

Abstract
Background
XM17 is a recombinant human follicle-stimulating hormone (follitropin alfa) for stimulation of multifollicular development in women undergoing controlled ovarian hyper-stimulation during assisted reproductive therapy and for treatment of anovulation. Manufactured using Chinese hamster ovary cells transfected with the human follicle-stimulating hormone gene, XM17 has an identical amino acid sequence to that of the human protein as well as to those of the other approved recombinant human follicle-stimulating hormone products. Glycosylation patterns may differ slightly between products. The objectives of this first-in-human study were to assess the safety, tolerability, pharmacokinetics, and dose-proportionality of single ascending subcutaneous doses of XM17 in healthy young female volunteers.
Methods
Endogenous follicle-stimulating hormone was downregulated by implanting a 1-month depot of goserelin acetate 3.6 mg on day 0 in eligible subjects. On day 14 of the experimental period, subjects received one of four ascending doses of XM17. Blood sampling to obtain the pharmacokinetic profile of XM17 was done at frequent intervals until 168 hours post-dose.
Results
Following downregulation of endogenous follicle-stimulating hormone to <4 IU/L, 40 subjects (of mean age 29±5.4 years) received single subcutaneous doses of 37.5 (n=4, pilot group), 75, 150, or 300 IU (n=12 each) of XM17. The mean serum concentration-time profiles of XM17 revealed dose-related increases in maximum concentration (Cmax) within 24 hours followed by monoexponential decay for the three higher dose levels. Slopes estimated by linear regression for Cmax and AUC0–168h were ~1.0 (0.9052 IU/L and 1.0964 IU·h/L, respectively). For each IU of XM17 administered, Cmax and AUC0–168h rose by 0.032 IU/L and 2.60 IU·h/L, respectively. Geometric mean elimination half-life ranged from 54 to 90 hours. No antibodies to XM17 were detected. The most common treatment-emergent adverse events were headache (12 events in eleven [27.5%] subjects) and dizziness (four events in four [10%] subjects); two subjects (5%) reported mild pain on touch at the injection site.
Conclusion
Single subcutaneous doses of XM17 up to 300 IU in healthy young women exhibited dose-proportional pharmacokinetics with good safety and tolerability.
Keywords: follitropin alfa, biosimilar, assisted reproductive technology, anovulation
Introduction
Therapeutic proteins used for a variety of clinical indications are manufactured by recombinant DNA methods, many of them using Chinese hamster ovary cells as the mammalian host.1–3 Proteins manufactured by recombinant techniques are regarded as safe, and eventually may replace plasma-derived or urine-derived therapeutic proteins as the standard of care.1,4,5 Assisted reproductive technology (ART) is one such therapeutic application for recombinant proteins, and specifically for recombinant human follicle-stimulating hormone (rhFSH).1,3,5
Clinical pregnancy rates and safety – in terms of avoiding ovarian hyperstimulation syndrome – with rhFSH are similar to those achieved with purified urinary gonadotropins;6 however, the recombinant protein offers some important advantages. First, the manufacturing techniques for rhFSH facilitate production of a consistent product,5 thereby improving the quality and availability of this product for clinical use.7 Although the risk for prion infection with urine-derived gonadotropic proteins is believed to be minimal,8 production and purification techniques for recombinant gonadotropins are sufficient to eliminate this risk.9 Concerns regarding immunological safety due to impurities are also minimized,3,5,10 and the tolerability of recombinant gonadotropins is better than that of products purified from urine.8 One drawback of recombinant proteins is the relatively high manufacturing cost, which is reflected in the acquisition price; however, some studies suggest that recombinant protein therapeutics are cost-effective.5,11
Any new rhFSH preparation brought to market will be expected to adhere to the highest manufacturing standards and exhibit an optimal pharmacokinetic (PK), pharmacodynamic, and safety profile. XM17 (Ovaleap®; follitropin alfa, Teva Pharmaceuticals Europe B.V., Utrecht, the Netherlands) is an rhFSH used in women for controlled ovarian hyper-stimulation undergoing ART procedures and for treatment of anovulation. XM17 has been approved by the European Medicines Agency as a biosimilar medicinal product to the already registered biological reference medicine (Gonal-f®, follitropin alfa, Merck Serono, Feltham, UK), providing another rhFSH option for ART patients and their caregivers. Manufactured using Chinese hamster ovary cells transfected with the human follicle-stimulating hormone gene, XM17 has an identical amino acid sequence and a similar glycosylation pattern to the human protein and to the reference product, as would be expected for a biosimilar approved by the European Medicines Agency.12 European Medicines Agency guidelines require that a manufacturer of a biosimilar product confirm comparable clinical performance – in terms of quality characteristics, biological activity, safety, and efficacy – to that of the reference product. Registered rhFSH products are generally well tolerated, with headache, ovarian cysts, abdominal pain, and local injection site reactions being the most frequent adverse events after subcutaneous administration.13–15 We conducted this first-in-human study to evaluate the safety, tolerability, PK, and dose-proportionality of single ascending doses of XM17.
Materials and methods
Study conduct
This was a single-center, Phase 1, open-label, parallel-group, single ascending dose study conducted at a Phase I clinical research unit in Germany from January to July 2008. The study was conducted according to the ethical principles specified in the Declaration of Helsinki (1996) and applicable Good Clinical Practice guidelines, and was approved by the responsible independent ethics committee in Germany.
Study objectives and parameters measured
This study was conducted in healthy female subjects after downregulation and achievement of ovarian quiescence by a gonadotropin-releasing hormone agonist. The primary objectives were to assess the safety, tolerability, and PK of a single, subcutaneous, low dose of 37.5 IU XM17 (pilot arm in the first four subjects) followed by an assessment of safety, tolerability, PK, and dose-proportionality of single subcutaneous ascending doses of 75, 150, or 300 IU XM17 in 36 additional subjects (n=12 at each dose). The study schema and schedule of events is shown in Figure 1.
Figure 1.

Study schema and schedule of events.
Abbreviations: ECG, electrocardiogram; FSH, follicle-stimulating hormone; PK, pharmacokinetic.
The primary PK endpoints were the Cmax (pre-dose corrected for baseline endogenous FSH measured on day 14) of XM17 in serum and the area under the concentration-time curve (AUC) from time 0 to 168 hours (AUC0–168h). Second ary endpoints included AUC from time 0 on dosing day 14 to last concentration measured (AUC0–t), AUC extrapolated to infinity (AUC0–∞), apparent terminal rate constant (λZ), apparent volume of distribution during the terminal phase (Vz/F, where F is fraction absorbed), time at which Cmax occurred (tmax), apparent terminal elimination half-life (t1/2), and the apparent clearance (CL/F) of XM17.
Safety was monitored in all subjects who received the study drug (safety population). Safety endpoints included: changes in subjects’ laboratory parameters (hematology, clinical chemistry, coagulation parameters, and urinalysis); changes in vital signs and electrocardiogram (ECG); incidence of adverse events and treatment-emergent adverse events (TEAEs); local tolerability at the injection site; presence of XM17 antibodies; and changes in the subjects’ physical or gynecological examinations.
Study protocol
Consenting subjects were screened for eligibility during a 14-day period. Key eligibility criteria were: young, healthy, nonpregnant women aged 18–39 years; body mass index 18–29 kg/m2 and weighing $50 kg; nonsmoker or moderate smoker (<10 cigarettes per day); use of combined oral contraceptives (COCs) for $3 months for contraceptive purposes; normal uterus and two normally functioning ovaries; agreement to use double-barrier contraceptive methods during the study; healthy according to medical history, physical examination, ECG, blood pressure, heart rate, blood laboratory profile including coagulation factors, and urinalysis; negative results for human immunodeficiency virus, hepatitis B virus, and hepatitis C virus antibodies and tests; and absence of polycystic ovary syndrome, class III or IV endometriosis, submucosal myoma uteri, history of endocrine abnormalities, and drug or alcohol abuse.
Gynecological investigations, including breast examination, transvaginal sonography, and a Papanicolaou test were performed at screening by an experienced gynecologist.
Based on previous reports, a gonadotropin-releasing hormone agonist (Zoladex® [goserelin acetate implant 3.6 mg, 1-month depot], AstraZeneca GmbH, Wedel, Germany) was administered to women (study day 0; corresponding to days 11–17 of the menstrual/COC cycle) who met the eligibility criteria to downregulate endogenous FSH secretion.3,16 Dosing of XM17 was started approximately 2 weeks after the gonadotropin-releasing hormone agonist was administered (Figure 1).
Subjects could start treatment with XM17 only when downregulation of plasma FSH to <4 IU/L was confirmed on day 10. To ensure contraceptive protection during the down-regulation process with an initial peak of FSH and luteinizing hormone levels,16 subjects took their own COCs for between 4 and up to a maximum of 7 days after beginning downregulation with goserelin acetate. No other prescribed concomitant medications were permitted beginning 4 weeks before XM17 administration (2 weeks for over-the-counter medications) until the final study visit. No grapefruit, grapefruit-containing or quinine-containing products, or alcoholic beverages were to be taken within 72 hours prior to XM17 administration. All subjects observed a 10-hour fast before XM17 dosing and for 4 hours post-dosing.
Subjects resided in the clinic from the morning of the day before dosing (day 13) until the morning of day 15. All subsequent visits, including the blood sampling visit 36 hours post-dosing, were ambulatory.
The first subject enrolled (pilot phase) received a single subcutaneous 37.5 IU dose of XM17 on day 14 and was observed for safety concerns; if no safety concerns arose, an additional three subjects were treated on the next or following days. Following completion of the pilot phase, single doses of XM17 were administered subcutaneously in stepwise ascending doses of 75, 150, and 300 IU XM17 to 12 subjects each. Dose-escalation was not to proceed when >50% of subjects treated at a particular dose had moderate or severe adverse events or when one subject had experienced a serious adverse event with a suspected relationship to XM17.
Blood sampling to determine the PK parameters of XM17 was performed pre-dose at −5 minutes and then at 0.5, 1, 2, 4, 6, 8, 10, 12, 16, 24, 36, 48, 72, 96, 120, 144, and 168 hours post-dose. Subjects were monitored pre-dose and at regular intervals post-dose for ECG and vital signs (pulse, blood pre ssure, temperature). Routine laboratory testing was performed on days 14, 15, and 21, and blood sampling for XM17 antibodies was performed on days 14 and 21. A second gynecological examination was performed at the final study visit, immediately after which subjects were to resume taking COCs. The study duration was 22–28 days for each subject.
XM17 drug product and drug concentration measurements
XM17 was manufactured according to Good Manufacturing Practice and relevant European Pharmacopoeia monographs. Vials of XM17 contained 1.0 mL solution for injection (600 IU XM17 per mL formulated with sodium phosphate, benzyl alcohol, and benzalkonium chloride, polysorbate 20, mannitol, and L-methionine, all meeting European Pharmacopoeia standards).
Serum concentrations of XM17 and downregulated endogenous FSH (measured pre-dose on day 14) were determined at MDS Pharma Services (Fehraltorf, Switzerland) using a validated Good Laboratory Practice electrochemiluminescence immunoassay technique.
Data analyses
PK endpoints for XM17 were calculated using noncompartmental methods to derive PK parameters. Calculations for the primary PK parameters were based on actual times, derived after subtracting pre-dose (baseline) endogenous FSH serum concentrations from concentrations obtained after dosing on day 14. If subtraction resulted in a value below zero, then that value was set to zero. Serum XM17 concentrations below the lower limit of quantification (LLOQ) were set to zero (if pre-dose) or to ½× LLOQ otherwise.
Selection of data points for calculation of λz were based on inspection of log-concentration versus time plots of subjects’ individual profiles. For all subjects, $3 of the last data points above the LLOQ were used to calculate λz. Cmax and corresponding tmax values were read directly from the concentration-time data.
All PK variables were estimated by noncompartmental analyses using WinNonLin software (version 5.01) and summarized using descriptive statistics. Geometric means were derived from the antilogarithm of the arithmetic mean of log-transformed (base e) values, defined only for values >0. Values #0 were omitted.
Linear regression analysis was applied to log-transformed Cmax and AUC0–168h (dependent variables) versus the log-transformed dose (independent variable). The statistical model was ln(parameter) = ln(α) + β × ln(dose) + random error; 95% confidence intervals (CIs) for the estimate of slope β were derived. If the 95% CI for slope β covered 1 (ie, β ≈1), then Cmax and AUC0–168h were plotted against dose and fitted by linear regression as follows: parameter = a × dose, with the 95% CI given for a.
Safety and tolerability were analyzed using descriptive reports of TEAEs (which included events reported up to study day 21), local tolerability, laboratory test results, ECGs, physical and gynecological examinations, and vital signs.
Results
Subjects
Seventy subjects were screened, and as planned, 40 young women (mean age 29±5.4 years) met the eligibility criteria, entered the study, and were treated with XM17. Since no subjects discontinued prematurely, all 40 subjects completed the study according to the protocol (PK population). The safety population was identical to the PK population.
Baseline characteristics of subjects in the dose groups (Table 1) were comparable, with the exception of subjects in the 150 IU dose group who had a lower mean body weight (61.2 kg) compared with the other dose groups (means, 66.8–71.0 kg).
Table 1.
Subject demographics and baseline characteristics (for pharmacokinetic and safety population)
| Overall (n=40) |
37.5 IU (n=4) |
75 IU (n=12) |
150 IU (n=12) |
300 IU (n=12) |
|
|---|---|---|---|---|---|
| Race, n (%) | |||||
| Caucasian | 39 (97.5) | 4 (100) | 12 (100) | 12 (100) | 11 (91.7) |
| Hispanic | 1 (2.5) | 0 | 0 | 0 | 1 (8.3) |
| Age (years) | |||||
| Mean (SD) | 29.0 (5.4) | 28.3 (6.8) | 28.5 (5.1) | 28.3 (5.3) | 30.5 (5.8) |
| Median (range) | 28.5 (20–39) | 26.0 (23–38) | 29.0 (21–39) | 26.5 (20–37) | 31.5 (20–38) |
| Height (cm) | |||||
| Mean (SD) | 168.4 (6.5) | 172.0 (6.4) | 171.3 (5.4) | 165.8 (6.4) | 166.9 (6.6) |
| Median (range) | 169.0 (153–180) | 174.0 (163–177) | 171.0 (161–180) | 164.0 (159–179) | 169.5 (153–174) |
| Weight (kg) | |||||
| Mean (SD) | 66.4 (10.2) | 71.0 (13.5) | 69.6 (10.6) | 61.2 (5.9) | 66.8 (11.1) |
| Median (range) | 64.4 (53.1–86.7) | 72.4 (55.3–83.8) | 67.5 (55.5–84.2) | 60.4 (53.1–70.5) | 62.6 (53.9–86.7) |
| BMI (kg/m2) | |||||
| Mean (SD) | 23.4 (2.9) | 23.9 (3.6) | 23.7 (2.9) | 22.3 (2.1) | 24.0 (3.3) |
| Median (range) | 22.4 (18.9–29.0) | 23.8 (20.7–27.2) | 22.4 (20.1–28.9) | 22.1 (19.3–25.4) | 24.0 (18.9–29.0) |
Abbreviations: IU, international unit; n, number of subjects within group; SD, standard deviation; BMI, body mass index; cm, centimeter; kg, kilogram; m, meter.
Pharmacokinetics of XM17
Both the geometric and arithmetic means of the pre-dose FSH-corrected primary PK parameters, Cmax and AUC0–168h, increased with the dose of XM17 (Table 2).
Table 2.
Primary and secondary pharmacokinetic parameters for XM17
| PK parameter | Statistic | 37.5 IU (n=4) |
75 IU (n=12) |
150 IU (n=12) |
300 IU (n=12) |
|---|---|---|---|---|---|
| Cmax (pre-dose corrected) in IU/L | Ncalc | 4 | 12 | 12 | 12 |
| Mean (SD) | 2.085 (1.019) | 2.329 (0.953) | 4.420 (0.652) | 9.835 (2.355) | |
| Geometric mean | 1.886 | 2.155 | 4.376 | 9.547 | |
| Median | 2.005 | 2.215 | 4.340 | 10.275 | |
| Min–max | 0.93–3.40 | 1.10–4.38 | 3.27–5.59 | 5.59–14.01 | |
| AUC0–168h (IU·h/L)a | Ncalc | 2 | 12 | 12 | 11 |
| Mean (SD) | 125.05 (141.35) | 204.24 (95.27) | 359.44 (75.69) | 792.05 (112.36) | |
| Geometric mean | 75.15 | 174.54 | 352.77 | 784.25 | |
| Median | 125.05 | 222.55 | 339.05 | 829.90 | |
| Min–max | 25.1–225.0 | 32.2–339.4 | 286.9–513.8 | 597.3–939.4 | |
| AUC0–t (IU·h/L) | Ncalc | 4 | 12 | 12 | 12 |
| Mean (SD) | 185.93 (113.87) | 204.35 (95.36) | 359.32 (75.69) | 839.04 (194.57) | |
| Geometric mean | 135.45 | 174.42 | 352.63 | 820.94 | |
| Median | 212.70 | 222.45 | 339.55 | 830.50 | |
| Min–max | 25.5–292.8 | 31.6–340.8 | 286.8–513.8 | 598.8–1,355.5 | |
| AUC0–∞ (IU·h/L) | Ncalc | 1 | 9 | 12 | 11 |
| Mean (SD) | 65.20 | 298.29 (202.61) | 448.20 (135.75) | 1,345.25 (809.14) | |
| Geometric mean | 65.20 | 222.01 | 430.35 | 1,203.26 | |
| Median | 65.20 | 267.20 | 414.75 | 1,023.50 | |
| Min–max | 65.2–65.2 | 38.3–579.7 | 297.6–687.1 | 628.9–3,599.9 | |
| λZ (1/hour)b | Ncalc | 1 | 9 | 12 | 11 |
| Mean (SD) | 0.0097 | 0.0108 (0.0062) | 0.0146 (0.0087) | 0.0102 (0.0071) | |
| Geometric mean | 0.0097 | 0.0095 | 0.0128 | 0.0077 | |
| Median | 0.0097 | 0.0102 | 0.0111 | 0.0090 | |
| Min–max | 0.0097–0.0097 | 0.0044–0.0243 | 0.0074–0.0368 | 0.0017–0.0215 | |
| VZ/F (L) | Ncalc | 1 | 9 | 12 | 11 |
| Mean (SD) | 59.2 | 41.4 (30.0) | 28.3 (8.0) | 36.1 (17.6) | |
| Geometric mean | 59.2 | 35.7 | 27.2 | 32.4 | |
| Median | 59.2 | 29.2 | 28.6 | 34.1 | |
| Min–max | 59.2–59.2 | 20.7–118.3 | 13.6–43.7 | 16.2–74.0 | |
| tmax (hours) | Ncalc | 4 | 12 | 12 | 12 |
| Mean (SD) | 122.4 (75.8) | 53.9 (64.4) | 22.1 (11.1) | 30.4 (35.9) | |
| Geometric mean | 79.8 | 30.6 | 19.7 | 22.9 | |
| Median | 155.8 | 24.0 | 24.0 | 24.0 | |
| Min–max | 10.0–168.0 | 10.0–168.3 | 10.0–47.6 | 10.1–143.2 | |
| t1/2 (hours) | Ncalc | 1 | 9 | 12 | 11 |
| Mean (SD) | 71.4 | 82.2 (40.7) | 60.2 (26.1) | 127.4 (120.1) | |
| Geometric mean | 71.4 | 73.1 | 54.1 | 90.1 | |
| Median | 71.4 | 67.6 | 63.2 | 77.0 | |
| Min–max | 71.4–71.4 | 28.6–156.6 | 18.9–93.7 | 32.3–408.4 | |
| CL/F (L/hour) | Ncalc | 1 | 9 | 12 | 11 |
| Mean (SD) | 0.575 | 0.520 (0.600) | 0.362 (0.102) | 0.270 (0.102) | |
| Geometric mean | 0.575 | 0.338 | 0.349 | 0.249 | |
| Median | 0.575 | 0.281 | 0.363 | 0.293 | |
| Min–max | 0.575–0.575 | 0.129–1.960 | 0.218–0.504 | 0.083–0.477 |
Notes:
Not evaluable in three subjects (two in the 37.5 IU group and one in the 300 IU group) because samples were taken earlier than scheduled at last time point;
in three subjects in the 37.5 IU group, three in the 75 IU group, and one in the 300 IU group, the λZ or any PK parameters derived from λZ were not estimable because an exponential decline over at least three data points could not be determined by inspection.
Abbreviations: IU, international unit; n, number of subjects within group; Ncalc, number of subjects used for calculation; L, litre; h, hour; SD, standard deviation; Cmax, maximum XM17 concentration; AUC0–168h, area under the XM17 concentration-time curve from time zero to 168 hours post-dose; AUC0–t, area under the XM17 concentration-time curve from time zero to the time of last observed concentration; AUC0–∞, area under the XM17 concentration time-curve from time zero to infinity; λZ, apparent terminal rate constant; Vz/F, apparent volume of distribution; tmax, time at which Cmax occurred; t1/2, apparent terminal elimination half-life; CL/F, apparent clearance.
The mean serum concentration-time profiles for XM17 following single ascending doses of XM17 in young women (Figure 2 [linear scale] and Figure 3 [log-linear scale]) were dose-proportional at the 75, 150, and 300 IU doses. Serum concentrations for these higher doses declined approximately monoexponentially until 120 hours post-dose and appeared to flatten thereafter. The concentration-time profile for the lowest dose (37.5 IU) began to rise approximately 36 hours post-dose and, for three of the four subjects, did not exhibit a terminal elimination phase. Accordingly, the PK parameters that were based on λZ (AUC0–∞, VZ/F, t1/2, and CL/F) were not estimable for the 37.5 IU dose.
Figure 2.

Mean (standard deviation) serum concentrations of XM17 after single ascending doses administered subcutaneously to healthy females. Values are corrected for pre-dose follicle-stimulating hormone levels (linear scale).
Figure 3.

Mean serum concentrations of XM17 after single ascending doses administered subcutaneously to healthy females. Values are corrected for pre-dose follicle-stimulating hormone levels (log-linear scale).
Absorption of XM17 was slow, with tmax values achieved from 10 to 168 hours post-dose (median of 24 hours in the three higher dose groups). The median tmax value in the lowest dose group (155.8 hours) appeared to be an artifact of the secondary rise observed in serum FSH concentration.
The geometric mean t½ in the three higher dose groups ranged from 54 to 90 hours (medians, 63–77 hours) without an apparent relationship to the XM17 dose. No dose-related differences were observed in the mean and median CL/F and Vz/F values (Table 2).
Dose linearity and proportionality
The slopes in a log-log linear (Power) model for pre-dose-corrected Cmax (IU/L) and AUC0–168h (IU·h/L) values were close to 1.0 (0.905 [95% CI 0.740–1.070] and 1.096 [95% CI 0.840–1.353], respectively), indicating dose proportionality.
In a simple linear model (not log-transformed, without intercept), the slope of the pre-dose-corrected regression line describing the proportionality of Cmax versus dose was 0.0322 (95% CI 0.0297–0.0347), predicting that, for every IU of XM17 administered, the Cmax increases by 0.0322 IU/L (Figure 4A). Similarly, the slope of the pre-dose-corrected regression line describing the proportionality of AUC0–168h versus dose was 2.596 (95% CI 2.429–2.763), predicting that, for every IU of XM17 administered, the AUC0–168h increases by ~2.596 IU·h/L (Figure 4B).
Figure 4.
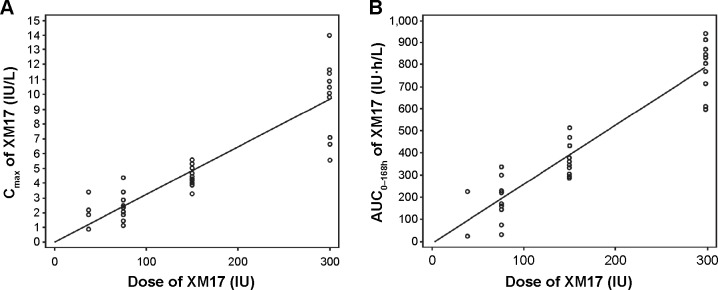
Individual pre-dose corrected values for XM17 Cmax (IU/L) (A) and AUC0–168h (IU·h/L) (B) versus XM17 dose (IU).
Abbreviations: Cmax, maximum XM17 concentration; AUC0–168h, area under the XM17 concentration-time curve from time zero to 168 hours post-dose.
Safety
All 40 subjects were included in the safety assessment, having each received both goserelin acetate to suppress endogenous FSH and a single dose of XM17. Adverse events were classified according to whether they occurred prior to or after administration of goserelin acetate, but before XM17 (baseline/run-in adverse events). They were classified as TEAEs if they occurred following administration of XM17 or increased in severity post-XM17 dosing.
Twenty-seven subjects (68%) experienced baseline/run-in adverse events that were not considered TEAEs. Twenty-one (53%) subjects experienced 33 TEAEs, of which 20 were deemed possibly related to XM17 administration. None were considered probably or definitely related to XM17.
Most TEAEs were mild (n=23), some moderate (n=10), and none severe. No serious adverse events were reported during the study, and no subject discontinued the study due to an adverse event or TEAE.
Headache (12 TEAEs in eleven subjects) and dizziness (four TEAEs in four subjects) were the most commonly reported TEAEs overall, and occurred with similar frequency among subjects in the three higher dose groups. Of these, headache and dizziness were considered by the investigators to be possibly related to XM17 in nine and two subjects, respectively. None of the other TEAEs (nasopharyngitis, gastroenteritis, hordeolum, urinary tract infection, lower abdominal pain, dyspepsia, acne, allergic dermatitis, hot flush, ear pain, injection site pain, drug hypersensitivity, pain in extremity, vulvovaginal pruritus) occurred in more than one subject per dose group.
Local tolerability of subcutaneous XM17 injection, assessed 1, 6, and 24 hours post-dosing, was good, with no reports of erythema or swelling at the injection site and two subjects reporting mild pain on touch, one at 1 hour (150 IU) and the other at 6 hours (300 IU) post-dose.
No XM17 antibodies were detected, and there were no clinically relevant laboratory, ECG, vital signs, and physical or gynecologic findings that would suggest any treatment-emergent effects.
Overall, XM17 was well tolerated, and the safety profile was comparable with published information available for Gonal-f®, an approved rhFSH.17
Discussion
Proteins manufactured by recombinant techniques are generally regarded as safe and may eventually replace human-derived proteins as the standard of care.1,4,5 Although these products are costly to make and add to the cost of ART procedures, evidence suggests that recombinant protein therapeutics may be cost-effective when compared with their human-derived counterparts.5,11
By and large, the surest method to improve the cost-effectiveness of medications used in ART and improve their accessibility for infertile couples is to simply make medications available at a lower cost. Having more products with clinically similar efficacy, safety, and tolerability will provide patients with options, and market competition will tend to drive down overall costs. Toward this end, introduction of other rhFSH products to treat human infertility should benefit ART patients and their caregivers.
Biosimilar products are similar to already authorized biological therapeutic agents and must undergo a comprehensive set of comparability tests to demonstrate their similarity with regard to physicochemical characteristics, efficacy, and safety. In this first-in-human Phase I study, we sought to demonstrate that XM17 is safe and well tolerated in healthy female subjects and that its PKs are dose-proportional.
Single ascending XM17 doses of 37.5, 75, 150, and 300 IU were successively administered to different subject groups after careful evaluation of safety and tolerability parameters at the lowest dose.
Mean maximum serum levels and AUC of XM17 increased with dose. Published clinical pharmacology data from le Cotonnec et al18 who administered single 150 IU subcutaneous doses of Gonal-f® to healthy downregulated female volunteers, reported a Cmax of 3 IU/L and an AUC0–∞ of 235 IU·h/L. These values were slightly lower than the present findings for XM17, ie, a Cmax of 4.42 IU/L and an AUC0–∞ of 448 IU·h/L. The tmax after subcutaneous injection of Gonal-f®150 IU was 16 hours,18 whereas the tmax for XM17 was 22 hours. However, the AUC0–∞ and other parameters derived from λZ, including t1/2, CL/F, and Vz/F, were not easily compared since the selection criteria for fitting the terminal elimination slope may have differed from the published study.
In the present study, a single subcutaneous dose of XM17 300 IU yielded a Cmax of 9.8±2.4 IU/L and an AUC0–t of 839.0±194.6 IU·h/L, which are very similar to data published for a 300 IU dose of Gonal-f® (Cmax 9.0±2.4 IU/L and AUClast 841±279 IU·h/L).19
Further, le Cotonnec et al3 reported that 150 IU of intravenously administered Gonal-f® had an apparent volume of distribution (noncompartmental analysis method) of 10 L and a clearance of 0.6 L/hour. Values for Vz/F and CL/F after subcutaneous administration were not reported. After 150 IU of subcutaneous XM17, the Vz/F was 28 L and the CL/F was 0.36 L/hour. Again, these parameters are not easily compared because of the different routes of administration and methods used to estimate the terminal elimination of rhFSH.
The authors of the present study recently completed a second study in which a direct comparison between XM17 and Gonal-f® was made using uniform and strict criteria to evaluate both products. All PK parameters (including t1/2 and AUC 200–∞ that rely on estimates of λZ) were nearly equal.
Importantly, the safety and tolerability profiles of XM17 at subcutaneous doses up to 300 IU in these healthy young women was very good compared with that known for Gonal-f®, the first approved follitropin alfa product with long-standing clinical experience.13 The Gonal-f ® summary of product characteristics lists headache, ovarian cysts, and injection site reactions, such as pain, erythema, hematoma, swelling, and/or irritation at the site of injection, as being common (>10% incidence).
In subjects treated with XM17, headache (27.5% of subjects) was the most frequently reported adverse event followed by dizziness in 10% of subjects. Headache and dizziness were deemed possibly attributable to XM17 in nine (22.5%) and two subjects (5%), respectively. Except for mild pain to touch in two subjects (5%), subcutaneous injection of XM17 was well tolerated, with no evidence of antibody development in any subject.
Single subcutaneous doses of XM17 up to 300 IU in healthy young women were associated with very good safety and tolerability and exhibited dose-proportional PKs. XM17, a European Medicines Agency-approved biosimilar to Gonal-f®, adds to the choices of infertility products and should eventually help lead to lower overall costs borne by individuals undergoing ART procedures.
Acknowledgments
The authors wish to thank Rebecca D Miles, PhD, of MedVal Scientific Information Services, LLC, Skillman, NJ, USA, for providing professional writing and editorial assistance. The professional writing and editorial assistance provided for this paper was funded by Teva Branded Pharmaceutical Products R&D, Inc. (Frazer, PA, USA). The study was sponsored by BioGeneriX AG, a member of the Teva group. Teva provided a full review of the article. The manuscript was prepared according to the International Society for Medical Publication Professionals’ “Good Publication Practice for Communicating Company-Sponsored Medical Research: the GPP2 Guidelines” and the International Committee of Medical Journal Editors’ “Uniform Requirements for Manuscripts Submitted to Biomedical Journals.” These data were presented at the European Society of Human Reproduction and Embryology, June 29 to July 2, 2014, Munich, Germany, and have previously been published in abstract form.21
Footnotes
Disclosure
AL, PB, and BG are employees of Merckle GmbH. BG was an employee of BioGeneriX AG. BioGeneriX AG (organization responsible for conducting the study) and Merckle GmbH are members of the Teva group.
References
- 1.Dhillon S, Keating GM. Lutropin alfa. Drugs. 2008;68(11):1529–1540. doi: 10.2165/00003495-200868110-00005. [DOI] [PubMed] [Google Scholar]
- 2.Durocher Y, Butler M. Expression systems for therapeutic glycoprotein production. Curr Opin Biotechnol. 2009;20(6):700–707. doi: 10.1016/j.copbio.2009.10.008. [DOI] [PubMed] [Google Scholar]
- 3.le Cotonnec JY, Porchet HC, Beltrami V, Khan A, Toon S, Rowland M. Clinical pharmacology of recombinant human follicle-stimulating hormone (FSH). I. Comparative pharmacokinetics with urinary human FSH. Fertil Steril. 1994;61(4):669–678. [PubMed] [Google Scholar]
- 4.Chambost H, Ljung R. Changing pattern of care of boys with haemophilia in western European centres. Haemophilia. 2005;11(2):92–99. doi: 10.1111/j.1365-2516.2005.01074.x. [DOI] [PubMed] [Google Scholar]
- 5.Daya S. Follicle-stimulating hormone in clinical practice: an update. Treat Endocrinol. 2004;3(3):161–171. doi: 10.2165/00024677-200403030-00004. [DOI] [PubMed] [Google Scholar]
- 6.van Wely M, Kwan I, Burt AL, et al. Recombinant versus urinary gonadotrophin for ovarian stimulation in assisted reproductive technology cycles. Cochrane Database Syst Rev. 2011;2:CD005354. doi: 10.1002/14651858.CD005354.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Recombinant Human FSH Product Development Group Recombinant follicle stimulating hormone: development of the first biotechnology product for the treatment of infertility. Hum Reprod Update. 1998;4(6):862–881. doi: 10.1093/humupd/4.6.862. [DOI] [PubMed] [Google Scholar]
- 8.Huirne JA, Lambalk CB, van Loenen AC, et al. Contemporary pharmacological manipulation in assisted reproduction. Drugs. 2004;64(3):297–322. doi: 10.2165/00003495-200464030-00005. [DOI] [PubMed] [Google Scholar]
- 9.Reichl H, Balen A, Jansen CA. Prion transmission in blood and urine: what are the implications for recombinant and urinary-derived gonadotrophins? Hum Reprod. 2002;17(10):2501–2508. doi: 10.1093/humrep/17.10.2501. [DOI] [PubMed] [Google Scholar]
- 10.Harrison S, Wolf T, Abuzeid MI. Administration of recombinant follicle stimulating hormone in a woman with allergic reaction to menotropin: a case report. Gynecol Endocrinol. 2000;14(3):149–152. doi: 10.3109/09513590009167674. [DOI] [PubMed] [Google Scholar]
- 11.Escobar MA. Health economics in haemophilia: a review from the clinician’s perspective. Haemophilia. 2010;16(Suppl 3):29–34. doi: 10.1111/j.1365-2516.2010.02257.x. [DOI] [PubMed] [Google Scholar]
- 12.European Medicines Agency Ovaleap (follitropin alfa) [assessment report] Jul 31, 2013. [Accessed February 25, 2015]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/002608/WC500152908.pdf.
- 13.Gonal-f® [summary of product characteristics] London, UK: European Medicines Agency; 2010. [Google Scholar]
- 14.Puregon [summary of product characteristics] London, UK: European Medicines Agency; 2006. [Google Scholar]
- 15.Bemfola (follitropin alfa) [summary of product characteristics] London, UK: European Medicines Agency; 2014. [Google Scholar]
- 16.Matta WH, Shaw RW, Burford GD. Endocrinologic and clinical evaluation following a single administration of a gonadotrophin-releasing hormone agonist (Zoladex), in a depot formulation, to premenopausal women. Fertil Steril. 1988;49(1):163–165. doi: 10.1016/s0015-0282(16)59670-8. [DOI] [PubMed] [Google Scholar]
- 17.Gonal-f® 75 IU (5.5 micrograms) powder and solvent for solution for injection [package insert] London, UK: Merck Serono Europe Ltd; 2012. [Google Scholar]
- 18.le Cotonnec JY, Porchet HC, Beltrami V, Khan A, Toon S, Rowland M. Clinical pharmacology of recombinant human follicle-stimulating hormone. II. Single doses and steady state pharmacokinetics. Fertil Steril. 1994;61(4):679–686. [PubMed] [Google Scholar]
- 19.Lugan I, Febbraro S, Lecuelle H, Papasouliotis O, Ho-Nguyena Q, Buraglio M. Bioequivalence of liquid and freeze-dried recombinant human follicle-stimulating hormone. Curr Med Res Opin. 2005;21(1):121–125. doi: 10.1185/030079904x18027. [DOI] [PubMed] [Google Scholar]
- 20.Gertz B, Lammerich A. Phase I Study to establish the bioequivalence of recombinant human follicle stimulating hormone (r-hFSH; XM17) to Gonal-f® (follitropin alfa) in downregulated healthy female subjects; Abstract presented at the 10th Congress of the European Society of Gynecology; September 18–21, 2013; Brussels, Belgium. [Google Scholar]
- 21.Gertz B, Bias P, Lammerich A. Phase I dose-ranging study of a recombinant human follicle-stimulating hormone (rhFSH) in healthy female volunteers. Hum Reprod. 2014;29(Suppl 1):i319–i320. Abstr P-486. [Google Scholar]