholar]
- 3.Fonseca C, Moreira JN, Ciudad CJ, Pedroso de Lima MC, Simões S. Targeting of sterically stabilised pH-sensitive liposomes to human T-leukaemia cells. Eur J Pharm Biopharm. 2005;59:359–366. doi: 10.1016/j.ejpb.2004.08.012. [DOI] [PubMed] [Google Scholar]
- 4.Ruefli AA, Smyth MJ, Johnstone RW. HMBA induces activation of a caspase-independent cell death pathway to overcome P-glycoprotein-mediated multidrug resistance. Blood. 2000;95:2378–2385. [PubMed] [Google Scholar]
- 5.Motomura S, Motoji T, Takanashi M, et al. Inhibition of P-glycoprotein and recovery of drug sensitivity of human acute leukemic blast cells by multidrug resistance gene (mdr1) antisense oligonucleotides. Blood. 1998;91:3163–3171. [PubMed] [Google Scholar]
- 6.Castranova V, Kang JH, Moore MD, Pailes WH, Frazer DG, Schwegler-Berry D. Inhibition of stimulant-induced activation of phagocytic cells with tetrandrine. J Leukoc Biol. 1991;50:412–422. doi: 10.1002/jlb.50.4.412. [DOI] [PubMed] [Google Scholar]
- 7.Fu L, Liang Y, Deng L, et al. Characterization of tetrandrine, a potent inhibitor of P-glycoprotein-mediated multidrug resistance. Cancer Chemother Pharmacol. 2004;53:349–356. doi: 10.1007/s00280-003-0742-5. [DOI] [PubMed] [Google Scholar]
- 8.Chen BA, Sun Q, Wang XM, et al. Reversal in multidrug resistance by magnetic nanoparticle of Fe3O4 loaded with adriamycin and tetrandrine in K562/A02 leukemic cells. Int J Nanomedicine. 2008;3:277–286. doi: 10.2147/ijn.s2714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yin Q, Shen J, Zhang Z, Yu H, Li Y. Reversal of multidrug resistance by stimuli-responsive drug delivery systems for therapy of tumor. Adv Drug Deliv Rev. 2013;65:1699–1715. doi: 10.1016/j.addr.2013.04.011. [DOI] [PubMed] [Google Scholar]
- 10.Moghassemi S, Hadjizadeh A. Nano-niosomes as nanoscale drug delivery systems: an illustrated review. J Control Release. 2014;185:22–36. doi: 10.1016/j.jconrel.2014.04.015. [DOI] [PubMed] [Google Scholar]
- 11.Wang Q, Sun Y, Zhang Z, Duan Y. Targeted polymeric therapeutic nanoparticles: design and interactions with hepatocellular carcinoma. Biomaterials. 2015;56:229–240. doi: 10.1016/j.biomaterials.2015.03.050. [DOI] [PubMed] [Google Scholar]
- 12.Wang Y, Liu P, Qiu L, et al. Toxicity and therapy of cisplatin-loaded EGF modified mPEG-PLGA-PLL nanoparticles for SKOV3 cancer in mice. Biomaterials. 2013;34(16):4068–4077. doi: 10.1016/j.biomaterials.2012.12.033. [DOI] [PubMed] [Google Scholar]
- 13.Guo LT, Chen BA, Liu R, et al. Biocompatibility assessment of Polyethylene Glycol-Poly L-Lysine-Poly Lactic-Co-Glycolic Acid nanoparticles in vitro and in vivo. J Nanosci Nanotechnol. 2015;15:3710–3719. doi: 10.1166/jnn.2015.9509. [DOI] [PubMed] [Google Scholar]
- 14.Bao W, Liu R, Wang Y, et al. PLGA-PLL-PEG-Tf-based targeted nanoparticles drug delivery system enhance antitumor efficacy via intrinsic apoptosis pathway. Int J Nanomedicine. 2015;10:557–566. doi: 10.2147/IJN.S75090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ahn CH, Chae SY, Bae YH, Kim SW. Synthesis of biodegradable multi-block copolymers of poly(L-lysine) and poly(ethylene glycol) as a non-viral gene carrier. J Control Release. 2004;97:567–574. doi: 10.1016/j.jconrel.2004.04.002. [DOI] [PubMed] [Google Scholar]
- 16.Bertram JP, Jay SM, Hynes SR, Robinson R, Criscione JM, Lavik EB. Functionalized poly(lactic-co-glycolic acid) enhances drug delivery and provides chemical moieties for surface engineering while preserving biocompatibility. Acta Biomater. 2009;5:2860–2871. doi: 10.1016/j.actbio.2009.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bertram JP, Rauch MF, Chang K, Lavik EB. Using polymer chemistry to modulate the delivery of neurotrophic factors from degradable microspheres: delivery of BDNF. Pharm Res. 2010;27:82–91. doi: 10.1007/s11095-009-0009-x. [DOI] [PubMed] [Google Scholar]
- 18.Zhou J, Patel TR, Fu M, Bertram JP, Saltzman WM. Octa-functional PLGA nanoparticles for targeted and efficient siRNA delivery to tumors. Biomaterials. 2012;33:583–591. doi: 10.1016/j.biomaterials.2011.09.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang H, Wang C, Chen B, Wang X. Daunorubicin-TiO2 nanocomposites as a “smart” pH-responsive drug delivery system. Int J Nanomedicine. 2012;7:235–242. doi: 10.2147/IJN.S27722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ren Y, Zhang H, Chen B, et al. Multifunctional magnetic Fe3O4 nanoparticles combined with chemotherapy and hyperthermia to overcome multidrug resistance. Int J Nanomedicine. 2012;7:2261–2269. doi: 10.2147/IJN.S29357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang W, Lu Z, Kong G, et al. Hepatitis B virus X protein accelerates hepatocarcinogenesis with partner survivin through modulating miR-520b and HBXIP. Mol Cancer. 2014;13:128. doi: 10.1186/1476-4598-13-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang F, Zhang W, Guo LT, et al. Gambogic acid suppresses hypoxia-induced hypoxia-inducible factor-1α/vascular endothelial growth factor expression via inhibiting phosphatidylinositol 3-kinase/Akt/mammalian target protein of rapamycin pathway in multiple myeloma cells. Cancer Sci. 2014;105:1063–1070. doi: 10.1111/cas.12458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li H, Takayama K, Wang S, et al. Addition of bevacizumab enhances antitumor activity of erlotinib against non-small cell lung cancer xenografts depending on VEGF expression. Cancer Chemother Pharmacol. 2014;74:1297–1305. doi: 10.1007/s00280-014-2610-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen YC, Wen S, Shang SA, Cui Y, Luo B, Teng GJ. Magnetic resonance and near-infrared imaging using a novel dual-modality nano-probe for dendritic cell tracking in vivo. Cytotherapy. 2014;16:699–710. doi: 10.1016/j.jcyt.2013.09.006. [DOI] [PubMed] [Google Scholar]
- 25.Xu X, Sabanayagam CR, Harrington DA, Farach-Carson MC, Jia X. A hydrogel-based tumor model for the evaluation of nanoparticle-based cancer therapeutics. Biomaterials. 2014;35:3319–3330. doi: 10.1016/j.biomaterials.2013.12.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Markman JL, Rekechenetskiy A, Holler E, Ljubimova JY. Nanomedicine therapeutic approaches to overcome cancer drug resistance. Adv Drug Deliv Rev. 2013;65(13–14):1866–1879. doi: 10.1016/j.addr.2013.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang X, Dong Y, Zeng X, et al. The effect of autophagy inhibitors on drug delivery using biodegradable polymer nanoparticles in cancer treatment. Biomaterials. 2014;35:1932–1943. doi: 10.1016/j.biomaterials.2013.10.034. [DOI] [PubMed] [Google Scholar]
- 28.Chen BA, Yin L, Cheng J, et al. Effect of D, L-threo-1-phenyl-2-decanoylamino-3-morpholino-1-propanol and tetrandrine on the reversion of multidrug resistance in K562/A02 cells. Hematology. 2011;16:24–30. doi: 10.1179/102453311X12902908411797. [DOI] [PubMed] [Google Scholar]
- 29.Du W, Fan Y, Zheng N, et al. Transferrin receptor specific nanocarriers conjugated with functional 7peptide for oral drug delivery. Biomaterials. 2013;34:794–806. doi: 10.1016/j.biomaterials.2012.10.003. [DOI] [PubMed] [Google Scholar]
- 30.Liu G, Shen H, Mao J, et al. Transferrin modified graphene oxide for glioma-targeted drug delivery: in vitro and in vivo evaluations. ACS Appl Mater Interfaces. 2013;5:6909–6914. doi: 10.1021/am402128s. [DOI] [PubMed] [Google Scholar]
- 31.Lindsay J, Esposti MD, Gilmore AP. Bcl-2 proteins and mitochondria – specificity in membrane targeting for death. Biochim Biophys Acta. 2011;1813:532–539. doi: 10.1016/j.bbamcr.2010.10.017. [DOI] [PubMed] [Google Scholar]
- 32.Gonzalez-Campora R, Davalos-Casanova G, Beato-Moreno A, et al. BCL-2, TP53 and BAX protein expression in superficial urothelial bladder carcinoma. Cancer Lett. 2007;250:292–299. doi: 10.1016/j.canlet.2006.10.011. [DOI] [PubMed] [Google Scholar]
- 33.Sriramoju B, Kanwar RK, Kanwar JR. Nanoformulated cell-penetrating survivin mutant and its dual actions. Int J Nanomedicine. 2014;9:3279–3298. doi: 10.2147/IJN.S60169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Altieri DC. Survivin, cancer networks and pathway-directed drug discovery. Nat Rev Cancer. 2008;8:61–70. doi: 10.1038/nrc2293. [DOI] [PubMed] [Google Scholar]
- 35.Beedholm-Ebsen R, van de Wetering K, Hardlei T, Nexø E, Borst P, Moestrup SK. Identification of multidrug resistance protein 1 (MRP1/ABCC1) as a molecular gate for cellular export of cobalamin. Blood. 2010;115:1632–1639. doi: 10.1182/blood-2009-07-232587. [DOI] [PubMed] [Google Scholar]
- 36.Murphy SH, Suzuki K, Downes M, et al. Tumor suppressor protein (p)53, is a regulator of NF-kappaB repression by the glucocorticoid receptor. Proc Natl Acad Sci U S A. 2011;108:17117–17122. doi: 10.1073/pnas.1114420108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Baud V, Karin M. Is NF-kappaB a good target for cancer therapy? Hopes and pitfalls. Nat Rev Drug Discov. 2009;8:33–40. doi: 10.1038/nrd2781. [DOI] [PMC free article] [PubMed] [Google Scholar]