

Summary
Transcriptional mediators of cell stress pathways, including HIF1α, ATF4, and p53, are key to normal development and play critical roles in disease, including ischemia and cancer. Despite their importance, mechanisms by which pathways mediated by these transcription factors interact with each other are not fully understood. In addressing the controversial role of HIF1α in cardiomyocytes (CMs) during heart development, we have discovered a mid-gestational requirement for HIF1α for proliferation of hypoxic CMs, involving metabolic switching and a complex interplay between HIF1α, ATF4 and p53. Loss of HIF1α resulted in activation of ATF4 and p53, the latter inhibiting CM proliferation. Bioinformatic and biochemical analyses revealed unexpected mechanisms by which HIF1α intersects with ATF4 and p53 pathways. Our results highlight previously undescribed roles of HIF1α and interactions between major cell stress pathways that could be targeted to enhance proliferation of CMs in ischemia, and may have relevance to other diseases, including cancer.
Introduction
To fulfill their critical blood pumping activity, cardiomyocytes (CMs) acquire profound structural and biochemical modifications during their maturation. Mature CMs are withdrawn from the cell cycle, display an intricate contractile apparatus and contain large numbers of mitochondria required to provide, via oxidative phosphorylation (OXPHOS), sufficient ATP for contractile activity. In contrast to their mitotically-withdrawn adult counterparts, fetal cardiomyocytes (fCMs) have the capacity to both contract and proliferate. Understanding the pathways that regulate proliferation of fCMs will allow for a better understanding of congenital heart disease, the most prevalent human birth defect, and may also provide potential pathways for triggering adult CM regeneration.
To identify regulators of fCM proliferation, we undertook an in silico screen which suggested that Hypoxia Inducible Factor 1α (HIF1α), a transcription factor (TF) activated by low oxygen, mediates a pro-proliferative response during cardiac development. In well-perfused tissues, HIF1α subunits are hydroxylated and targeted for proteasomal degradation via VHL-mediated ubiquitination. Hypoxia (when [O2] < ~5%) disrupts HIF1α ubiquitination, leading to its stabilization and association with HIF1β (ARNT) to promote a pro-survival response based on induction of genes involved in processes such as energy metabolism and angiogenesis (Denko, 2008; Pugh and Ratcliffe, 2003). HIF1α is typically activated in poorly vascularized solid tumors and mutations in Vhl that disrupt HIF1α degradation cause von Hippel-Lindau disease, an inherited disorder characterized by predisposition to several tumors (Denko, 2008).
Under conditions of prolonged exposure to hypoxia, pro-survival effects of HIF1α can be blocked by activation of p53, a TF that promotes cell death and senescence (Schmid et al., 2004). p53 blocks cell cycle, at least in part, by inducing expression of members of the CIP/KIP family (Schmid et al., 2004). Proteins of this family (p21, p27 and p57) inhibit cyclin-dependent kinases and block G1/S progression (Kochilas et al., 1999). Activation of p53 blocks HIF1α signaling by at least two distinct mechanisms: p53-induced degradation of HIF1α, and competition for the limiting transcriptional co-activator, p300 (Schmid et al., 2004). On the other side of this antagonistic relationship, HIF1α transcriptional activity prevents post-translational activation of p53 by inducing expression of Mif (Fingerle-Rowson et al., 2003; Hudson et al., 1999; Welford et al., 2006).
Hypoxic stress also results in activation of PERK and consequent increased translation of ATF4, another major stress-responsive TF that plays a role in cancer by regulating redox balance, protein folding, amino acid uptake and metabolism and angiogenesis (Fels and Koumenis, 2006; Han et al., 2013; Harding et al., 2003; Roybal et al., 2005). Despite being induced in response to similar stimuli, how HIF1α and ATF4 pathways might mutually regulate each other remains unexplored.
In addition to a prominent role in tumorigenesis, oxygen levels play critical roles in cardiogenesis, as highlighted by the recent finding that hyperoxia, reactive oxygen species (ROS), and ensuing DNA damage induce neonatal CM cell cycle withdrawal (Puente et al., 2014). However, the role of hypoxia and HIF1α in mammalian cardiac development has been controversial and, thus, poorly understood. Three Hif1a null alleles, all targeting exon 2 that encodes the HIF1α DNA binding domain, were independently generated by distinct groups (reviewed by Dunwoodie, 2009). Regardless of the allele considered, mutant embryos die around E10 and display severe vascular and placental defects. Interestingly, phenotypic coherence is lost when cardiac phenotypes are analyzed. Two of the Hif1a null alleles produce mutants with underdeveloped hearts, while a third allele, Hif1atm1Jhu, produces mutants exhibiting myocardial hyperplasia (Dunwoodie, 2009).
The early lethality of global Hif1a KOs precludes study of potential roles of HIF1α in later stages of cardiogenesis. Furthermore, severe placental and vascular defects of global KOs make it difficult to ascertain whether reported cardiac defects are a primary or secondary phenotype. The generation of a fourth Hif1a allele in which exon 2 is flanked by loxP sites enabled spatial and temporal regulation of Hif1a ablation (Ryan et al., 2000). Using this floxed allele and the Mlc2v-Cre line, two groups independently ablated Hif1a in ventricular CMs. Here again, results were inconsistent. When two floxed Hif1a alleles were used, conditional knockouts did not present any developmental abnormalities (Huang et al., 2004). In contrast, when the breeding strategy included one floxed Hif1a allele and the hyperproliferative Hif1atm1Jhu null allele, mutant embryos exhibited myocardial hyperplasia and lethality between E11.5 and E12 (Krishnan et al., 2008).
Driven by our bioinformatics results, and to resolve current discrepancies in the literature, we investigated the role of HIF1α in murine cardiogenesis. Our results revealed previously undescribed spatiotemporal dynamics of HIF1α nuclear localization in developing myocardium that critically regulate a specific window of fCM proliferation prior to completion of cardiac angiogenesis. Using the Hif1a floxed allele (Ryan et al., 2000), we found that efficient depletion of HIF1α in fCMs could be achieved only by utilizing an early-acting Cre, Nkx2-5-Cre. Ablation of Hif1a with Nkx2-5-Cre resulted in embryonic lethality between E14.5 and E17.5, with mutant hearts exhibiting hypoplastic ventricles and ventricular septal defects (VSDs).
RNA-seq and genomewide ChIP-seq analyses of mutant and control hearts demonstrated that HIF1α regulated fCM proliferation by activation or repression of key downstream target genes. As observed in other contexts, we have found that, in fCMs, HIF1α coordinately represses and activates genes to effect metabolic switching to a glycolytic state, providing biosynthetic substrates that facilitate proliferation (Vander Heiden et al., 2009). Of note, we also defined a role for HIF1α in promoting cell proliferation by direct repression of cell cycle inhibitors of the CIP/KIP family. Additionally, our findings have revealed interplay between HIF1α and p53 pathways during cardiogenesis, as well as previously unsuspected interactions between HIF1α and the ATF4 cellular stress response. These interactions between major cellular stress pathways may be targeted to enhance proliferation of CMs in settings of pediatric cardiac hypoplasia or in adult ischemic disease and are likely to have relevance in the setting of other diseases, including cancer.
Results
Bioinformatics and comparative genomics analyses suggest HIF1α as a candidate regulator of fCM proliferation
We developed a two-step bioinformatics and comparative genomics analysis to identify TFs that might play a role in heart development and adult cardiac remodeling. We compared 148 Affymetrix microarrays of mouse ventricular tissue isolated from multiple physiological and pathological situations (Table S1) and clustered transcripts to identify those exhibiting similar expression dynamics (Figure 1A). Cluster 1 grouped genes highly expressed in developing heart, down-regulated perinatally, and re-induced, to a limited extent, in pathological situations. Functional annotation of this cluster revealed significant enrichment for transcripts encoding proteins involved in cell cycle regulation, DNA replication and cytokinesis (Figure 1A).
Figure 1. Bioinformatics prediction of transcription factors that regulate fetal cardiomyocyte proliferation.
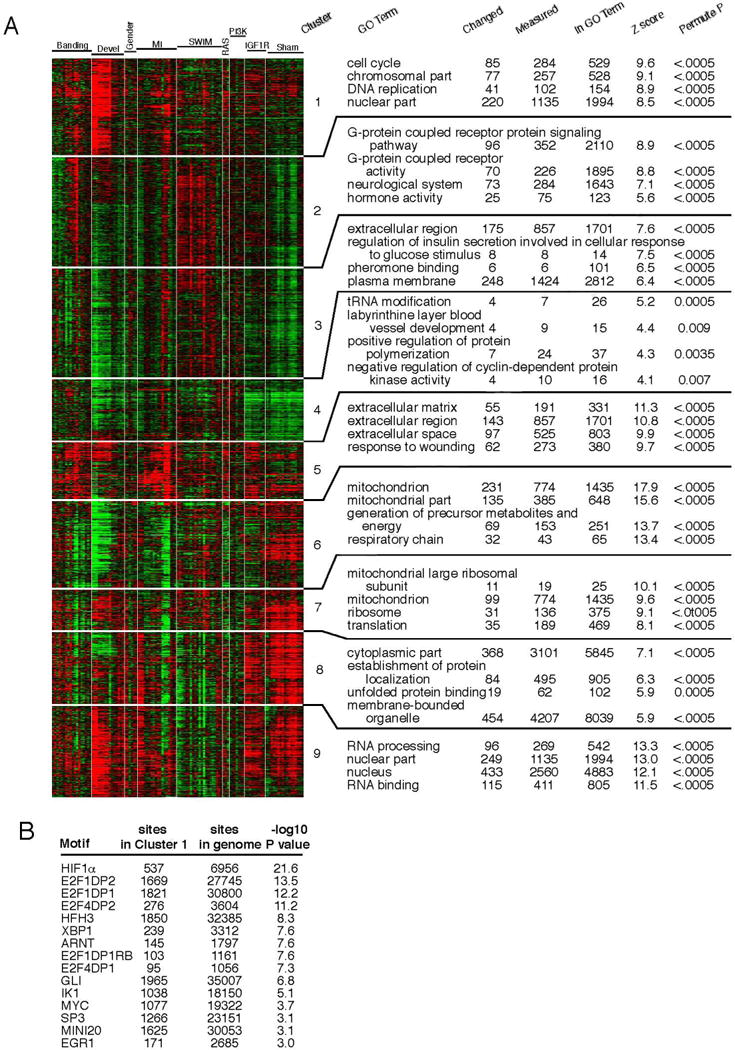
A) Based on common expression trends, transcripts differentially expressed in cardiac development or adult remodeling (physiological and/or pathological) were grouped into 9 distinct clusters. Enriched Gene Ontology (GO) terms revealed biological processes associated with each cluster. Cluster 1 contained genes associated with cell cycle, strongly expressed in development and attenuated perinatally. B) Analysis for enrichment of evolutionarily conserved TF binding sites within the proximal promoters of all Cluster 1 genes, revealing candidate TF regulators of fCM proliferation. See also Table S1.
Whole Genome RVista was then used to predict which TFs might control expression of these genes (Dubchak et al., 2013). For this analysis, the 5kb proximal promoter regions of all Cluster 1 genes were scanned for statistical enrichment of evolutionarily conserved (human/mouse) TF binding sites. Several TF binding motifs were highly enriched in these promoters, suggesting that corresponding binding TFs might regulate fCM proliferation (Figure. 1B). From these, HIF1α was the most significantly enriched binding site (P<10−22). HIF1α has been shown to promote, via Vegf induction, angiogenesis in adult hypertrophic remodeling (Sano et al., 2007) and in zebrafish is required for CM proliferation post ventricular amputation (Jopling et al., 2012). However, the role of HIF1α during cardiogenesis remains highly controversial (Dunwoodie, 2009).
Distinct temporal and spatial subcellular localization of HIF1α in fCMs
To investigate if HIF1α protein is present and therefore possibly playing a role in fCMs, immunohistochemistry for HIF1α was performed on embryonic mouse hearts at several stages of development. Antibody specificity was tested in global Hif1a KOs. A null allele of Hif1a, designated Hif1aΔ, was generated by floxing-out the conditional Hif1a allele (Ryan et al., 2000) and, as expected, breeding of Hif1aΔ/WT heterozygotes produced no viable Hif1aΔ/Δ progeny. Analysis of timed pregnancies revealed no homozygous mutants from E10.5 onwards. E9.5 mutants were much smaller than littermates, exhibiting reduced proliferation, markedly increased apoptosis of neuronal and mesenchymal tissues and an extremely small, unlooped heart (Figure S1 A–F). Histological analyses of these mutants with anti-HIF1α antibody produced no detectable signal, in contrast to results from wildtype littermates, demonstrating antibody specificity (Figure S1C and D).
The recently looped E9.5 heart is composed of an inner layer of endocardial cells surrounded by a single layer of fCMs. At this stage, robust levels of HIF1α protein were detected in the developing heart, but, intriguingly, most HIF1α was retained outside the nucleus, regardless of the cell type considered (Figure 2A–C).
Figure 2. Spatiotemporal dynamics of HIF1α protein during cardiogenesis.
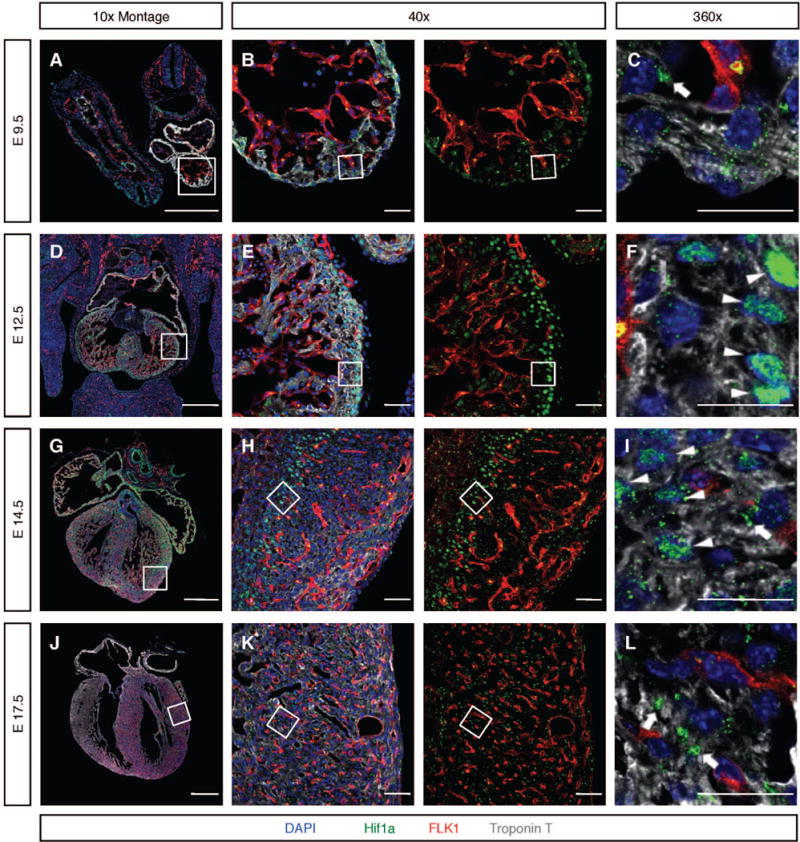
A–C) In E9.5 fCMs, HIF1α was retained in extra-nuclear aggregates (arrow in C). D–F) At E12.5, HIF1α accumulated in nuclei of fCMs located in non-vascularized areas of the myocardium, including fCMs of the inner core of the interventricular septum (IVS), and outermost layers of the compact wall (arrowheads) G–I) At E14.5, the coronary vascular system has colonized most of the ventricular free walls. At this stage, most fCMs displayed cytoplasmic HIF1α (arrow in I) and only a small fraction of fCMs located between the endocardium and the leading edge of the progressing coronary vasculature exhibited significant levels of nuclear HIF1α (boxed area in H and arrowheads in I). J–L) At E17.5, the coronary vascular system has colonized the entire heart and HIF1α was no longer detectable in the nucleus of fCMs, being present only in cytoplasmic aggregates (arrows in L). To allow better visualization of the location of HIF1α relative to the coronary endothelium, all 40× images are shown in two distinct panels: a four-color merge and a red and green only merge. Boxed areas are imaged in higher magnifications. Bars represent 500μm in the 10× montages and 20 μm in all other panels. See also Figure S1.
At E12.5 the interventricular septum (IVS) is relatively well developed and the ventricular free walls are composed of two distinct domains, the trabeculae and the compact zone. At this stage, robust levels of HIF1α were detected in nuclei of fCMs located in the core of the developing septum and outermost layers of the compact zone, but not in nuclei of trabecular fCMs or fCMs located in the innermost layers of compact myocardium (Figure 2D–F).
From E12.5 endothelial precursors of the coronary vasculature progressively invade the myocardial wall, connecting to the aorta around E14.0 to perfuse developing myocardial walls with oxygenated blood (Figure S1G and H). This angiogenic process is gradual and takes place in an epicardial-to-endocardial direction. At E14.5, in regions of the myocardial wall adjacent to endocardium or containing coronary vasculature, myocardial HIF1α was observed outside the nucleus in cytoplasmic aggregates (Figure 2G–I). Nuclear HIF1α, however, was consistently observed in fCMs within non-vascularized regions of myocardium (arrowheads in Figure 2I). At E17.5 the coronary vascular system has colonized the entire heart and HIF1α was detected in cytoplasmic aggregates (Figure 2J–L).
Correlation between timing of perfusion of the coronary wall (as assessed by fluorescent microsphere injection, Figure S1G–H) and spatiotemporal dynamics of nuclear accumulation of HIF1α in fCMs (Figure 2), strongly suggested availability of oxygenated blood as a major regulator of HIF1α subcellular localization during cardiogenesis, without excluding other factors from playing a role in this process. Importantly, at developmental timepoints in which strong nuclear HIF1α could be detected, nuclear presence of this TF correlated with high fCM proliferation rates (Figure S1I–L). Notably, fCMs were the only cell type of the developing heart that displayed robust nuclear localization of HIF1α. Epicardium, endocardium, and mesenchymal cells of the cushions exhibited accumulation of HIF1α in cytoplasmic clusters.
Early myocardial ablation of Hif1a produces severe cardiac defects and embryonic lethality
Following our bioinformatics prediction of a role for HIF1α in promoting fCM proliferation and the observation of nuclear HIF1α in highly proliferative subsets of midgestational fCMs, we investigated the potential role of this TF in fCM proliferation. As we had observed HIF1α protein present in heart from as early as E9.5 and as HIF1α is stabilized in hypoxic conditions, we hypothesized that early and efficient myocardial ablation is critical for effective loss of HIF1α in fCMs. Commonly used myocardial Cres are Nkx2-5-Cre, αMhc-Cre, or Mlc2v-Cre, with expression of each Cre being activated at distinct developmental timepoints. As demonstrated in Figure S2 A–F, we found that the Nkx2-5-Cre was the only Cre that effectively activated a reporter throughout the myocardium by E8.5. At the same stage, αMhc-Cre or Mlc2v-Cre produced much weaker and mosaic reporter activation. We utilized both αMhc-Cre and Nkx2-5-Cre to ablate the floxed Hif1a allele (Ryan et al., 2000) in fCMs. To ensure the most efficient recombination possible, the null Hif1aΔ allele, generated by germline ablation of the floxed allele, was included in all crosses.
Results revealed that αMhc-Cre; Hif1aΔ/fl mutants were viable, born at expected Mendelian ratios, and displayed normal survival at 3 months (Table 1). In contrast, ablation of Hif1a driven by Nkx2-5-Cre resulted in embryonic lethality, with only 3 out of an expected 21 mutants found dead at P0, and no mutants recovered at weaning. Consistent with these results, histological analyses of E13.5 embryos revealed that Nkx2-5-Cre, but not αMhc-Cre, resulted in effective depletion of HIF1α protein in developing fCMs (Figure S2 G–J). At E13.5, even in Nkx2-5-Cre; Hif1aΔ/fl mutants (hereafter simply designated as Hif1a cKOs), only 73% (8 out of 11) of the mutants displayed a complete knockout, with the remaining 27% exhibiting reduced, but not absent, HIF1α (Figure S2 I and J).
Table 1.
List of Cre lines used to promote conditional KO of Hif1a, their target cellular populations, and resulting genotype frequencies at birth.
| Genotype Distribution (%) at birth | |||||||
|---|---|---|---|---|---|---|---|
| Cre Positive | Cre Negative | ||||||
| Cre | Target Population | Number of Litters | Number of Animals | Hif1aΔ/fl | Hif1aWT/fl | Hif1aΔ/fl | Hif1aWT/fl |
| αMhc | Myocardium | 9 | 71 | 32.39 | 26.76 | 18.31 | 22.54 |
| Nkx2-5 | Myocardium | 15 | 83 | 3.61* | 27.71 | 34.94 | 33.73 |
| Wt1 | Epicardium and Derivatives | 7 | 41 | 26.83 | 24.39 | 26.83 | 21.95 |
| Tie2 | Endocardium; Cushions; Endothelium | 7 | 57 | 24.56 | 31.58 | 24.56 | 19.30 |
Pups found dead at P0.
In comparison with stage-matched control littermates, E12.5 Hif1a cKOs with efficient HIF1α protein depletion displayed smaller IVS (Figure 3A). At E14.5, in addition to prominent VSDs, mutant hearts exhibited thinner ventricular walls (Figure 3A–C). These defects were accompanied by a significant decrease in the rate of fCM proliferation as assessed by EdU incorporation (Figure 3D). Quantification of EdU-positive fCMs demonstrated that in wild-type septa and left ventricular compact myocardium approximately 30% of fCMs underwent DNA synthesis through the course of a two-hour period. In mutant hearts these rates significantly decreased to 18.35 +/− 1.33% in the septum and 22.76 +/− 1.87% (P<0.01) in the ventricular compact wall (Figure 3E). No changes in apoptosis were detected by cleaved Caspase 3 in mutant hearts (Figure 3F). Consequent to these defects, the majority of Hif1a cKOs died between E14.5 and E17.5 (Table 2). At E17.5, out of 12 expected, only 3 (25%) mutants were recovered alive, all exhibiting abnormal ventricular architecture (Figure S2K–N). Similar frequencies of embryos with incomplete HIF1α depletion at E13.5 and Hif1a cKOs recovered at E17.5 (27% and 25%, respectively) suggested that mutants that reached this late gestational stage were embryos in which Nkx2-5-Cre activity had not resulted in effective depletion of HIF1α.
Figure 3. Efficient ablation of HIF1α in fCMs results in hypoplasia and arrested cardiac development.
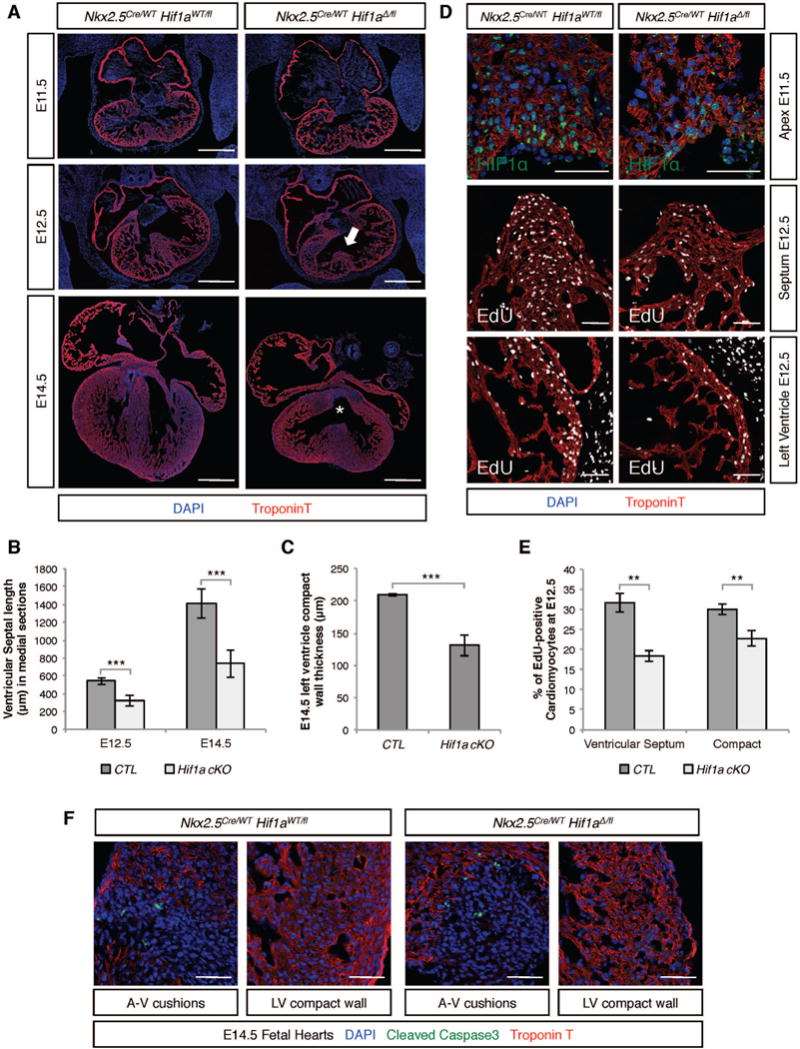
A) At E12.5, efficient depletion of HIF1α mediated by Nkx2-5-Cre resulted in shortened interventricular septa (IVS) (arrow in middle panels). At E14.5, (bottom panels), mutant hearts exhibited ventricular septal defects (asterisk) and hypoplastic ventricles. B) Quantification of septal length in medial sections of control and cKO hearts. C) Quantification of left ventricular wall thickness in E14.5 hearts. D) E11.5 was the first stage at which a clear decrease in HIF1α protein levels was identified in Hif1 cKO hearts (top panels). At E12.5, lack of HIF1α resulted in markedly lower numbers of EdU positive nuclei (two hour pulse) in the IVS (middle panel) and compact ventricular myocardium (lower panel). E) Quantification of proliferative defects in distinct compartments of E12.5 hearts as a ratio of EdU positive fCMs/Total fCMs. F) Apoptotic mesenchymal cells were equally abundant in the atrioventricular (A–V) cushions of control and mutant hearts, but no evidence of apoptotic fCMs was detected in the compact wall of either genotype. Bars represent 500μm in A and 50 μm in all other panels. LV=left ventricle. B, C and E: data represented as mean ± SD; ***P<0.001; **P<0.01. See also Figure S2.
Table 2.
Genotype frequencies, at distinct stages of embryonic development, for conditional KO of Hif1a by Nkx2-5-Cre.
| Genotype Distribution (%) | ||||||
|---|---|---|---|---|---|---|
| Stage | Number of Litters | Number of animals | Cre Positive | Cre Negative | ||
| Hif1aΔ/fl | Hif1aWT/fl | Hif1aΔ/fl | Hif1aWT/fl | |||
| E12.5 | 18 | 129 | 22.48 | 33.33 | 24.81 | 19.38 |
| E13.5 | 6 | 52 | 21.15 | 15.38 | 36.54 | 26.92 |
| E14.5 | 5 | 34 | 29.41 | 23.53 | 23.53 | 23.53 |
| E17.5 | 7 | 49 | 6.12 | 32.65 | 24.49 | 36.73 |
| Total | 36 | 264 | ||||
As Nkx2-5-Cre ablates HIF1α protein in fCM and non-fCM populations within the heart, it was possible that non-fCM-specific effects could contribute to the lethal phenotype. However, the absence of nuclear accumulation of HIF1α in non-fCM cell populations throughout the relevant developmental period suggested that the observed phenotype resulted from a fCM-autonomous requirement for HIF1α. Supportive of this conclusion, we utilized Wt1-Cre and Tie2-Cre to generate epicardial- and endocardial-specific Hif1a conditional knockouts, neither resulting in lethality (Table 1).
Gene expression profiling of Hif1a cKO hearts
To identify molecular pathways responsible for reduced fCM proliferation in Hif1a cKOs, we performed RNA sequencing (RNA-seq) analysis of RNA extracted from E12.5 hearts of Nkx2-5WT/Cre; Hif1aΔ/fl mutants and Nkx2-5WT/Cre; Hif1aWT/fl littermate controls. From this analysis, 14502 protein coding genes were expressed in both mutant and control hearts. Of these, 494 and 957 were significantly (|Fold| > 20% and P<0.05) up- or down-regulated, respectively, in mutant hearts relative to controls (Figure 4A and Table S2A and B). Clustering of differentially expressed genes by molecular function using the Reactome database enabled an overview of known cellular pathways most severely altered in mutant hearts. Consistent with known roles of HIF1α, some of the most significantly downregulated categories in mutant hearts were associated with glycolysis and glucose metabolism (Figure 4B and Table S2F). Less expected was observed enrichment for processes associated with extracellular matrix (ECM) organization.
Figure 4. Identification of direct HIF1α targets in E12.5 fCMs.
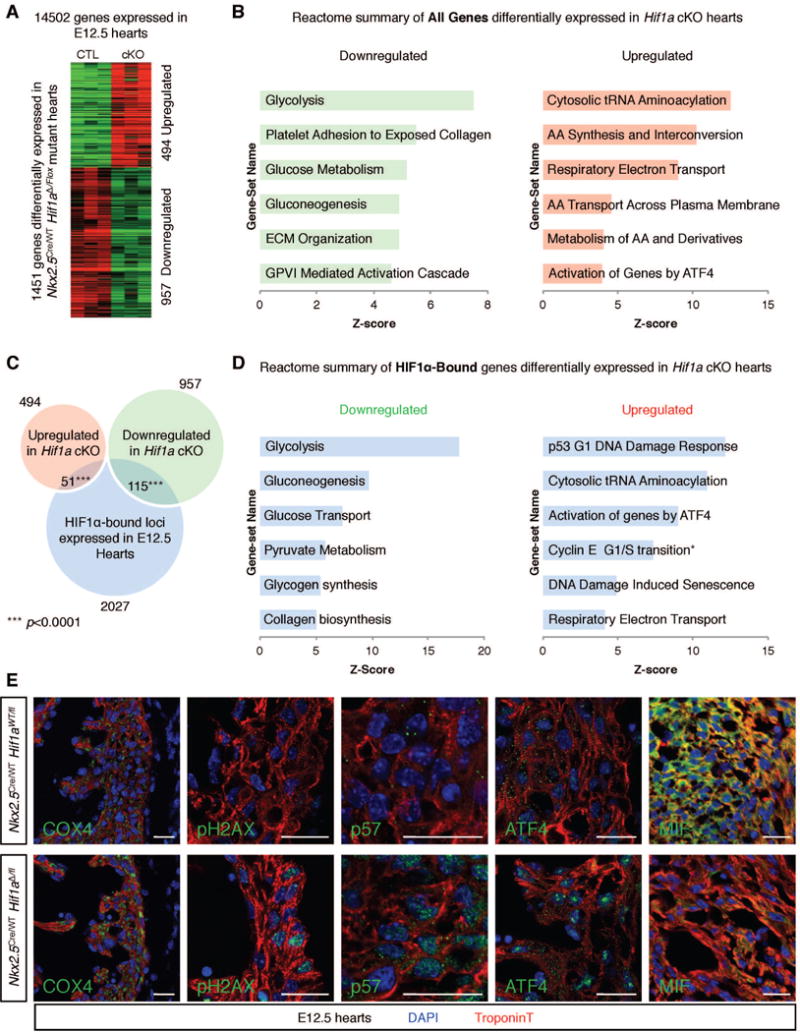
A) Comparison of control and Hif1α cKO E12.5 cardiac transcriptomes (n=3 for each genotype) unveiled a total of 1451 genes modulated in mutant hearts. B) REACTOME database functional clustering of genes down- and up-regulated in cKOs, highlighting cellular processes most significantly affected in mutant hearts. C) Overlay of RNA-seq and ChIP-seq results revealed 166 likely direct targets of HIF1α. D) REACTOME functional clustering of these genes allowed for identification of cellular functions directly regulated by HIF1α. *category containing the G1/S transition inhibitor p21. E) Immunostaining validation of RNA-seq results in the left ventricular free wall or IVS (MIF) of E12.5 hearts. Bars represent 20 μm. AA=amino acid; ECM=extracellular matrix; GPVI=glycoprotein VI. See also Figure S3 and Tables S2, S3 and S4.
Similar analysis of upregulated genes (Figure 4B and Table S2F) demonstrated striking enrichment of categories related to tRNA aminoacylation, amino acid synthesis and interconversion, amino acid transport, unfolded protein response and respiratory electron transport.
Identification of direct targets of HIF1α in E12.5 hearts
Having determined a wide array of genes displaying altered expression upon HIF1α depletion, direct HIF1α targets were predicted by integrating transcriptomic results with genome-wide assessment of HIF1α binding sites in wild-type E12.5 hearts (ChIP-seq). Two independent ChIP-seq experiments resulted in 17289 peaks significantly enriched over input reads, indicating putative HIF1α binding sites across the genome (Table S3). Motif analysis of these peaks demonstrated that HIF1α motifs were the most significantly enriched sequence (p=10−281), indicating successful pulldown of HIF1α-bound loci.
Intragenic peaks with a minimum peak score of 3.5 were assigned to the protein-coding gene with the nearest transcriptional start site. Filtering for genes actively expressed in E12.5 hearts resulted in a list of 2027 genes (Table S2C). To identify direct HIF1α targets (genes bound by HIF1α and modulated upon HIF1α depletion) this list was compared with the list of 1451 genes regulated in mutant hearts. This analysis suggested 115 genes directly activated and 51 genes directly repressed by HIF1α (Figure 4C and Table S2D,E). The fact that HIF1α activated more than twice as many genes as it repressed confirmed this TF acts preferentially as a transcriptional activator in fetal heart. Thirty percent of downregulated and 39.2% of upregulated HIF1α targets did not contain a minimal HIF binding motif in the associated peak (Figure S3A), suggesting that HIF1α-mediated gene regulation is not always dependent on the presence of a classic hypoxia-responsive element (HRE), consistent with previous observations (Tanimoto et al., 2010).
To gain insight into co-factors other than HIF1β that might be cooperating with HIF1α to either activate or repress gene expression, the vicinities (+/− 100 bp) of HIF1α ChIP-seq peaks corresponding to targets modulated in Hif1a cKO hearts were screened for enrichment in known transcription factor binding sequences (Table S4). This analysis was independently performed for peaks with and peaks without HRE. Among peaks containing an HRE, enrichment in SMAD binding sites was observed regardless of the nature of regulation promoted by HIF1α. Enrichment in binding sites for Sp1, a known HIF1α partner (Miki et al., 2004), was observed in peaks corresponding to genes activated by HIF1α. Binding sites for ATF4 were the most enriched motif in peaks corresponding to targets repressed by HIF1α.
Among peaks devoid of an HRE, a significant enrichment in binding sites for members of the ETS family was observed, regardless of the nature of regulation exerted by HIF1α. Interestingly, among peaks corresponding to genes activated by HIF1α, a significant enrichment in binding sites of cardiac specific transcription factors (MEF2c, MEF2a and GATA4) was observed, suggesting that HIF1α may associate with tissue specific transcription factors to activate expression of direct targets.
HIF1α directly regulates glycolysis, OXPHOS and collagen fibrillogenesis
Perhaps not surprisingly, in view of known roles of HIF1α in other cell types (Denko, 2008), functional annotation of direct targets positively regulated by HIF1α (Figure 4D) revealed a pivotal role in the regulation of energy metabolism. Examination of direct targets listed in Table S2D revealed that HIF1α regulated the glycolytic potential of fCMs by direct activation of 13 enzymes or carriers directly involved in each step of glycolysis (Slc2a3, Hk2, Pfkl, Pfkp, Aldoa, Aldoc, Gapdh, Tpi1, Pgk1, Pgam1, Eno1, Pkm2 and Ldha, Figure S3B and C). In addition to the critical role in promoting glycolysis, detailed analysis of direct HIF1α targets modulated in cKO hearts (Table S2D,E) demonstrated that HIF1α acted at multiple levels to reduce mitochondrial biomass and regulate OXPHOS potential of hypoxic fCMs (Table S2G). These included activation of the Mxi1 gene, encoding a known inhibitor of C-MYC-mediated mitochondrial biogenesis (Zhang et al., 2007). HIF1α-mediated induction of glycolysis and reduction in OXPHOS primes hypoxic fCMs to adapt their energy metabolism to low oxygen tension. These adaptations are critical for sustained ATP production and to avoid excessive ROS production by oxygen-depleted mitochondria (Semenza, 2013). Comparison of control and mutant E12.5 left ventricular compact walls by immunohistochemistry confirmed that Hif1a cKO fCMs failed to adapt their OXPHOS potential to hypoxic conditions, displaying increased mitochondrial biomass (anti-COX IV staining) and increased DNA damage, as revealed by anti-pH2AX staining (Figure 4E).
Consistent with recent observations in fibroblasts (Gilkes et al., 2013), our results indicated that, in fCMs, HIF1α directly promoted expression of two genes encoding enzymes required for collagen fiber assembly: Plod2 and P4ha1.
Interestingly, although our ChIP-seq data suggested that HIF1α bound to Vegf and Epo loci, RNA-seq indicated that the expression of these genes was not changed in Hif1a cKO hearts (Figure S3D).
HIF1α directly represses genes encoding cell cycle inhibitors and effectors of a cell stress response
Of direct relevance to observed fCM hypoplasia in Hif1a cKOs, functional clustering of the 51 direct targets upregulated in mutants, thus normally repressed by HIF1α in WT hearts, resulted in a significant enrichment for genes associated with cell cycle regulation (Figure 4D). Examination of the complete list (Table S2E) suggested HIF1α directly repressed expression of three well characterized cell cycle inhibitors: Cdkn1a (p21), Cdkn1c (p57) and Tob2. Further analysis of direct HIF1α targets upregulated in mutant hearts also demonstrated enrichment for genes encoding critical effectors of the unfolded protein response: Atf4, Herpud1 and Ppp1r15a (Figure 4D and Table S2E). Immunofluorescence analysis of the compact ventricular wall of E12.5 hearts confirmed upregulation and nuclear accumulation of p57 and ATF4 in Hif1a cKO fCMs (Figure 4E).
Multiple genes aberrantly upregulated in Hif1a cKO hearts are ATF4 targets
ChIP-seq results showed that HIF1α binds to the Atf4 locus in fCMs and RNA-seq and immunostaining analyses demonstrated that Hif1a cKO hearts displayed increased transcription of Atf4 mRNA and increased levels of ATF4 protein, placing Atf4 as a direct target of transcriptional repression by HIF1α. To examine the contribution of ATF4-mediated transcription to the aberrant transcriptome of Hif1a cKO hearts, we directly compared our RNA-seq results with previously published ChIP-seq data for ATF4. No cardiomyocyte specific ATF4 ChIP-seq is available. However, as ATF4 targets similar sets of genes in multiple cellular contexts (Kilberg et al., 2009), we compared our RNA-seq results with ATF4 targets as defined by ChIP-seq performed in mouse embryonic fibroblasts (Han et al., 2013). Out of 494 genes upregulated in Hif1a cKO hearts, 77 corresponded to loci previously reported to be bound by ATF4 (Figure 5A and Table S5A). Considering the total of 413 loci bound by ATF4 (ChIP-seq), one would expect the presence, just by chance, of 12 ATF4 targets in any list of 494 genes randomly generated from the 14502 genes expressed in E12.5 hearts. The observed 77 genes represent a very significant enrichment in ATF4 targets amongst genes upregulated in Hif1a cKOs (P<0.0001) and Reactome analysis of these revealed striking similarity to results for all genes upregulated in Hif1a mutant hearts (Figure 4B and 5B), suggesting that ATF4-mediated transcription contributes significantly to the aberrant mutant transcriptome. Notably, out of the 77 direct ATF4 targets upregulated in Hif1a cKO hearts, 17 were also bound by HIF1α (Table S5E), suggesting that, in addition to directly blocking Atf4 expression, HIF1α may also directly regulate direct targets of ATF4 activation. Consistent with the general perception of ATF4 as a transcriptional activator, and not a repressor (Han et al., 2013), overlap between genes downregulated in Hif1a cKO hearts and ATF4 targets was not significant (15 genes observed versus 23 expected by chance, Figure 5A).
Figure 5. Intersections between HIF1α, ATF4 and p53.
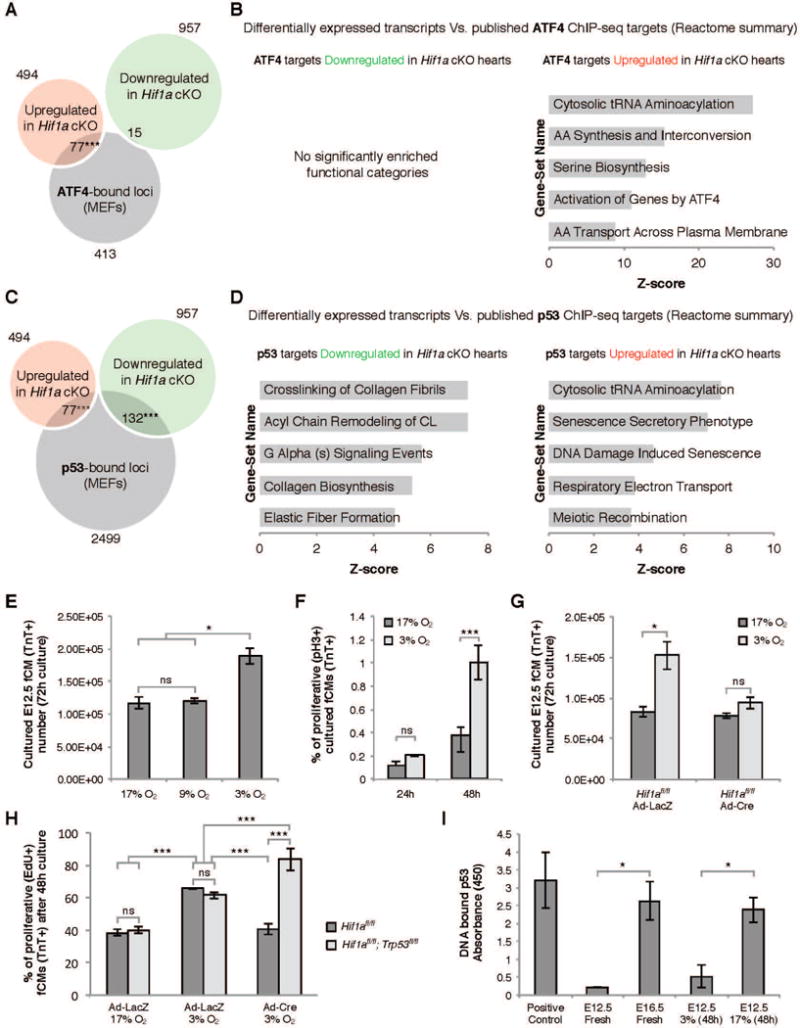
A) Overlap between genes modulated in Hif1a cKO hearts and ATF4 ChIP-seq data revealed a significant enrichment in ATF4 targets amongst upregulated genes. B) REACTOME functional clustering of ATF4 targets modulated in Hif1a cKO hearts. C) Overlap between genes modulated in Hif1a cKO hearts and p53 ChIP-seq data revealed a significant enrichment in p53 targets amongst both down and upregulated genes. D) REACTOME functional clustering of p53 targets modulated in Hif1a cKO hearts. E) Ex vivo culture of primary E12.5 ventricular cells in different oxygen concentrations revealed that hypoxia (3% O2) promoted an increase in fCM number. This effect was associated with increased proliferation of TroponinT-positive fCMs (F) and was dependent on HIF1α (G). H) Blunted proliferation resulting from absence of HIF1α was further demonstrated by quantification of EdU-positive fCMs and could be rescued by the simultaneous ablation of HIF1α and p53. I) Elisa quantification of p53-DNA binding revealed that during cardiogenesis p53 and HIF1α activity inversely correlated. E–I: data represented as mean ± SD; *P<0.05; ***P<0.05. See also Figure S4 and Table S5.
Intersection between HIF1α and p53 pathways
Functional clustering of direct targets repressed by HIF1α using Reactome was enriched in classes associated with signaling downstream of p53 (Figure 4D), suggesting a regulatory interaction between these two transcription factors. In contrast to ATF4, p53 did not appear to be transcriptionally regulated by HIF1α, as its mRNA levels were unchanged in Hif1a cKO hearts and our ChIP-seq assay provided no evidence for HIF1α binding to the Trp53 locus or vicinity. However, the gene encoding MIF, a well-characterized inhibitor of p53 post-translational activation (Fingerle-Rowson et al., 2003; Hudson et al., 1999; Welford et al., 2006), was directly bound by HIF1α (Figure S3E) and downregulated in Hif1a cKO hearts, both at RNA and protein levels (Table S2D, Figure S3E and Figure 4E). The combined actions of DNA damage (Schmid et al., 2004) and Mif downregulation are likely to trigger ectopic p53 activation.
Following a similar informatics strategy to the one described for ATF4, we looked for significant enrichment of direct targets of p53 in the list of 1451 genes modulated in Hif1a cKO hearts by comparing our RNA-seq results with previously published ChIP-seq data for p53 in mouse embryonic fibroblasts (Kenzelmann Broz et al., 2013). Consistent with known activity of p53 as a transcriptional repressor and activator (Schmid et al., 2004), this analysis resulted in significant (P<0.0001) intersection of direct targets of p53 with genes that were both upregulated (77 observed versus 39 expected by chance) and downregulated (132 observed versus 77 expected by chance) in Hif1a cKOs (Figure 5C and Table S5B and C).
Targets of p53 upregulated in Hif1a cKO hearts corresponded to functional classes associated with tRNA aminoacylation, senescence and respiratory electron transport (Figure 5D). Examination of the full gene list (Table S5B) suggested that, in HIF1α-depleted fCMs, activated p53 may promote transcription of genes encoding four cell cycle blockers: p21, p57, GADD45α and TOB2. Of these, p21 and GADD45α are well-established targets activated by p53 (Schmid et al., 2004).
Targets of p53 downregulated in Hif1a cKO hearts showed enrichment in categories containing ECM proteins (Col1a1, Col5a2, Col27a1, Fbln5, Lama2, Lamc2) and their remodeling enzymes (Lox, Plod2, P4ha1, Adamts2) (Figure 5D). Whether these genes are direct targets of repression by p53 has not been previously investigated, but our data is consistent with this being the case. Importantly, overlap between HIF1α and p53 targets (Table S5F) highlighted the fact that both TFs seem to regulate critical targets involved in cell cycle control (Cdkn1a, Cdkn1c, Tob2) and ECM fiber assembly (Plod2, P4ha1).
HIF1α and p53 exert rheostatic control over fCM proliferation
The foregoing analyses suggested that the hypoplastic phenotype of Hif1a cKOs, was, at least in part, owing to molecular dynamics between HIF1α and p53. To gain mechanistic insight into these interactions, we established a primary culture system to mimic in vivo conditions. E12.5 mouse ventricles were dissociated into single cell suspensions, cultured for 72 hours in different oxygen conditions and assessed for proliferation. Culture in hypoxia (3% oxygen), but not 9% or 17% oxygen, resulted in a significant increase in fCM numbers (Figure 5E) that was associated with an increase in the percentage of proliferating (pH3-positive) TroponinT-positive fCMs (Figure 5F).
Cells were then isolated from E12.5 Hif1afl/fl ventricles and transduced with control LacZ-expressing adenovirus (Ad-LacZ) or Cre-expressing adenovirus (Ad-Cre) to ablate Hif1a ex vivo. qPCR and immunocytochemistry analyses demonstrated that Ad-Cre efficiently excised the floxed Hif1a allele, promoting depletion of HIF1α protein and molecular alterations that recapitulated those observed in Hif1a cKO hearts, including ATF4 upregulation and increased mitochondrial content and DNA damage (Figure S4A–K). Increase in fCM numbers induced by culture in 3% O2 was observed in Ad-LacZ, but not in Ad-Cre transduced cells, demonstrating that the hypoxia-induced proliferation of fCMs was HIF1α dependent (Figure 5G). This observation was confirmed by evaluation of mitotic rates following a 48-hour culture in the presence of 2.5 μM EdU. Approximately 38% of Ad-LacZ treated fCMs exhibited evidence of DNA replication when cultured in 17% O2. Culture in 3% O2 significantly increased this to 63% and Ad-Cre mediated Hif1a depletion reverted the rate to 40% (Figure 5H). Importantly, simultaneous knockout of Hif1a and Trp53 restored fCM proliferation, indicating that HIF1α drives proliferation in hypoxia, at least in part, by antagonism of p53 activity (Figure 5H). Similar results were obtained in siRNA knockdown experiments (Figure S4L–M).
The idea that HIF1α promotes proliferation in hypoxia by preventing p53 activation was further supported by ELISA quantification of p53-DNA binding activity in extracts from developing hearts at distinct stages (Figure 5I). These data revealed that, during cardiogenesis, p53 activity inversely correlated with nuclear localization of HIF1α (Figure 2), with p53-DNA binding being 10 fold higher at E16.5 than at E12.5. Interestingly, culture of E12.5 fCMs for 48h in 3% O2 resulted in p53 activity levels comparable to those found in freshly isolated E12.5 hearts, whereas culture in 17% O2 increased p53 activity 5-fold, to levels similar to those observed in E16.5 hearts (Figure 5I).
Discussion
Hypoxia-mediated signaling has been a target of extensive analyses in multiple contexts, but the role of HIF1α in mammalian cardiogenesis has remained controversial. Here, we presented a series of experiments that demonstrated that HIF1α is critical for mid-gestational proliferation of a distinct subset of fCMs. Prompted by an in silico screen (Figure 1), we described a highly dynamic pattern of HIF1α accumulation in the nucleus of a subset of fCMs from E11.5 to E15 (Figure 2), providing a detailed examination of the localization of HIF1α at a subcellular level in embryonic heart and unveiling a direct correlation between nuclear HIF1α and the lack of an available blood supply (Figure S1G and H).
The null Hif1aΔ allele used in our studies was generated by germline excision of an exon encoding the DNA binding domain, with no exogenous DNA sequences retained in the engineered locus. Homozygous Hif1aΔ/Δ embryos exhibited myocardial hypoplasia, further supporting the idea that the hyperplastic heart observed utilizing the Hif1atm1Jhu allele might be an artifact arising from the retention of a PGK-Neo cassette, an element known to introduce significant phenotypic variability (Dunwoodie, 2009).
Efficient cardiac-specific HIF1α depletion was only achieved using a very early myocardial recombinase (Nkx2-5-Cre) and resulted in decreased fCM proliferation, hypoplastic ventricles and VSDs (Figure 3). In contrast, ablation with a later-acting Cre, αMhc-Cre, did not result in effective depletion of HIF1α protein, with the result that embryos with “mutant” genotypes were born at expected ratios and able to survive to adulthood.
Results from our characterization of control and mutant transcriptomes, in conjunction with HIF1α ChIP-seq analyses (Figure 4), showed that, in fCMs, HIF1α acted preferentially as a transcriptional activator (115 direct targets downregulated in mutant hearts), but also worked as a direct transcriptional repressor (51 direct targets upregulated in mutant hearts). Interestingly, in 33% of candidate direct targets, HIF1α binding to DNA was independent of an HRE. This percentage is similar to ones reported in previous HIF1α CHIP-seq studies (Tanimoto et al., 2010). In addition to direct target regulation, HIF1α can indirectly regulate gene expression programs by inducing transcription of molecules (Mxi1 and Mif) that repress the activity of other TFs (C-MYC and p53, respectively) (Welford et al., 2006; Zhang et al., 2007). Schematic summaries of mechanistic interactions are provided in Figures 6 and S5. Although intersection of HIF1α ChIP-seq with RNA-seq data yielded major pathways directly regulated by HIF1α in fCMs, as confirmed by immunostaining of tissue sections and cultured fCMs, it is possible that some genes with altered expression in Hif1a cKOs as detected by our RNA-seq dataset may reflect secondary alterations in non-fCM transcriptomes, owing to crosstalk between affected fCMs and their neighboring cells.
Figure 6. Model for interactions between HIF1α, ATF4 and p53 in the response to hypoxic stress.
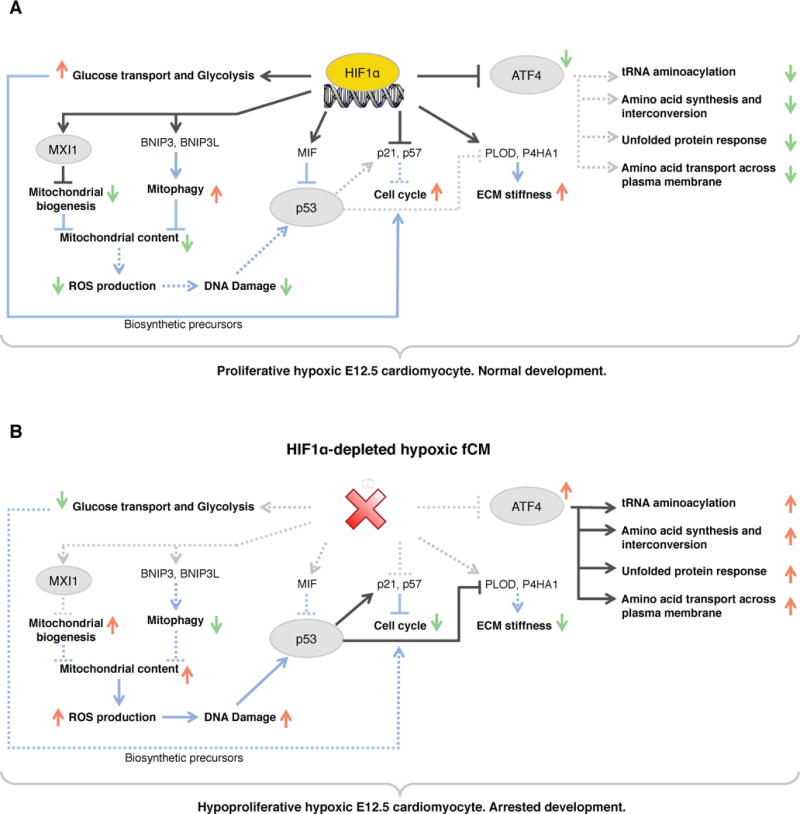
A) In hypoxic wild-type fCMs, HIF1α potentiates proliferation by regulating a myriad of cellular functions: carbohydrate metabolism, ECM deposition, OXPHOS, cell cycle, p53 and ATF4 signaling. B) In the absence of HIF1α, hypoxic fCMs failed to adapt their metabolism to low oxygen, ectopically activated ATF4 and p53 pathways and upregulated expression of inhibitors of cell cycle. As consequence, these cells adopted a quiescent phenotype that lead to arrested cardiac development. Red arrows represent upregulation and green arrows represent downregulation. TFs are circled and cellular processes are bold. Black lines represent transcriptional regulation and blue lines represent post-translational interactions or effects. Dashed lines represent inactive interactions. See Figure S5 for a detailed list of all genes involved in the cellular processes highlighted in this diagram.
p53 and proteins of the Retinoblastoma (RB) and E2F families play a critical role in controlling cell cycle. In the specific context of cardiogenesis, ectopic activation of p53 in fCMs results in hypoplasia and arrested development (Grier et al., 2006), whereas inactivation of RB and p130 results in excessive proliferation in adult heart (MacLellan et al., 2005). Our datasets revealed that HIF1α did not directly regulate transcription of genes encoding these proteins. However, HIF1α inhibited p53 post-translational activation by inducing MIF accumulation and by suppressing DNA damage (Figure 4E). These observations were supported by ELISA quantification of p53/DNA binding, revealing an inverse correlation between presence of HIF1α and p53 activity, both in vivo and in vitro. In primary cell cultures, knockout of Trp53 could restore proliferation of HIF1α depleted fCMs (Figure 5H), further suggesting that relative levels of active HIF1α and p53 exert rheostatic control over fCM proliferation. p53 blocks cell cycle, at least in part, by inducing expression of members of the CIP/KIP family. Interestingly, our analyses revealed that, in fCMs, HIF1α and p53 shared the genes encoding p21 and p57 as common targets, albeit regulating them in opposite directions. HIF1α bound to DNA in the vicinity of Cdkn1a (p21) and Cdkn1c (p57) and HIF1α depletion resulted in increased expression of these genes, demonstrating direct repression by HIF1α in fCMs. Previous reports have shown that p21/Cdkn1a is activated by HIF1α in human cancer cell lines (Koshiji et al., 2004), revealing that effects of HIF1α on Cdkn1a transcription are context dependent. p57 is abundant in trabecular fCMs of mid-gestational heart and absent from fCMs of the compact layer (Kochilas et al., 1999). Increased levels of p57 protein were evident in compact layer fCMs of Hif1a cKO hearts (Figure 4E), providing a direct link between HIF1α depletion and decreased fCM proliferation.
In hypoxia, mitochondrial oxidative phosphorylation is inefficient and generates ROS (Semenza, 2013). To avoid detrimental effects of these compounds, HIF1α establishes a gene expression program that promotes reduction of mitochondrial biomass, limits OXPHOS and prevents ROS production (Table S2G). fCMs of Hif1a cKOs had higher mitochondrial content and increased DNA damage, likely consequent to increased ROS production. Concomitantly to repressing OXPHOS, HIF1α promoted expression of glycolytic enzymes, allowing for sustained ATP production in hypoxia. It is widely accepted that proliferative cells preferentially use glycolysis for energy production, as this process generates metabolic products required for biosynthesis prior to cell division (Vander Heiden et al., 2009). Thus, blunted expression of glycolytic genes observed in Hif1a cKO hearts is likely to contribute to the observed hypoproliferative phenotype.
Another significant outcome of our study was to highlight critical interactions between HIF1α and the transcription factor ATF4. HIF1α and ATF4 are both activated by hypoxia and regulate downstream gene programs aimed at cellular adaptation to environmental stresses. The two TFs converge on the induction of angiogenesis, sharing Vegf as common target (Roybal et al., 2005), however, analysis of their downstream targets clearly indicates that their response strategies are distinct. Whereas HIF1α adapts energy metabolism to low oxygen preventing entry into a crisis situation, ATF4 works to mitigate the consequences of an ongoing metabolic crisis. To do so, ATF4 promotes expression of genes associated with functions such as protein folding, amino acid uptake and metabolism and activates pathways that promote biosynthesis of glutathione, a powerful reducing agent that neutralizes intracellular peroxides (Harding et al., 2003).
Our results demonstrated that, in hypoxic fCMs, HIF1α directly binds and represses expression of Atf4, exerting a brake on the crisis response. Hypoxia has previously been shown to activate ATF4 translation via PERK (Fels and Koumenis, 2006) but, to our knowledge, this is the first report of direct transcriptional regulation of Atf4 by HIF1α. Additionally, our analyses revealed enrichment in ATF4 binding sites within ChIP-seq peaks corresponding to genes repressed by HIF1α (Table S4) and suggested HIF1α binds and regulates a substantial proportion of genes activated downstream of ATF4 (Table S5E). These observations suggest a complex interplay between both TFs and, similar to the HIF1α/p53 dichotomy, detailed titration of the relative impact of HIF1α and ATF4 on common target expression will be the subject of future studies.
Hyperoxia has recently been shown to promote neonatal CM cell cycle withdrawal (Puente et al., 2014) and in zebrafish hypoxia and HIF1α are involved in CM proliferation post ventricular amputation (Jopling et al., 2012). Here, we have shown that HIF1α directly regulates energy metabolism, cell cycle, ECM deposition, p53 and ATF4 signaling to promote proliferation of a distinct subset of fCMs located within non-vascularized regions of the midgestational heart, reiterating that an environmental variable (oxygen) can play a critical role in the regulation of CM proliferation. Several adult heart pathologies are of an ischemic nature, and HIF1α has previously been shown to regulate angiogenesis in adult cardiac hypertrophy via induction of Vegf, a function countered by p53-induced degradation of HIF1α (Sano et al., 2007). It will be of future interest to study the extent to which the non-angiogenic pathways described here may play a role in such pathological conditions, to gain greater insight into the therapeutic potential of targeting HIF1α and intersecting pathways to harness the proliferative potential of adult hypoxic CMs.
Experimental Procedures
Animal procedures and phenotypic analyses
All animal care was in compliance with NIH guidelines and institutional guidelines at the University of California, San Diego. The floxed Hif1a allele (Ryan et al., 2000) and all other transgenic mouse lines used have been previously described and are listed in Supplemental Experimental Procedures. To ensure efficient recombination, the null Hif1aΔ allele, generated by germline ablation of the floxed allele, was included in all crosses. Standard immunohistochemistry techniques, described in detail in Supplemental Experimental Procedures, followed by confocal microscopy imaging were used for detection of endogenous HIF1α protein in cryosections of embryonic hearts and for phenotypic characterization of mutant hearts. Timing of coronary perfusion by aortic blood flow was determined by injection of fluorescent microspheres (Life Technologies) into the left ventricle of E13.5 and E14.5 embryos and assessment of microsphere presence within developing coronary vessels. Proliferation of fCMs in vivo and ex vivo was inferred by quantification of EdU (Life Technologies) incorporation. An unpaired Student’s t test was employed to assess statistical significance of quantitative differences between controls and Hif1a cKOs.
Bioinformatic analyses and Molecular Biology
The list of publically available microarrays used in our initial bioinformatics screen, as well as the strategy for clustering genes exhibiting similar expression dynamics can be found in Supplemental Experimental Procedures. Analysis of enrichment in known TF binding sequences in the promoter of genes grouped in each cluster was performed using Whole Genome RVista (Dubchak et al., 2013).
For trancriptome analyses, total RNA was extracted from E12.5 hearts using Trizol (Life Technologies). For each genotype, three independent replicates, each one corresponding to a single E12.5 heart, were sequenced on an Illumina HiSeq2000. Reads were mapped using TopHat2 and Bowtie2 and differentially expressed transcripts identified using AltAnalyze. For identification of genomic regions bound by HIF1α, nuclei were isolated from E12.5 hearts previously cross-linked in 1.5% formaldehyde. Cross-linked chromatin was sonicated to an average size of 200bp and DNA-HIF1α complexes precipitated using an anti-HIF1α antibody (Novus). 10 ng of ChIP DNA from two biological replicates were used to create standard Illumina sequencing libraries and processed for deep sequencing. Reads were mapped using Bowtie2 and peak calling performed using Homer. Extensive descriptions of these procedures and informatics analyses are provided in Supplemental Experimental Procedures. Resulting RNA-seq and ChIP-seq datasets can be accessed at GEO using accession numbers GSE61209 and GSE61247. ELISA evaluation of p53-DNA binding activity was performed using the TransAM p53 kit (Active Motif).
Isolation and culture of E12.5 ventricular cells
E12.5 mouse ventricles were dissociated into single cell suspensions using Trypsin and cultured in 17% or 3% oxygen. Quantification of fCM proliferation was achieved by flow cytometry using antibodies against TroponinT (Thermo Scientific) and phosphoHistone3 (Millipore). Gene knockout was achieved using cells isolated from homozygous floxed Hif1a or double homozygous floxed Hif1a;Trp53 embryos and treatment with Cre or LacZ (control) expressing adenoviruses. For gene knockdown, cells were transfected with siRNAs targeting Hif1a or Trp53 mRNAs using Dharmafect I Transfection Reagent (Dharmacon).
Supplementary Material
Acknowledgments
NGC received a doctoral fellowship (SFRH/BD/32983/2006) from the Portuguese Foundation for Science and Technology. This work was supported by an American Heart Association grant to ACZ (10SDG2630130), an NIH grant to ACZ and AM (PO1 HL098053-04), and NIH grants to MSK (DK092062 and DK094729), JC (R01HL66100 and R01HL106968) and SME (HL117649 and HL119967). Imaging was performed at the UCSD Neuroscience Microscopy Facility supported by the NIH grant P30 NS047101.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions
NGC, SME and ACZ were responsible for experimental design, data interpretation and manuscript elaboration. NGC, ACZ JS, IA, PC and LH performed experimental work. Bioinformatic analyses were done by ACZ and NS. RSJ, MSK, AM, JC, MN provided reagents and contributed to data interpretation.
References
- Denko NC. Hypoxia, HIF1 and glucose metabolism in the solid tumour. Nat Rev Cancer. 2008;8:705–713. doi: 10.1038/nrc2468. [DOI] [PubMed] [Google Scholar]
- Dubchak I, Munoz M, Poliakov A, Salomonis N, Minovitsky S, Bodmer R, Zambon AC. Whole-Genome rVISTA: a tool to determine enrichment of transcription factor binding sites in gene promoters from transcriptomic data. Bioinformatics. 2013;29:2059–2061. doi: 10.1093/bioinformatics/btt318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunwoodie SL. The Role of Hypoxia in Development of the Mammalian Embryo. Dev Cell. 2009;17:755–773. doi: 10.1016/j.devcel.2009.11.008. [DOI] [PubMed] [Google Scholar]
- Fels DR, Koumenis C. The PERK/eIF2alpha/ATF4 module of the UPR in hypoxia resistance and tumor growth. Cancer Bio & Ther. 2006;5:723–728. doi: 10.4161/cbt.5.7.2967. [DOI] [PubMed] [Google Scholar]
- Fingerle-Rowson G, Petrenko O, Metz CN, Forsthuber TG, Mitchell R, Huss R, Moll U, Muller W, Bucala R. The p53-dependent effects of macrophage migration inhibitory factor revealed by gene targeting. Proc Natl Acad Sci USA. 2003;100:9354–9359. doi: 10.1073/pnas.1533295100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilkes DM, Bajpai S, Chaturvedi P, Wirtz D, Semenza GL. Hypoxia-inducible factor 1 (HIF-1) promotes extracellular matrix remodeling under hypoxic conditions by inducing P4HA1, P4HA2, and PLOD2 expression in fibroblasts. J Bio Chem. 2013;288:10819–10829. doi: 10.1074/jbc.M112.442939. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Grier JD, Xiong S, Elizondo-Fraire AC, Parant JM, Lozano G. Tissue-specific differences of p53 inhibition by Mdm2 and Mdm4. Mol Cell Bio. 2006;26:192–198. doi: 10.1128/MCB.26.1.192-198.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han J, Back SH, Hur J, Lin YH, Gildersleeve R, Shan J, Yuan CL, Krokowski D, Wang S, Hatzoglou M, et al. ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat Cell Biol. 2013;15:481–490. doi: 10.1038/ncb2738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding HP, Zhang Y, Zeng H, Novoa I, Lu PD, Calfon M, Sadri N, Yun C, Popko B, Paules R, et al. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol Cell. 2003;11:619–633. doi: 10.1016/s1097-2765(03)00105-9. [DOI] [PubMed] [Google Scholar]
- Huang Y, Hickey RP, Yeh JL, Liu D, Dadak A, Young LH, Johnson RS, Giordano FJ. Cardiac myocyte-specific HIF-1α deletion alters vascularization, energy availability, calcium flux, and contractility in the normoxic heart. FASEB J. 2004;18:1138–1140. doi: 10.1096/fj.04-1510fje. [DOI] [PubMed] [Google Scholar]
- Hudson JD, Shoaibi MA, Maestro R, Carnero A, Hannon GJ, Beach DH. A proinflammatory cytokine inhibits p53 tumor suppressor activity. J Exp Med. 1999;190:1375–1382. doi: 10.1084/jem.190.10.1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jopling C, Sune G, Faucherre A, Fabregat C, Izpisua Belmonte JC. Hypoxia induces myocardial regeneration in zebrafish. Circulation. 2012;126:3017–3027. doi: 10.1161/CIRCULATIONAHA.112.107888. [DOI] [PubMed] [Google Scholar]
- Kenzelmann Broz D, Spano Mello S, Bieging KT, Jiang D, Dusek RL, Brady CA, Sidow A, Attardi LD. Global genomic profiling reveals an extensive p53-regulated autophagy program contributing to key p53 responses. Genes & Dev. 2013;27:1016–1031. doi: 10.1101/gad.212282.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilberg MS, Shan J, Su N. ATF4-dependent transcription mediates signaling of amino acid limitation. Trends in endocrinology and metabolism: TEM. 2009;20:436–443. doi: 10.1016/j.tem.2009.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kochilas LK, Li J, Jin F, Buck CA, Epstein JA. p57Kip2 expression is enhanced during mid-cardiac murine development and is restricted to trabecular myocardium. Ped Res. 1999;45:635–642. doi: 10.1203/00006450-199905010-00004. [DOI] [PubMed] [Google Scholar]
- Koshiji M, Kageyama Y, Pete EA, Horikawa I, Barrett JC, Huang LE. HIF-1alpha induces cell cycle arrest by functionally counteracting Myc. EMBO J. 2004;23:1949–1956. doi: 10.1038/sj.emboj.7600196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan J, Ahuja P, Bodenmann S, Knapik D, Perriard E, Krek W, Perriard JC. Essential Role of Developmentally Activated Hypoxia-Inducible Factor 1α for Cardiac Morphogenesis and Function. Circ Res. 2008;103:1139–1146. doi: 10.1161/01.RES.0000338613.89841.c1. [DOI] [PubMed] [Google Scholar]
- MacLellan WR, Garcia A, Oh H, Frenkel P, Jordan MC, Roos KP, Schneider MD. Overlapping roles of pocket proteins in the myocardium are unmasked by germ line deletion of p130 plus heart-specific deletion of Rb. Mol Cell Bio. 2005;25:2486–2497. doi: 10.1128/MCB.25.6.2486-2497.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miki N, Ikuta M, Matsui T. Hypoxia-induced activation of the retinoic acid receptor-related orphan receptor alpha4 gene by an interaction between hypoxia-inducible factor-1 and Sp1. J Bio Chem. 2004;279:15025–15031. doi: 10.1074/jbc.M313186200. [DOI] [PubMed] [Google Scholar]
- Puente BN, Kimura W, Muralidhar SA, Moon J, Amatruda JF, Phelps KL, Grinsfelder D, Rothermel BA, Chen R, Garcia JA, et al. The oxygen-rich postnatal environment induces cardiomyocyte cell-cycle arrest through DNA damage response. Cell. 2014;157:565–579. doi: 10.1016/j.cell.2014.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pugh CW, Ratcliffe PJ. Regulation of angiogenesis by hypoxia: role of the HIF system. Nat Med. 2003;9:677–684. doi: 10.1038/nm0603-677. [DOI] [PubMed] [Google Scholar]
- Roybal CN, Hunsaker LA, Barbash O, Vander Jagt DL, Abcouwer SF. The oxidative stressor arsenite activates vascular endothelial growth factor mRNA transcription by an ATF4-dependent mechanism. J Bio Chem. 2005;280:20331–20339. doi: 10.1074/jbc.M411275200. [DOI] [PubMed] [Google Scholar]
- Ryan HE, Poloni M, McNulty W, Elson D, Gassmann M, Arbeit JM, Johnson RS. Hypoxia-inducible Factor-1α Is a Positive Factor in Solid Tumor Growth. Cancer Res. 2000;60:4010–4015. [PubMed] [Google Scholar]
- Sano M, Minamino T, Toko H, Miyauchi H, Orimo M, Qin Y, Akazawa H, Tateno K, Kayama Y, Harada M, et al. p53-induced inhibition of Hif-1 causes cardiac dysfunction during pressure overload. Nature. 2007;446:444–448. doi: 10.1038/nature05602. [DOI] [PubMed] [Google Scholar]
- Schmid T, Zhou J, Brune B. HIF-1 and p53: communication of transcription factors under hypoxia. J Cell Mol Med. 2004;8:423–431. doi: 10.1111/j.1582-4934.2004.tb00467.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenza GL. HIF-1 mediates metabolic responses to intratumoral hypoxia and oncogenic mutations. J Clin Inv. 2013;123:3664–3671. doi: 10.1172/JCI67230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanimoto K, Tsuchihara K, Kanai A, Arauchi T, Esumi H, Suzuki Y, Sugano S. Genome-wide identification and annotation of HIF-1alpha binding sites in two cell lines using massively parallel sequencing. HUGO J. 2010;4:35–48. doi: 10.1007/s11568-011-9150-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welford SM, Bedogni B, Gradin K, Poellinger L, Broome Powell M, Giaccia AJ. HIF1alpha delays premature senescence through the activation of MIF. Genes & Dev. 2006;20:3366–3371. doi: 10.1101/gad.1471106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Gao P, Fukuda R, Kumar G, Krishnamachary B, Zeller KI, Dang CV, Semenza GL. HIF-1 inhibits mitochondrial biogenesis and cellular respiration in VHL-deficient renal cell carcinoma by repression of C-MYC activity. Cancer Cell. 2007;11:407–420. doi: 10.1016/j.ccr.2007.04.001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.